

Abstract
Molecular adaptation, as characterized by the detection of positive selection, was quantified in a number of genes from different human immunodeficiency virus type 1 (HIV-1) group M subtypes, group O, and an HIV-2 subtype using the codon-based maximum-likelihood method of Yang and coworkers (Z. H. Yang, R. Nielsen, N. Goldman, and A. M. K. Pedersen, Genetics 155:431-449, 2000). The env gene was investigated further since it exhibited the strongest signal for positive selection compared to those of the other two major HIV genes (gag and pol). In order to investigate the pattern of adaptive evolution across env, the location and strength of positive selection in different HIV-1 sequence alignments was compared. The number of sites having a significant probability of being positively selected varied among these different alignment data sets, ranging from 25 in HIV-1 group M subtype A to 40 in HIV-1 group O. Strikingly, there was a significant tendency for positively selected sites to be located at the same position in different HIV-1 alignments, ranging from 10 to 16 shared sites for the group M intersubtype comparisons and from 6 to 8 for the group O to M comparisons, suggesting that all HIV-1 variants are subject to similar selective forces. As the host immune response is believed to be the dominant driving force of adaptive evolution in HIV, this result would suggest that the same sites are contributing to viral persistence in diverse HIV infections. Thus, the positions of the positively selected sites were investigated in reference to the inferred locations of different epitope types (antibody, T helper, and cytotoxic T lymphocytes) and the positions of N and O glycosylation sites. We found a significant tendency for positively selected sites to fall outside T-helper epitopes and for positively selected sites to be strongly associated with N glycosylation sites.
A detailed appreciation of the extremely high diversity of human immunodeficiency virus (HIV), the causative agent of AIDS, has resulted from the extensive sequencing and phylogenetic analysis of viral genes and gene fragments over the last decade and a half (12). In addition, phylogenetic analysis of HIV and related simian immunodeficiency virus (SIV) strains has revealed a relatively recent simian origin for HIV (HIV type 1 [HIV-1] and HIV-2) from SIV-infected primates (6, 8). More specifically, the origin of HIV-2 is linked to SIVsm-infected sooty mangabeys in West Africa, and the origin of HIV-1 is linked to SIVcpz-infected chimpanzees in Central Africa. In the case of HIV-1, at least three independent cross-species transmission events need to be postulated to account for the three most divergent HIV-1 lineages (designated groups M, N, and O), whereas seven independent events are required to account for the seven HIV-2 lineages (designated subtypes A to G) (8).
Within HIV-1 group M, nine major subtypes (A to D, F to H, J, and K) have been designated, as have 14 circulating recombinant forms (CRF01 to CRF14) (12, 24). Interestingly, recent studies have identified diversity within HIV-1 group O equivalent to that exhibited by group M (25, 33), despite the fact that almost all group O infections are restricted to Cameroon or to individuals with strong links to that region. Although there is phylogenetic substructure within group O phylogenies, distinct group M-like subtypes are not apparent (25). This is not too surprising, given that the prominence of the group M subtypes is strongly linked to founder events in the course of the HIV-AIDS pandemic that occurred outside the Democratic Republic Congo region (23). Analogous founder events have not occurred in the case of group O, as these types of infection have remained strongly associated with one geographic location, Cameroon. The third HIV-1 group, N, also remains restricted to Cameroonian residents, and to date only five infections have been conclusively documented (3).
The development of candidate vaccines specific to different HIV lineages (7) demands a thorough investigation of the consistency of the selective environment, which is presumed to be due primarily to the host immune responses (15, 22, 39) to divergent HIVs. Evidence for adaptive evolution has been found previously among HIV sequences from intra- and interpatient studies (4, 29, 30, 38, 40). Early studies involved the pairwise comparison of synonymous (silent, dS) and nonsynonymous (amino acid changing, dN) substitutions between protein-coding DNA sequences. The dN/dS ratio, ω, was then used to measure the difference between these two rates of substitution such that an ω value less than 1 corresponds to purifying (negative) selection, an ω value of 1 corresponds to neutral evolution (absence of selection), and an ω value greater than 1 indicates adaptive evolution (positive selection) (reviewed in reference 37). The pairwise approach to quantifying adaptive evolution assumes that all sites are prone to the same selective pressure, making such tests very conservative. In reality, positively selected sites normally occur in a background of negatively selected sites within a functional protein.
The problem of resolving positively selected sites against this background of negative selection has been solved in a maximum-likelihood (ML) and Bayesian statistical framework (for a review, see reference 37). First, the ML method determines whether positive selection is present by evaluating a series of models with or without a class of positively selected sites. Second, if the favored model includes positive selection, a Bayesian analysis assigns each amino acid site a “posterior probability” of being conserved, neutral, or positively selected.
Here, we focus on positively selected sites that were inferred by using the codon-based method (38), and we determine the extent to which their locations and the intensity of their selection overlap among different HIV lineages. We first quantified positive selection in the major HIV genes (gag, pol, and env) for the three HIV-1 group M subtypes (A, B, and C) and for HIV-2 subtype A. Since env exhibited the strongest signal for positive selection, the location of sites in env with a high probability of being under positive selection was compared across different HIV data sets corresponding to sequence alignments of HIV-1 group M subtypes A through D, group O, and an HIV-2 subtype. The hypothesis that phylogenetically divergent HIV lineages are subject to similar selective pressures was tested by determining whether the occurrence of positively selected sites at the same locations was statistically significant and whether the strength of selection was similar. On the assumption that sites are positively selected primarily as a consequence of pressure from the immune system (15, 22, 39), our results have some interesting consequences for vaccine design, as they suggest the possibility of cross-subtype and -group immunogenicity. We investigated whether the immune response, as represented by experimentally defined epitopes or the positions of N and O glycosylation (13, 28), could account for the observed distribution of the positively selected sites. We found a significant tendency for positively selected sites to fall outside T-helper epitope regions and for positively selected sites to be strongly associated with N glycosylation sites.
MATERIALS AND METHODS
Data sets.
The data sets used in this computer-based study each correspond to a sequence alignment for a given genomic region (gag, pol, or env) and HIV group or subtype. A total of 22 data sets were analyzed and named A through V (Table 1) for convenience. Most of the data sets were retrieved as an alignment of sequences from the 2000 release of the Los Alamos National Laboratory HIV Sequence Database (12), except for the group O sequences composing data set M, which was retrieved directly from GenBank (33) and aligned with CLUSTALW (http://www.ebi.ac.uk/clustalw). Known intersubtype recombinants, gap-containing sites, and stop codons were excluded (17) from each data set. Moreover, since the models used for positive selection analysis are codon based and assume that a synonymous substitution is always synonymous, all portions of the data set consisting of overlapping reading frames were excluded. The 22 data sets used in this study (Table 1) are the data sets for which enough sequences and sites were available for effective selection analysis (1, 2).
TABLE 1.
Data sets used in this studya
| Data set | Lineage | No. of seq. | No. of codons | Gene | Source |
|---|---|---|---|---|---|
| A | HIV-1 M:A | 11 | 404 | gag | LANL |
| B | HIV-1 M:B | 35 | 425 | gag | LANL |
| C | HIV-1 M:C | 17 | 418 | gag | LANL |
| D | HIV-2 A | 12 | 386 | gag | LANL |
| E | HIV-1 M:A | 13 | 838 | pol | LANL |
| F | HIV-1 M:B | 33 | 913 | pol | LANL |
| G | HIV-1 M:C | 16 | 911 | pol | LANL |
| H | HIV-2 A | 12 | 916 | pol | LANL |
| I | HIV-1 M:A | 16 | 578 | env | LANL |
| J | HIV-1 M:B | 30 | 578 | env | LANL |
| K | HIV-1 M:C | 30 | 578 | env | LANL |
| L | HIV-1 M:D | 15 | 578 | env | LANL |
| M | HIV-1 O | 30 | 621 | env | GenBank |
| N | HIV-2 A | 22 | 679 | env | LANL |
| O | HIV-1 M:A | 20 | 415 | env-gp120 | LANL |
| P | HIV-1 M:B | 20 | 433 | env-gp120 | LANL |
| Q | HIV-1 M:C | 20 | 423 | env-gp120 | LANL |
| R | HIV-2 A | 20 | 460 | env-gp120 | LANL |
| S | HIV-1 M:A | 19 | 232 | env-gp41 | LANL |
| T | HIV-1 M:B | 30 | 233 | env-gp41 | LANL |
| U | HIV-1 M:C | 30 | 237 | env-gp41 | LANL |
| V | HIV-2 A | 22 | 193 | env-gp41 | LANL |
Each data set is an alignment of nucleotide sequences of a given HIV subtype or group and a given gene. The number of sequences (No. of seq.) and sites (No. of codons) in each alignment are indicated as well as the source: the 2000 release of the Los Alamos National Laboratory (LANL) HIV Sequence Database (12) and GenBank (33). Positive selection was analyzed for each of the data sets. Statistical analyses on the positively selected sites were performed for the env data sets (I to N).
Selection analyses.
Positive selection analysis was performed on each of the 22 data sets in Table 1. For each data set, the PAUP* package (27) was first used to build an ML tree for selection analysis using the HKY85+Γ model of nucleotide substitution with optimal values for the TS/TV rate ratio and the shape parameter (α) of a gamma distribution (with eight categories) of rate variation among sites, both determined during tree construction. The ML method of Yang and coworkers (38) utilized codon-based models that incorporate statistical distributions to account for variable ω ratios among codons. Efficient determination of sites under positive selection requires implementation of only six models of codon substitution (M0, M1, M2, M3, M7, and M8) out of the original 14 models (for further details, see reference 38 and http://www.bioinf.man.ac.uk/∼robertson/supplementary-material [appendix A]). Briefly, null models M0, M1, and M7 do not allow for the existence of positively selected sites because ω ratios are fixed or estimated between the bounds 0 and 1, whereas models M2, M3, and M8 account for positive selection by using parameters that estimate ω to be greater than 1. The significance of positive selection can be confirmed with a likelihood ratio test (LRT) between null models and those able to account for positive selection. An LRT is performed by taking twice the difference in log likelihood between nested models and comparing the result to a χ2 distribution with degrees of freedom equivalent to the difference in the number of parameters between the models. Models M0 and M1 are both nested with M2 and M3, M2 is nested with M3, and M7 is nested with M8. All the model comparisons (M0 versus M2, M1 versus M2, M0 versus M3, M1 versus M3, M2 versus M3, and M7 versus M8) gave similar results, and for the sake of simplicity we focus on the results of models M7 and M8. M7 uses a discrete (10 classes) beta distribution to model sites with ω ratios between the bounds 0 and 1. For each class i (1 ≤ i ≤ 10) of the beta distribution, the value of the ωi ratio and the proportion (pi) of sites belonging to this class are estimated by maximizing the likelihood. M8 adds two additional parameters to model M7 such that p11 can account for a positively selected class of sites where ω11 is not constrained by the beta distribution and is allowed to be greater than 1. Once positively selected sites have been shown to exist, i.e., if model M7 is rejected in favor of M8 by the LRT, a Bayesian approach (for which the p1 to p11 values are used as a prior distribution) is used to infer the posterior probability that site i belongs to one of the 11 ω classes: 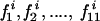 . Models were implemented using the CODEML program of the PAML package, version 3.1 (36).
. Models were implemented using the CODEML program of the PAML package, version 3.1 (36).
Statistical analysis of sites identified as positively selected.
A “shared-position” statistic and Monte Carlo simulations were used to test whether putative positively selected sites (defined as those having a p11 value of greater than 0.95 when ω11 is greater than 1 for model M8) tend to occur at the same positions in data sets I to N (H1) more often than would be expected by chance (H0). The shared-position statistic used is the count of the match between the positions of positively selected sites in one data set and the positions of positively selected sites in another data set. As this test depends on the quality of the alignment among the diverse data sets, the result should be conservative.
To study the “strength” of positive selection, we defined for each site, i, the weighted mean ω value as 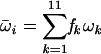 as previously implemented (7). For eachk=1 pair of data sets, we tested whether the strength of positive selection was significantly different (H1), as opposed to being equivalent (H0), by using a paired Wilcoxon rank sum test with a continuity correction applied to the normal approximation for the P values (26). Only shared sites having a weighted mean ω value greater than 1 in the two data sets being compared were included. Note that the positively selected sites with a weighted ω value greater than 1 are not necessarily identified as positively selected by model M8 at the 95% level. The latter sites identified at the 95% level by M8 will be a subset of the former weighted sites. The paired Wilcoxon rank sum test was repeated only for those shared sites identified by M8 at the 95% level.
as previously implemented (7). For eachk=1 pair of data sets, we tested whether the strength of positive selection was significantly different (H1), as opposed to being equivalent (H0), by using a paired Wilcoxon rank sum test with a continuity correction applied to the normal approximation for the P values (26). Only shared sites having a weighted mean ω value greater than 1 in the two data sets being compared were included. Note that the positively selected sites with a weighted ω value greater than 1 are not necessarily identified as positively selected by model M8 at the 95% level. The latter sites identified at the 95% level by M8 will be a subset of the former weighted sites. The paired Wilcoxon rank sum test was repeated only for those shared sites identified by M8 at the 95% level.
Finally, Monte Carlo simulations were again used to test a null hypothesis (H0) that sites of positive selection are not associated with the positions of epitope regions, or sites of glycosylation, against the alternative hypotheses (H1) that the positively selected sites are associated with the location of the epitope regions (or various combinations of the three types of regions) or the positions of the glycosylation sites in the different data sets. An additional hypothesis (H2) that the positive selected sites tend to fall outside the defined epitope regions (or various combinations of the three types of regions) was also tested against H0. The epitope regions are experimentally defined and correspond to antibody (Ab), cytotoxic T-cell (CTL), and helper T-cell immune response data available from the Los Alamos National Laboratory HIV Immunology Database (11). As the majority of epitope mapping has focused on subtype B-infected individuals (11), only the positively selected sites identified in data set J were tested. For each data set, the positions of the N and O glycosylation sites were predicted using the NetNGlyc (R. Gupta, E. Jung, and S. Brunak, unpublished data) and NetOGlyc (9) programs, respectively.
For all Monte Carlo simulations, 9,999 repetitions proved to be enough to reach an asymptotic state. The programs used to implement the Monte Carlo simulations are available upon request from M. Choisy.
RESULTS
Mean ω values for gag, pol, and env.
The results for the mean ω values (assuming the same value for ω at all sites) for the genes gag, pol, and env, and for the individual subunits of env (gp120 and gp41), are shown for HIV-1 group M subtypes A, B, and C and for HIV-2 subtype A in Fig. 1. Except for the group M subtype A, B, and C results for gp120 and subtype B for gp41, all ω values are less than 1, indicating that the majority of sites are subject to purifying selection. The effect of purifying selection is particularly strong in the gag and pol genes but is much weaker in the envelope region, which is not surprising given that env codes for the envelope surface proteins, which are the most exposed to the immune system. Note that despite the low mean ω values in the gag and pol genes, positive selection can still occur at a minority of sites, but this signal can be averaged out by M0 and pairwise methods. For example, others have previously found a comparable ω value (0.196) for the pol gene of a subtype B alignment as well as strong evidence for adaptive evolution (38). The contrast in mean ω ratios between gag and pol compared to that of the env regions indicates that the env region contains more positively selected sites than do the other genes. Within the env region, positive selection appears to be particularly strongly associated with the gp120 subunit, coding for the extramembrane envelope protein.
FIG. 1.

Mean ω ratios in gag, pol, env, env-gp120 and env-gp41 for HIV-1 group M subtypes A, B, and C, and HIV-2 subtype A (data sets A to K and N to V in Table 1). The mean ω ratios are calculated by averaging the results over all of the sites and are obtained from model M0. The numbers above the bars indicate the number of sequences and the number of codons in each data set. For example, “11/404” above the first gag bar indicates that there were 11 sequences and 404 codons in the gag HIV-1 group M subtype A data set (called data set A in Table 1).
Identification of positively selected sites across env.
A comparative analysis of HIV-1 group M subtypes A, B, C, and D; group O; and HIV-2 subtype A in the envelope region (data sets I to N in Table 1) was carried out in order to identify specific positively selected sites. All models that were able to detect positive selection (M2, M3, and M8) identified a positively selected class (ω > 1) and rejected those models that were unable to account for positive selection (M0, M1, and M7). For the sake of clarity, only results for M8 are presented in Table 2 (results for the other models are available from http://www.bioinf.man.ac.uk/∼robertson/supplementary-material (appendixes B and C). M2 and M8 identified the same positively selected sites when taking posterior probabilities greater than the 95% level using the Bayesian approach. M3 identified all of the sites identified by M2 and M8 and several more. We consider the sites identified by M8 only, as M3 has the potential to overestimate the number of positively selected sites (2, 38). The number of positively selected sites identified by model M8 was 22 for HIV-2 subtype A, between 30 and 35 for HIV-1 M subtypes, and 40 for HIV-1 O (Table 2). Figure 2 shows the location of these putative positively selected sites across the multiple alignment of data sets I to N. Positively selected sites are not restricted to the variable regions (V1 to V5) of env, a finding that supports previous work that used a maximum-parsimony-based method to identify amino acid sites that were potentially under the influence of positive selection in an HIV-1 subtype B alignment of sequences (35).
TABLE 2.
Positive selection in the env genea
| Data set | Lineage | Mean ω | 11th class | No. of sites | P |
|---|---|---|---|---|---|
| I | HIV-1 M:A | 0.690 | 4.702 | 33 | <0.001 |
| J | HIV-1 M:B | 0.623 | 4.009 | 35 | <0.001 |
| K | HIV-1 M:C | 0.610 | 4.463 | 33 | <0.001 |
| L | HIV-1 M:D | 0.568 | 3.821 | 30 | <0.001 |
| M | HIV-1 O | 0.590 | 3.992 | 40 | <0.001 |
| N | HIV-2 A | 0.444 | 3.568 | 25 | <0.001 |
Mean ω was calculated by averaging over all the sites. The 11th class is from model M8, and the number of sites refers to those found to be under positive selection over the 95% level. P is the probability resulting from the likelihood ratio test between M7 and M8. Significant results (P < 0.05) are indicated in boldface type.
FIG. 2.

Positions of positively selected sites across env for HIV-1 group M subtypes A, B, C, and D; group O; and HIV-2 subtype A (data sets I to N in Table 1). Each data set analyzed by CODEML is represented by one sequence, with the sites included in the analysis indicated with boldface type. Sites identified as being positively selected with a posterior probability of more than 95% are shaded. Notations above the sequences divide env into the gp120 and gp41 subunits and show the position of vpu and the second rev exon with the beginning and end of regions. The positions of the five variable regions V1 to V5 are indicated. Sites critical for CD4 binding are identified by a vertical bar, and sites implicated in the CXCR4 to CCR5 receptor switch are indicated with an * above the sequences. The numbers 2, 3, and 4 below the sequences indicate the number of data sets for which positive selection was identified at that site. The representative sequences for HIV-1 group M subtypes A, B, C, and D; group O; and HIV-2 subtype A are MA246, MBC18, BU910112, 84ZR085, ANT70, and CBL21, respectively.
Comparison of the locations of positively selected sites.
The null hypothesis that there is no association of the position of sites of positive selection among data sets I to N was rejected by using the shared-position statistic and Monte Carlo simulations for the majority of pairwise comparisons (Table 3). The exception was HIV-2 subtype A, which showed only a significant association with the position of positively selected sites with the HIV-1 group M subtype A data set. This result indicates that different HIV-1 group M subtypes and group O contain sites in env that are under similar selective pressures.
TABLE 3.
Monte Carlo simulations testing the association of sites of positive selection between data setsa
| Data set | Lineage and value type | Values for data set and lineage
|
||||
|---|---|---|---|---|---|---|
| I | J | K | L | M | ||
| HIV-1 M:A | HIV-1 M:B | HIV-1 M:C | HIV-1 M:D | HIV-1 O | ||
| J | HIV-1 M:B | |||||
| E | 2.018 | |||||
| O | 13 | |||||
| P | 0.001 | |||||
| K | HIV-1 M:C | |||||
| E | 1.990 | 2.015 | ||||
| O | 16 | 15 | ||||
| P | 0.001 | 0.001 | ||||
| L | HIV-1 M:D | |||||
| E | 1.760 | 1.793 | 1.741 | |||
| O | 10 | 14 | 15 | |||
| P | 0.001 | 0.001 | 0.001 | |||
| M | HIV O | |||||
| E | 1.016 | 0.996 | 0.889 | 0.848 | ||
| O | 7 | 7 | 8 | 6 | ||
| P | 0.001 | 0.001 | 0.001 | 0.001 | ||
| N | HIV-2 A | |||||
| E | 0.633 | 0.722 | 0.571 | 0.511 | 0.729 | |
| O | 3 | 1 | 2 | 2 | 1 | |
| P | 0.024 | 0.535 | 0.104 | 0.091 | 0.539 | |
E, expected value from a random distribution; O, observed value; and P, level of significance at which E is different from O. Significant results (P < 0.05) are indicated in boldface type.
Comparison of the strength of positive selection.
The result just described seemingly contradicts the finding by Gaschen and coworkers (7) that group M subtypes B and C undergo different evolutionary pressures in the C2V3 region of env; this result is presumed to be due to different antigenic exposure patterns being exhibited by different subtypes. To investigate this possibility further, we plotted for each of the I to N data sets the weighted ω ratio for each site (see Materials and Methods) that had a value greater than 1 (Fig. 3). When sites that had a weighted ω value greater than 1 were tested among different data sets with a paired Wilcoxon ranked sum test (Table 4), the comparison was significant for the strength of selection differing between HIV-1 subtypes B and C, a result that is in agreement with that of Gaschen and coworkers (7). This was also the case for the comparison between HIV-1 subtypes A and B and between HIV-1 group O and HIV-2 subtype A. However, no other comparisons were significant, indicating that generalizations about differences in the strength of selection between diverse HIV data sets should not be made based on the available data. For the subset of sites with a weighted ω value greater than 1 and that were identified as positively selected by model M8 at the 95% level, the comparisons between HIV-1 subtypes A and B, B and C, and between HIV-1 group O and HIV-2 were significant (P = 0.0001, 0.0282, and 0.0156, respectively; data available at http://www.bioinf.man.ac.uk/∼robertson/supplementary-material [appendix D]).
FIG. 3.

The weighted mean ω ratio greater than 1 at each codon position in the env data sets comparing HIV-1 group M subtypes A, B, C, and D; group O; and HIV-2 subtype A (data sets I to N in Table 1). The weighted mean ω value for each site is calculated by multiplying ω by the posterior probability for each class under M8 and summing the results (see Materials and Methods). The positions of the five variable regions V1 to V5 are indicated.
TABLE 4.
Paired Wilcoxon ranked sum test to determine differences in the strength of positive selection between data setsa
| Data set | Lineage | Values for data set and lineage
|
||||
|---|---|---|---|---|---|---|
| I | J | K | L | M | ||
| HIV-1 M:A | HIV-1 M:B | HIV-1 M:C | HIV-1 M:D | HIV-1 O | ||
| J | HIV-1 M:B | |||||
| Z | 3.8266 | |||||
| N | 69 | |||||
| P | 0.0001 | |||||
| K | HIV-1 M:C | |||||
| Z | 1.2150 | −2.1945 | ||||
| N | 67 | 62 | ||||
| P | 0.2244 | 0.0282 | ||||
| L | HIV-1 M:D | |||||
| Z | 0.6009 | −1.1021 | −0.4077 | |||
| N | 46 | 54 | 51 | |||
| P | 0.5479 | 0.2704 | 0.6835 | |||
| M | HIV-1 O | |||||
| Z | 0.3652 | −1.853 | −1.0934 | 0.0000 | ||
| N | 23 | 22 | 26 | 18 | ||
| P | 0.7149 | 0.0639 | 0.2742 | 1.0000 | ||
| N | HIV-2 A | |||||
| Z | −0.4001 | −1.0193 | 1.8347 | 0.8293 | 2.2819 | |
| N | 11 | 10 | 10 | 9 | 7 | |
| P | 0.6891 | 0.3081 | 0.0665 | 0.4069 | 0.0225 | |
Z is the statistic and N is the number of sites with ω greater than one. Significant differences (P < 0.05) are indicated in boldface type. A continuity correction is applied to the normal approximation for the P values.
Association of positively selected sites with epitope regions and glycosylation sites.
None of the Monte Carlo tests used to investigate whether the positively selected sites of the env gene of subtype B (data set J in Table 1) had a tendency to be associated with experimentally defined Ab, CTL, T-helper epitopes, or combinations of these were significant (Table 5). However, the reciprocal investigation, which tested whether positively selected sites had a tendency to fall between the different epitope regions, was significant for the T-helper and CTL-T-helper combination (P < 0.05), while the significance of CTL alone was marginal (P = 0.053) (Table 5).
TABLE 5.
Correlation of positively selected sites with epitopes in the env gene of HIV-1 group M subtype B (data set J)a
| Epitope(s) | NINb | OINc | EINd | PINe | NOUTf | OOUTg | EOUTh | POUTi |
|---|---|---|---|---|---|---|---|---|
| Ab | 370 | 18 | 22.17 | 0.946 | 208 | 17 | 12.53 | 0.072 |
| CTL | 394 | 19 | 23.82 | 0.976 | 184 | 16 | 11.12 | 0.053 |
| Th | 499 | 26 | 30.16 | 0.989 | 79 | 9 | 4.78 | 0.028 |
| Ab and CTL | 507 | 30 | 30.64 | 0.726 | 71 | 5 | 4.17 | 0.401 |
| Ab and Th | 537 | 33 | 32.40 | 0.496 | 41 | 2 | 1.90 | 0.612 |
| CTL and Th | 524 | 27 | 31.67 | 0.999 | 54 | 8 | 2.92 | 0.018 |
The first three rows correspond to the three epitope types analyzed separately (Ab, antibody; CTL, cytotoxic T-cell and Th, T-helper responses), and the remaining rows refer to combinations of these epitope types analyzed together.
Number of sites targeted by epitopes from the HIV Immunology Database.
Observed number of identified positively selected sites that fall inside the epitope regions.
Expected number of positively selected sites in the epitope regions as calculated by Monte Carlo simulations.
Significance level at which OIN differs from EIN.
Number of sites that have not been identified in the HIV Immunology Database to be targeted by an epitope.
Observed number of identified positively selected sites that fall outside the epitope regions.
Expected number of positively selected sites that fall outside the epitope region as calculated by Monte Carlo simulations.
Significance level at which OOUT differs from EOUT, significant values (P < 0.05) are indicated in boldface type.
For the test of the association of N glycosylation sites identified in each HIV-1 data set (ranging from 22 to 39) with the identified positively selected sites, a significant association (P < 0.05) was found for all comparisons (Table 6). Note that N glycosylation sites were not identified in the HIV-2 data set. Between two and six of the N glycosylation sites were conserved in all sequences of the HIV-1 data sets. The number of O glycosylation sites for the HIV-1 M:A, HIV-1 M:B, HIV-1 M:C, HIV-1 M:D, group O, and HIV-2 A data sets was 2, 2, 4, 1, 8, and 0, respectively. No associations (P ≥ 0.05) were found for the test of the association of O glycosylation sites identified in each HIV-1 data set with the putative positively selected sites (results not shown).
TABLE 6.
Association of positively selected sites with sites of N glycosylation in env
| Data set | Lineage | No. of sites of N glyca | No. of conserved N glycb | No. observedc | No. expectedc | P valued |
|---|---|---|---|---|---|---|
| I | HIV-1 M:A | 28 | 5 | 11 | 1.64 | 0.001 |
| J | HIV-1 M:B | 27 | 2 | 5 | 1.62 | 0.019 |
| K | HIV-1 M:C | 30 | 2 | 7 | 1.69 | 0.002 |
| L | HIV-1 M:D | 22 | 5 | 4 | 1.17 | 0.023 |
| M | HIV-1 O | 39 | 6 | 13 | 2.46 | 0.001 |
Total number of N glycosylation sites in the data set.
Number of N glycosylation sites that are conserved across all sequences of the data set.
The observed number of associations between positively selected sites and sites of N glycosylation is compared to the expected number of associations between positively selected sites and those of N glycosylation (as calculated from the mean of the Monte Carlo simulated distribution).
Significant results (P < 0.05) are indicated in boldface type.
Finally, the locations of sites implicated in the binding of the envelope glycoprotein to the CD4 receptor molecules (18, 32) and of sites implicated in the receptor switch from CCR5 to CxCR4 tropism (20) are indicated in Fig. 2, and neither associates with any of the positively selected sites identified. The finding that sites involved in chemokine binding are apparently not under the influence of positive selection may be due to our comparison of viral sequence data from several different infected individuals rather than from the viral population of one individual, while CD4 binding sites are presumably under the influence of purifying selection.
DISCUSSION
As vaccine candidates are being designed to target different HIV-1 group M subtypes, it is important to investigate how the immune system responds to different HIV-1 strains. Assuming that the immune response is providing the evolutionary pressure for the majority of adaptive evolution observed in the HIV genome (15, 22, 39), we have quantified positive selection in HIV-1 group O, different group M subtypes, and HIV-2 subtype A sequence alignments. The majority of positive selection was found to occur in the envelope region of the genome as opposed to the gag or pol (Fig. 1) region, thereby confirming the results of previous studies (4, 30, 34, 35).
Further analysis of env revealed that a proportion of the sites that were identified as positively selected (ranging from 25 in the HIV-1 M group subtype A data set to 40 in the HIV-1 group O data set) were at the same positions in the different data sets (Fig. 2). We believe that only the immune response could be driving this propensity of HIV to exhibit adaptive molecular evolution to such an extent. Furthermore, for the HIV-1 group M comparisons, between 10 and 16 sites were shared depending on the subtypes compared (Table 3). On the assumption that the immune response provides the evolutionary pressure for amino acid change at these sites (15, 22, 39), the finding that positively selected sites are shared between divergent HIV-1 lineages suggests that the immune response may be targeting the same viral regions in the different groups and subtypes, thus raising the possibility of cross-subtype or -group immunogenicity.
However, it has been reported previously (7) that the strength of selection at positively selected sites in the C2V3 region of group M subtypes B and C is different. We made the same observation here, not only for the C2V3 region but also for the entire env gene, and we moreover show that the finding is statistically significant (Table 4). Importantly, we found that (i) there is a tendency for the position of positively selected sites to be correlated among different HIV-1 data sets and that (ii) there can be, at the same time, a difference in the strength of selection for some comparisons; these findings are not mutually exclusive. This is true because selection may be acting at the same sites but to differing extents, or, alternatively, the different strengths of selection might be predominantly at the sites that are not correlated among the different data sets. Nevertheless, as most comparisons of the strength of selection are not significant, generalizations about differences among diverse HIV data sets should not be made based on the available data. Indeed, differences in the strength of selection may be due to other factors such as the predominant form of transmission in a given subtype or the amount of diversity in that subtype.
To explicitly test the assumption that the majority of adaptive evolution observed in the HIV envelope is due to the immune response, we then investigated the potential of the different types of immune response (Ab, CTL, and T helper) to account for the location of the positively selected sites. This comparison is relatively crude because the epitopes that can be recognized in different individuals and their frequencies in different populations will vary. Ideally, viral sequences would be analyzed that are from a distinct population in conjunction with information concerning the types of epitope that could be recognized, as has been done for comparisons between HLA haplotypes and polymorphisms present in viral sequences (15). Also, some of the positively selected sites may be relevant to nonlinear epitopes, which are difficult to detect since they are formed by protein tertiary structure bringing distant sites into proximity. For example, a “glycan shield” model (28) has been proposed, which suggests that linear epitopes in regions essential for viral fitness and that are unable to tolerate mutation can be protected from neutralizing antibodies by the bound carbohydrates. Mutations of gp120 would cause the permanent rearrangement of the carbohydrates, thus creating a moving protective shield around epitopes that are unable to tolerate mutation, as they are in functionally conserved regions. Despite these limitations, a significant result was found (Table 5) for the tendency of the T-helper and possibly for the CTL epitope regions to not include positively selected sites. This result might be explained by a finding that CTL epitopes are more concentrated in relatively conserved regions across the HIV genome, whereas positively selected sites will have a tendency to be detected in the more variable regions (39). Alternatively, positively selected sites may correspond to proteolytic cleavage sites such that mutation in the epitope-flanking residues alters intracellular processing, thereby permitting CTL escape (39).
We also investigated the predicted positions of the N glycosylation sites with respect to the positions of the identified positively selected sites. The significance of N glycosylation sites is that they allow the binding of carbohydrates to the viral envelope to mask viral protein epitopes from the immune response (5, 19, 28, 31). The bound carbohydrates are large molecules contributing to half of the molecular mass of gp120 (5) and that are linked to the gp120 protein on N glycosylation sites, and, to a lesser extent, sites of O glycosylation. They are thought to play an important role for the stability of the gp120 molecule (19), for CCR5 and CXCR4 coreceptor utilization (21), and for escape from the immune defense (5, 10). The relatively rapid turnover of mutations of the gp120 protein may also induce continual conformational changes, and such a constantly moving structure may help to distort epitopes and prevent antibody binding (13). In accordance with previous reports (5), between 22 and 39 N glycosylation sites were predicted (Table 6) for the different HIV-1 data sets. When the N glycosylation sites present in the HIV-1 data sets were considered, Monte Carlo simulations indicated that these sites are significantly associated with putative positively selected sites. These findings of a correlation among positively selected sites but not of the location of Ab epitope regions is consistent with the glycan shield model of viral escape (28). Interestingly, no N glycosylation sites were detected in the HIV-2 data set, despite glycosylation for HIV-2 being previously reported (14). This finding seems to reflect the very low number of such sites in HIV-2 strains. HIV-2 is apparently less virulent than HIV-1 (8), possibly due to HIV-2 being less antigenic, thus accounting for the lack of N glycosylation sites.
In our opinion, potential correlations of adaptive molecular evolution among divergent HIVs, such as those we have detected here, warrant further investigation, as they are indicative of possible shared antigenicity. Given the unquestionable need for an HIV-AIDS vaccine to elicit an immune response against multiple group M subtypes, specifically in Africa where multiple subtypes frequently cocirculate, a hypothetical vaccine cocktail that would include antigens from a number of genomic regions is clearly worth investigating. The present preoccupation with consensus and ancestral sequences targeted at an individual subtype as optimal immunogens (for an example, see references 7 and 16) make limited or no specific attempts to elicit immune responses that may be cross-reactive to different subtypes. In addition, the most antigenic viral regions will be embedded in sequence of differing immunogenic potential. Thus, there is a possibility that constructing a consensus sequence from the genetic material of circulating viruses would result in the least optimal antigenic regions being included in the vaccine. This result would occur because the consensus sequence would represent optimal genetic material for immune escape because it would have sequences from multiple viruses that have successfully escaped the immune response. Furthermore, neither a consensus sequence nor a reconstructed ancestral sequence (due to ongoing recombination within individuals [with or without superinfection] and positive selection resulting in escape mutants with the same convergent amino acid changes) can represent any virus that has ever existed and so may lack important properties that could be of immunogenic importance in a potential vaccine (for example, folding). In conclusion, if we are to control HIV, we must understand its evolution and conceive appropriate intervention strategies accordingly.
Acknowledgments
We thank Bénédicte Lafay, Andrew Rambaut, Simon Lovell, Jay Taylor, Mike Worobey, and Eddie Holmes for helpful comments and discussion.
We also thank the Wellcome Trust (which provided assistance through their Biodiversity program while D.L.R. was at the Department of Zoology, University of Oxford, where this work was begun), the National Institutes of Health (AIDS training grant number AI07384), and the CNRS for funding (M.C. is supported by a Bourse Docteur Ingénieur from the CNRS-Région Languedoc Roussillon).
REFERENCES
- 1.Anisimova, M., J. P. Bielawski, and Z. Yang. 2002. Accuracy and power of the Bayes prediction of amino acid sites under positive selection. Mol. Biol. Evol. 19:950-958. [DOI] [PubMed] [Google Scholar]
- 2.Anisimova, M., J. P. Bielawski, and Z. Yang. 2001. Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol. Biol. Evol. 18:1585-1592. [DOI] [PubMed] [Google Scholar]
- 3.Ayouba, A., S. Souquieres, B. Njinku, P. M. Martin, M. C. Muller-Trutwin, P. Roques, F. Barre-Sinoussi, P. Mauclere, F. Simon, and E. Nerrienet. 2000. HIV-1 group N among HIV-1-seropositive individuals in Cameroon. AIDS 14:2623-2625. [DOI] [PubMed] [Google Scholar]
- 4.Bonhoeffer, S., E. C. Holmes, and M. A. Nowak. 1995. Causes of HIV diversity. Nature 376:125. [DOI] [PubMed] [Google Scholar]
- 5.Botarelli, P., B. A. Houlden, N. L. Haigwood, C. Servis, D. Montagna, and S. Abrignani. 1991. N-glycosylation of HIV-gp120 may constrain recognition by T lymphocytes. J. Immunol. 147:3128-3132. [PubMed] [Google Scholar]
- 6.Gao, F., E. Bailes, D. L. Robertson, Y. Chen, C. M. Rodenburg, S. F. Michael, L. B. Cummins, L. O. Arthur, M. Peeters, G. M. Shaw, P. M. Sharp, and B. H. Hahn. 1999. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature 397:436-441. [DOI] [PubMed] [Google Scholar]
- 7.Gaschen, B., J. Taylor, K. Yusim, B. Foley, F. Gao, D. Lang, V. Novitsky, B. Haynes, B. Hahn, T. Bhattacharya, and B. Korber. 2002. Diversity considerations in HIV-1 vaccine selection. Science 296:2354-2360. [DOI] [PubMed] [Google Scholar]
- 8.Hahn, B. H., G. M. Shaw, K. M. De Cock, and P. M. Sharp. 2000. AIDS as a zoonosis: scientific and public health implications. Science 287:607-614. [DOI] [PubMed] [Google Scholar]
- 9.Hansen, J. E., O. Lund, N. Tolstrup, A. A. Gooley, K. L. Williams, and S. Brunak. 1998. NetOglyc: prediction of mucin type O-glycosylation sites based on sequence context and surface accessibility. Glycoconj. J. 15:115-130. [DOI] [PubMed] [Google Scholar]
- 10.Huang, X. L., J. J. Barchi, F. D. T. Lung, P. P. Roller, P. L. Nara, J. Muschik, and R. R. Garrity. 1997. Glycosylation affects both the three-dimensional structure and antibody binding properties of the HIV-1IIIB BP120 peptide RP135. Biochemistry 36:10846-10856. [DOI] [PubMed] [Google Scholar]
- 11.Korber, B., C. Brander, B. Haynes, R. Koup, C. Kuiken, J. P. Moore, B. D. Walker, and D. I. Watkins. 2000. HIV molecular immunology. Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, N.Mex.
- 12.Kuiken, C., B. Foley, B. Hahn, P. A. Marx, F. McCutchan, J. W. Mellors, J. L. Mullins, S. Wolinsky, and B. Korber. 2000. HIV sequence compendium. Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, N.Mex.
- 13.Kwong, P. D., M. L. Doyle, D. J. Casper, C. Cicala, S. A. Leavitt, S. Majeed, T. D. Steenbeke, M. Venturi, I. Chaiken, M. Fung, H. Katinger, P. W. Parren, J. Robinson, D. Van Ryk, L. Wang, D. R. Burton, E. Freire, R. Wyatt, J. Sodroski, W. A. Hendrickson, and J. Arthos. 2002. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 420:678-682. [DOI] [PubMed] [Google Scholar]
- 14.Liedtke, S., R. Geyer, and H. Geyer. 1997. Host-cell-specific glycosylation of HIV-2 envelope glycoprotein. Glycoconj. J. 14:785-793. [DOI] [PubMed] [Google Scholar]
- 15.Moore, C. B., M. John, I. R. James, F. T. Christiansen, C. S. Witt, and S. A. Mallal. 2002. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 296:1439-1443. [DOI] [PubMed] [Google Scholar]
- 16.Nickle, D. C., M. A. Jensen, G. S. Gottlieb, D. Shriner, G. H. Learn, A. G. Rodrigo, and J. I. Mullins. 2003. Consensus and ancestral state HIV vaccines. Science 299:1515-1518. [DOI] [PubMed] [Google Scholar]
- 17.Nielsen, R., and Z. Yang. 1998. Likelihood models for detecting positively selected amino-acid sites and applications to the HIV-1 envelope gene. Genetics 148:929-936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pantophlet, R., E. Ollmann Saphire, P. Poignard, P. W. Parren, I. A. Wilson, and D. R. Burton. 2003. Fine mapping of the interaction of neutralizing and nonneutralizing monoclonal antibodies with the CD4 binding site of human immunodeficiency virus type 1 gp120. J. Virol. 77:642-658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Papandreou, M. J., T. Idziorek, R. Miquelis, and E. Fenouillet. 1996. Glycosylation and stability of mature HIV envelope glycoprotein conformation under various conditions. FEBS Lett. 379:171-176. [DOI] [PubMed] [Google Scholar]
- 20.Pillai, S., B. Good, D. D. Richman, and J. Corbeil. 2003. A new perspective on V3 phenotype prediction. AIDS Res. Hum. Retrovir. 19:145-149. [DOI] [PubMed] [Google Scholar]
- 21.Pollakis, G., S. Kang, A. Kliphuis, M. I. M. Chalaby, J. Goudsmit, and W. A. Paxton. 2001. N-linked glycosylation of the HIV type 1 gp120 envelope glycoprotein as a major determinant of CCR5 and CXCR4 coreceptor utilization. J. Biol. Chem. 276:13433-13441. [DOI] [PubMed] [Google Scholar]
- 22.Price, D. A., P. J. R. Goulder, P. Klernerman, A. K. Sewell, P. J. Easterbrook, M. Troop, C. R. M. Bangham, and R. E. Phillips. 1997. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc. Natl. Acad. Sci. USA 94:1890-1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rambaut, A., D. L. Robertson, O. G. Pybus, M. Peeters, and E. C. Holmes. 2001. Phylogeny and the origin of HIV-1. Nature 410:1047-1048. [DOI] [PubMed] [Google Scholar]
- 24.Robertson, D. L., J. P. Anderson, J. A. Bradac, J. K. Carr, B. Foley, R. K. Funkhouser, F. Gao, B. H. Hahn, M. L. Kalish, C. Kuiken, G. H. Learn, T. Leitner, F. E. McCutchan, S. Osmanov, M. Peeters, D. Pieniazek, M. Salminen, P. M. Sharp, S. Wolinsky, and B. Korber. 2000. HIV-1 nomenclature proposal. Science 288:55-57. [DOI] [PubMed] [Google Scholar]
- 25.Roques, P., D. L. Robertson, S. Souquiere, F. Damond, A. Ayouba, I. Farfara, C. Depienne, E. Nerrienet, D. Dormont, F. Brun-Vezinet, F. Simon, and P. Mauclere. 2002. Phylogenetic analysis of 49 newly derived HIV-1 group O strains: high viral diversity but no group M-like subtype structure. Virology 302:259-273. [DOI] [PubMed] [Google Scholar]
- 26.Sokal, R. R., and F. J. Rohlf. 1981. Biometry. W. H. Freeman and Company, New York, N.Y.
- 27.Swofford, D. L. 2000. PAUP*: phylogenetic analysis using parsimony (* and other methods). Version 4.0b6. Sinauer Associates, Sunderland, Mass.
- 28.Wei, X., J. M. Decker, S. Wang, H. Hui, J. C. Kappes, X. Wu, J. F. Salazar-Gonzalez, M. G. Salazar, J. M. Kilby, M. S. Saag, N. L. Komarova, M. A. Nowak, B. H. Hahn, P. D. Kwong, and G. M. Shaw. 2003. Antibody neutralization and escape by HIV-1. Nature 422:307-312. [DOI] [PubMed] [Google Scholar]
- 29.Woelk, C. H., and E. C. Holmes. 2002. Reduced positive selection in vector-borne RNA viruses. Mol. Biol. Evol. 19:2333-2336. [DOI] [PubMed] [Google Scholar]
- 30.Wolinsky, S. M., B. T. Korber, A. U. Neumann, M. Daniels, K. J. Kunstman, A. J. Whetsell, M. R. Furtado, Y. Cao, D. D. Ho, and J. T. Safrit. 1996. Adaptive evolution of human immunodeficiency virus-type 1 during the natural course of infection. Science 272:537-542. [DOI] [PubMed] [Google Scholar]
- 31.Wu, L., N. P. Gerard, R. Wyatt, H. Choe, C. Parolin, N. Ruffing, A. Borsetti, A. A. Cardoso, E. Desjardin, W. Newman, C. Gerard, and J. Sodroski. 1996. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature 384:179-183. [DOI] [PubMed] [Google Scholar]
- 32.Wyatt, R., P. D. Kwong, E. Desjardins, R. W. Sweet, J. Robinson, W. A. Hendrickson, and J. G. Sodroski. 1998. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 393:705-711. [DOI] [PubMed] [Google Scholar]
- 33.Yamaguchi, J., A. S. Vallari, P. Swanson, P. Bodelle, L. Kaptue, C. Ngansop, L. Zekeng, L. G. Gurtler, S. G. Devare, and C. A. Brennan. 2002. Evaluation of HIV type 1 group O isolates: identification of five phylogenetic clusters. AIDS Res. Hum. Retrovir. 18:269-282. [DOI] [PubMed] [Google Scholar]
- 34.Yamaguchi, Y., and T. Gojobori. 1997. Evolutionary mechanisms and population dynamics of the third variable envelope region HIV within single hosts. Proc. Natl. Acad. Sci. USA 94:1264-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamaguchi-Kabata, Y., and T. Gojobori. 2000. Reevaluation of amino acid variability of the human immunodeficiency virus type 1 gp120 envelope glycoprotein and prediction of new discontinuous epitopes. J. Virol. 74:4335-4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang, Z. 1997. PAML: a program package for the phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13:555-556. [DOI] [PubMed] [Google Scholar]
- 37.Yang, Z., and J. P. Bielawski. 2000. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 15:496-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang, Z. H., R. Nielsen, N. Goldman, and A. M. K. Pedersen. 2000. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yusim, K., C. Kesmir, B. Gaschen, M. M. Addo, M. Altfeld, S. Brunak, A. Chigaev, V. Detours, and B. T. Korber. 2002. Clustering patterns of cytotoxic T-lymphocyte epitopes in human immunodeficiency virus type 1 (HIV-1) proteins reveal imprints of immune evasion on HIV-1 global variation. J. Virol. 76:8757-8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zanotto, P. M., E. G. Kallas, R. F. de Souza, and E. C. Holmes. 1999. Genealogical evidence for positive selection in the nef gene of HIV-1. Genetics 153:1077-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]