

Abstract
Glucocorticoids are used for treating preterm neonatal infants suffering from life-threatening lung, airway, and cardiovascular conditions. However, several studies have raised concerns about detrimental effects of postnatal glucocorticoid administration on the developing brain leading to cognitive impairment, cerebral palsy, and hypoplasia of the cerebellum, a brain region critical for coordination of movement and higher-order neurological functions. Previously, we showed that glucocorticoids inhibit Sonic hedgehog–Smoothened (Shh-Smo) signaling, the major mitogenic pathway for cerebellar granule neuron precursors. Conversely, activation of Shh-Smo in transgenic mice protects against glucocorticoid-induced neurotoxic effects through induction of the 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) pathway. Here, we show that systemic administration of a small-molecule agonist of the Shh-Smo pathway (SAG) prevented the neurotoxic effects of glucocorticoids. SAG did not interfere with the beneficial effects of glucocorticoids on lung maturation, and despite the known associations of the Shh pathway with neoplasia, we found that transient (1-week-long) SAG treatment of neonatal animals was well tolerated and did not promote tumor formation. These findings suggest that a small-molecule agonist of Smo has potential as a neuroprotective agent in neonates at risk for glucocorticoid-induced neonatal cerebellar injury.
INTRODUCTION
More than 60,000 very low birth weight (<1.5 kg) preterm neonates are born yearly in the United States. Such infants suffer from a range of neurological impairments such as intracranial hemorrhage, damage to gray and white matter, cerebral palsy, and learning and behavioral problems (1–3). Cerebellar hypoplasia is common in such infants (2), is a hallmark of severe neurological injury (4–7), and is associated with cognitive and affective disturbances, including autistic syndrome (8). The cerebellum is responsible for coordination of movement and higher-order relay functions of the brain (7). The most abundant cell type in the cerebellum is the granule neuron precursor (CGNP) (9–11). In humans, CGNP expansion starts at 11 weeks of gestational age and is robust in the third trimester of pregnancy, between 24 and 40 weeks of gestational age, during which deficient growth and foliation can result in cerebellar hypoplasia (12). Human cerebellar growth continues until at least 1 year of age (13). The analogous period of robust CGNP growth during mouse development is postnatal days 0 (P0) to 14 (P14), and functional studies have demonstrated that Sonic hedgehog (Shh) signaling is the prime driver of this process (11, 14). Shh activates the transmembrane receptor protein Smoothened (Smo) to up-regulate the target genes Gli1 and N-myc, which are essential for CGNP cell cycle progression (15, 16). Mutations that activate the Shh-Smo pathway in CGNPs result in the cerebellar tumor medulloblastoma in mouse and humans (17, 18).
For several decades, glucocorticoids (GCs) have been used in pre-term infants for the treatment of life-threatening chronic lung disease, as well as for cardiovascular conditions (for example, hypotension) (4, 7). In the immature lung, GCs promote surfactant production and lung epithelial differentiation (19). However, numerous studies have shown that GCs can have detrimental effects on the development of the brain, leading to impaired cognition, cerebral palsy, and cerebellar hypoplasia (5–7, 11, 20). 11β-Hydroxysteroid dehydrogenase type 2 (11β-HSD2) catalyzes the interconversion of the endogenous physiological GCs cortisol and corticosterone to the inert 11-ketometabolites cortisone and 11-dehydrocorticosterone, respectively (21, 22). The synthetic GC prednisolone is also inactivated by 11β-HSD2 (23). 11β-HSD2 is expressed by the cerebellar anlage commencing at embryonic day 12.5 (E12.5) in mice (24), and its function is necessary for normal cerebellar development (22, 24).
GCs generally regulate gene expression through binding to the intracellular mineralocorticoid and/or GC receptor (25) or by directly affecting intracellular signaling pathways for protein stability and cell metabolism (26). Although the molecular basis of GC-induced neurotoxicity in the developing brain remains incompletely understood, we have shown that neonatal administration (P0 to P7) of GCs in mice inhibits mitogenic Shh signaling in CGNPs through destabilization of Gli and N-myc proteins (20). Conversely, Shh-Smo signaling can antagonize neurotoxic effects of GC signaling in CGNPs. Forced expression of a constitutively active form of Smo (SmoM2) in transgenic mice induced up-regulation of 11β-HSD2 and prevented neurotoxic effects of prednisolone in the neonatal cerebellum (20). Nevertheless, such a transgenic approach to protect the cerebellum against GC-induced damage is not clinically feasible and carries the additional problem that the animals eventually succumb to medulloblastoma (18).
New and practical strategies are needed to assuage the negative effects of postnatal GC administration on developing brain structures, such as the cerebellum, while preserving their desirable therapeutic profile in the developing lung. We therefore tested whether systemic administration of a small molecular agonist of the Shh-Smo pathway (SAG) could antagonize GC-induced cerebellar injury while demonstrating safety and preserving the beneficial effects of GCs to promote lung maturation.
RESULTS
SAG antagonizes GC’s antiproliferative effects in CGNP primary cultures
We and others have previously described a small-molecule chlorobenzo[b]thiophene derivative (SAG) that binds and activates Smo (27, 28). To assess the direct activity of SAG on CGNPs, we used primary cultures derived from P4 wild-type mice. SAG treatment promoted strong proliferation of CGNPs with a peak activity at 120 nM that was similar to that of the biologically active N-terminal fragment of Shh (ShhN) (Fig. 1A). Both dexamethasone and prednisolone significantly inhibited ShhN-induced CGNP proliferation (Fig. 1B). We chose the GC concentrations 40 μM (dexamethasone) and 120 nM (prednisolone) on the basis of previous findings showing that these are the maximum GC concentrations that affect CGNP proliferation without causing apoptosis (20). These GCs did not significantly inhibit the SAG-induced effects shown in Fig. 1A (Fig. 1B), suggesting that direct activation of Smo with SAG can counteract GC antagonism of Shh-driven CGNP proliferation.
Fig. 1.
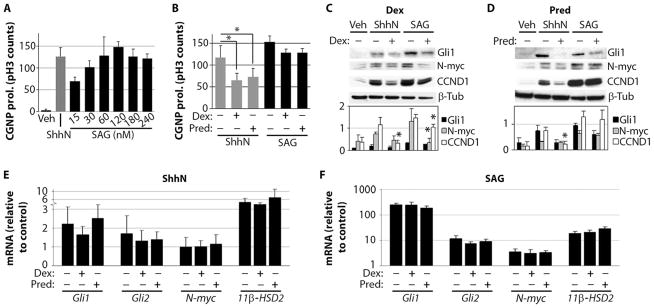
SAG antagonizes GC effects on cultured primary CGNPs. (A) CGNP proliferation (prol.) stimulated by various SAG concentrations (15 to 240 nM) compared with ShhN (3 μg/ml) and vehicle (Veh) after 24 hours in vitro (n = 4). (B) Effects of dexamethasone (Dex) (40 μM) and prednisolone (Pred) (120 nM) on ShhN-induced (P < 0.001, ANOVA with Tukey’s post hoc; n = 5) and 120 nM SAG–induced CGNP cultures (no significant change). (C and D) Western blots of protein lysates prepared from CGNPs treated with vehicle, ShhN, or 120 nM SAG in the presence or absence of 40 μM dexamethasone (C) [Gli1: P < 0.005, ANOVA; N-myc: P < 0.02, ANOVA (Tukey’s post hoc: SAG versus SAG + dexamethasone, P = 0.02); CCND1: P < 0.005, ANOVA (Tukey’s post hoc: ShhN versus ShhN + dexamethasone, P < 0.0001; SAG versus SAG + dexamethasone, P = 0.05)] or 120 nM prednisolone (D) [Gli1: P < 0.03, ANOVA; N-myc: P < 0.001, ANOVA; CCND1: P < 0.001, ANOVA (Tukey’s post hoc: ShhN versus ShhN + prednisolone, P = 0.04)] for 24 hours (n = 3). Signal intensity of the bands is illustrated in the histograms below. (E and F) Total RNA was isolated from CGNP cultures treated with vehicle, ShhN (E), or 120 nM SAG (F) after 24 hours in the presence or absence of 40 μM dexamethasone or 120 nM prednisolone (n = 3; no significant changes). Asterisks indicate significant changes using Tukey’s post hoc test.
Previously, we reported that GC treatment reduces levels of protein, but not mRNA, for Shh proliferative targets in CGNPs (20), indicating that GCs act downstream of Smo activation in the Shh pathway. In keeping with this conclusion, in ShhN-treated CGNP cultures, prednisolone and dexamethasone significantly reduced levels of cyclin D1 (CCND1) protein (Fig. 1, C and D), which serve as a marker of G1 cell cycle progression. Although levels of Gli and N-myc proteins were reduced by GC treatment (prednisolone or dexamethasone), differences were not significant. GCs did not significantly change mRNA up-regulation of the Shh transcriptional targets Gli1, Gli2, N-myc, or 11β-HSD2 (Fig. 1E), as expected. In SAG-treated cultures, we found that the 11β-HSD2–sensitive GC prednisolone did not significantly reduce levels of N-myc or CCND1 protein levels, whereas dexamethasone caused significant reductions of N-myc and CCND1 (Fig. 1, C and D), consistent with previous results in SmoM2 transgenic CGNPs (20). SAG also was more potent than ShhN in up-regulating expression of RNA for Gli1, Gli2, N-myc, and 11β-HSD2 in vitro (Fig. 1, E and F). Together, these findings indicate that SAG can function as an effective antagonist of GC-induced inhibition of CGNP proliferation. These effects may be a result of the relatively potent induction of Smo signaling gene targets by SAG compared to Shh itself.
SAG activates a Gli-luciferase reporter in vivo
We next used CGNPs derived from P4 Gli-luciferase (29) mice and established that SAG could activate a Shh signaling transgene reporter in vitro (Fig. 2A). As a prelude to analysis of the effect of neonatal SAG administration on cerebellar development, we investigated whether SAG could cross the blood-brain barrier to activate Shh target genes in vivo. We treated Gli-luciferase pups at P11 with various doses of SAG (5.6, 14, and 25.2 μg/g) and, after 4 hours, administered luciferin substrate to visualize reporter activity in the brain and cerebellum. We observed a dose-response profile with maximal activation of the reporter at a dosage of 25.2 μg/g (Fig. 2, B and C), and this effect was confirmed by up-regulation of mRNA transcripts for the endogenous Shh targets Gli1 and N-myc (Fig. 2D). These findings show that SAG can cross the blood-brain barrier to activate Shh transcriptional targets in vivo.
Fig. 2.

SAG activates Gli-luciferase reporter transgene in CGNPs in vitro and in vivo. (A) Gli-luciferase (Gli-luc) reporter expression in primary CGNPs treated with ShhN (3 μg/ml) and SAG (120 nM) for 24 hours in vitro (n = 8). (B) P11 Gli-luciferase mice were injected intraperitoneally with SAG (0, 5.6, 14.0, or 25.2 μg/g) (in saline) and killed after 4 hours. The brains and ears were dissected, placed in luciferin (0.4 mg/ml)/phosphate-buffered saline for 35 min, and then imaged in the Xenogen IVIS detector for 4 min. A photographic image was taken onto which the pseudocolor image representing the spatial distribution of photon count was projected. FB, forebrain; CB, cerebellum. (C) Luciferase levels in the cerebellum quantified in photons per second at various SAG doses. (D) qRT-PCR analysis of SAG effects on the Smo targets Gli1 and N-myc in vivo. SAG dosages (14.0 and 25.2 μg/g) induced significantly increased N-myc levels over a dosage of 5.6 μg/g (P < 0.05, ANOVA with Tukey’s post hoc; n = 2). No significant differences were seen between 14.0 and 25.2 μg/g. Asterisks indicate significant changes with the Tukey’s post hoc test.
Because SAG effects were similar with the 14 or 25.2 μg/g dose, we chose an intermediate dose of 20 μg/g, which we termed the “treatment dose.” We also used primary CGNP cultures as a bioassay and determined a second “high dose” by using a free-base form of SAG that exhibits greater potency. The free-base form of SAG was about seven times more potent than the treatment dose of SAG, as measured by its ability to stimulate proliferation of CGNPs (Fig. 3C), likely reflecting its higher purity and greater bioavailability. This form was chosen for high-dose testing to maintain similar volumes of injected drug and/or vehicle. The treatment and high dosages of SAG were next assessed for potential toxic effects in mice.
Fig. 3.

Results of toxicity studies with SAG at treatment and high doses. (A) Histological analysis of the cerebella of the 6-month-old control and SAG-HD (SAG high dose), and P28 control and SAG-TD (SAG treatment dose)–treated animals. (B) Immunocytochemistry for the proliferation marker Ki67 in the cerebella of P28 control and SAG-TD–treated animals. DAPI, 4′,6-diamidino-2-phenylindole. (C) Determination of treatment and high dose of SAG using CGNP proliferation in vitro. Histogram represents the ratio of the number of pH3-positive cell cultures using free-base SAG versus the salt form of SAG at 1 nM (n = 2). (D and E) Growth curves of vehicle, SAG-TD, prednisolone, prednisolone + SAG-TD (D), and SAG-HD–treated (E) mice.
Daily administration of SAG at treatment or high doses is not tumorigenic
The Shh pathway is etiologic in several types of human cancer (30), including the cerebellar tumor medulloblastoma (17). To assess whether the Smo agonist SAG showed toxicity and/or tumor formation, we first administered to pups the treatment dose of SAG (20 μg/g, n = 6) daily from P0 to P7 (Table 1). At 1 month of age, we performed the modified-SHIRPA (31) protocol to analyze the behavioral and morphological effects on the treated animals. As shown (Table 2), no motor or behavioral changes were detected. All animals were asymptomatic and showed normal weight gain at 2 months of age, at which point they were euthanized for inspection of all viscera (heart, lungs, kidneys, pancreas, liver, intestine, and spleen) and brain (Table 1 and Fig. 3A). In four treated animals and three controls, complete blood counts were done at 2 months of age; these showed no signs of leukemia or abnormal hematological counts of leukocytes, erythrocytes, or thrombocytes. Solid tumors were undetectable in any organ system including forebrain and cerebellum (Table 1 and Fig. 3, A and B).
Table 1.
Summary of SAG toxicity studies in neonatal mice. All mice were treated (intraperitoneally) with SAG treatment dose (SAG-TD) (20 μg/g per day), SAG high dose (SAG-HD) (140 μg/g per day), or vehicle (daily) (P0 to P7 or P1 to P8). Mice were sacrificed at P9, P14, and P28, and at 2, 4, or 6 months of age for necropsy and comprehensive pathological analysis as indicated. Y, yes; N, no; ND, not done.
| Group | Age | n | Treatment (P0 to P8) | Gross tumors? (Y/N) | Histological findings | Precancerous lesions “EGL rests” in cerebellum? | Complete blood count |
|---|---|---|---|---|---|---|---|
| 1 | P9 | 3 | Vehicle | N | Unremarkable | ND | ND |
| P9 | 3 | SAG-HD | N | Intestinal hyperplasia (one of three animals) | ND | ND | |
| 2 | P14 | 1 | Vehicle | N | Unremarkable | ND | ND |
| P14 | 1 | SAG-HD | N | Intestinal hyperplasia | ND | ND | |
| 3 | P28 | 3 | Vehicle | N | Unremarkable | ND | ND |
| P28 | 3 | SAG-TD | N | Unremarkable | None | ND | |
| P28 | 3 | SAG-TD + prednisolone | N | Unremarkable | None | ND | |
| 4 | 2 months | 3 | Vehicle | N | ND | ND | Within normal limits |
| 2 months | 4 | SAG-TD | N | ND | ND | Within normal limits | |
| 2 months | 2 | SAG-TD | N | Unremarkable | ND | ND | |
| 5 | 4 months | 2 | Vehicle | N | Unremarkable | ND | ND |
| 4 months | 1 | SAG-HD | N | Unremarkable | None | ND | |
| 6 | 6 months | 1 | Vehicle | N | Unremarkable | None | ND |
| 6 months | 1 | SAG-HD | N | Unremarkable | None | ND |
Table 2.
Clinical data (cyanosis assessment and modified-SHIRPA test) on SAG-TD– and SAG-HD–treated mice. All the pups treated with SAG-HD were monitored every day for cyanosis. The color of the skin is indicated in the table for each day of injection. The SAG-TD– and SAG-HD–treated animals were tested at 1 and 6 months of age, respectively, for behavioral, morphological, or performance changes using the modified-SHIRPA protocol. N, normal; NP, not present; P, present.
| Age
|
1 month
|
6 months
|
||||
|---|---|---|---|---|---|---|
| Treatment (P0 to P8) | Vehicle | SAG-TD | SAG-TD + prednisolone | Vehicle | SAG-HD | |
| n | Males | 1 | 2 | 1 | 1 | 1 |
| Females | 2 | 1 | 2 | — | — | |
| Category | Tests | |||||
| Coat | Coat color; hair length; hair morphology; skin color; piloerection | N | N | N | N | N |
| Morphology | Tail, pinna, head, limb, whisker, and teeth morphology; palpebral closure | N | N | N | N | Abnormal head morphology |
| Posture | Body position; body, limb, and abdominal tone; visual placing; tail and pelvic elevation; trunk curl | N | N | N | N | N |
| Activity | Spontaneous and locomotor activity; wire maneuver; negative geotaxis; gait | N | N | N | N | Poor negative geotaxis |
| Behavior | Fear; irritability; aggression; vocalization; bizarre behavior; convulsions; provoked biting; tremor | NP | NP | NP | NP | NP |
| Reflexes | Limb grasping; grip strength; pinna and corneal reflex; toe pinch; touch escape; transfer arousal; contact righting | P | P | P | P | Semi-effective grip |
| Body releases | Defecation; urination; lacrimation; salivation | N | N | N | N | N |
| Heart rate | N | N | N | N | N | |
| Respiration rate | N | N | N | N | N | |
| Body weight (g) | Males | 29.3 | 29.1 ± 0.6 | 28.9 | 48.5 | 28 |
| Females | 26.0 ± 1.113 | 25 | 25.1 ± 0.4 | — | — | |
| Body length (mm) | Males | 105 | 100 ± 0 | 105 | 120 | 90 |
| Females | 102.5 ± 3.5 | 95 | 100 ± 0 | — | — | |
| Tail length (mm) | Males | 95 | 95 ± 7.1 | 85 | 75 | 80 |
| Females | 90.0 ± 7.1 | 90 | 95 ± 0 | — | — | |
| Age analysis
|
P1
|
P2
|
P3
|
P4
|
P5
|
P6
|
P7
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Treatment (P0 to P8) | Vehicle | SAG-HD | Vehicle | SAG-HD | Vehicle | SAG-HD | Vehicle | SAG-HD | Vehicle | SAG-HD | Vehicle | SAG-HD | Vehicle | SAG-HD |
| n | 6 | 8 | 5 | 8 | 4 | 7 | 4 | 7 | 4 | 7 | 3 | 7 | 3 | 7 |
| Skin color | Pink | Pink | Pink | Pink | Pink | Pink | Pink | Pink | Pink | Pink | Pink | Pink | Pink | Pink |
We next analyzed animals treated with the high-dose form of SAG daily from P1 to P8 at P9 (n = 3, group 1; Table 1), P14 (n = 1, group 2; Table 1), and 4 to 6 months (n = 3, groups 5 and 6; Table 1). No gross or microscopic evidence of tumor formation was found in the brain or other viscera of any treated animal. Before medulloblastoma formation in tumor-prone Ptc+/− mice, preneoplastic rests of cells are readily identified in the outer layers of the cerebellum as early as 3 weeks of age (32). As shown (Fig. 3A and Table 1), we saw no such precancerous cells in the cerebellum of animals at P28 or 4 to 6 months after administration of the treatment or high dose of SAG.
In summary, we found no evidence of cancers of blood or solid organs in any of the 14 animals studied at ages greater than 1 month, nor was there evidence of pretumorigenic lesions of medulloblastoma. We noted that SAG treatment at the high dose resulted in relatively poor long-term growth (Fig. 3E), which might relate to intestinal hyperplasia caused by high-dose SAG in some cases (Table 1, groups 1 and 2). At the treatment dose, SAG did not cause any such growth or intestinal abnormalities (Fig. 3D and Table 1). Together, these findings indicated that transient (1 week) administration of SAG at a treatment dose was well tolerated in neonatal mice. Our studies do not rule out potential side effects of longer-term SAG administration.
SAG administration does not interfere with beneficial GC effects on pulmonary development
In preterm neonates at risk for respiratory distress syndrome, GCs are given pre- or postnatally to promote lung maturation and surfactant protein production as well as to reduce inflammation. Because SAG is antagonistic to GC activity in the cerebellum, we investigated its effects on neonatal mouse lung to determine whether SAG interfered with GC beneficial effects in lung. SAG administered at treatment dose alone or in combination with prednisolone did not detectably alter amounts of the differentiation markers surfactant proteins A (SP-A) or B (SP-B) (Fig. 4A). When given at high dose, SAG alone appeared to promote lung hyperplasia but did not inhibit expression of the differentiation markers SP-A and SP-B (Fig. 4B). Moreover, animals that received SAG at treatment dose or high dose did not exhibit respiratory symptoms such as cyanosis or increased respiratory rate (Table 2).
Fig. 4.

SAG treatment has no detrimental effect on lung maturation. (A and B) Phase-contrast analysis (upper panels) and immunocytochemistry (lower panels) for the differentiation markers SP-B SP-A and nuclear stain DAPI on P9-inflated lungs treated with vehicle, prednisolone, SAG-TD, prednisolone + SAG-TD (A), and SAG-HD (B). (C) qRT-PCR analysis for SP-B, SP-A, 11β-HSD1, 11β-HSD2, Gli1, and Gli2 expression in the P9 lung of SAG-HD–treated mice (n = 3; not significant). Data in (C) are expressed relative to values obtained with vehicle treatment (control), which was set at 1. (D) qRT-PCR for SP-B on human fetal lung explant cultures untreated (Waymouth medium) or treated with dexamethasone (10 nM) or DCI (10 nM dexamethasone, 0.1 mM 8-bromo-cAMP, and 0.1 mM isobutylmethylxanthine together) in the presence or absence of SAG (120 nM) (see Materials and Methods). SAG does not alter the expression level of SP-B (P < 0.005, ANOVA; Tukey’s post hoc: Way-mouth medium versus Waymouth medium + SAG, P > 0.05; dexamethasone versus dexamethasone + SAG, P > 0.05; DCI versus DCI + SAG, P > 0.05; vehicle versus DCI, P < 0.05, n = 3), which is differentially induced by GCs (43). Data are expressed relative to control levels, which were set at 1. β-Actin was used as a reference gene to calculate SP-B expression levels (n = 3). Scale bars, 100 μm [(A) and (B)].
We next used quantitative polymerase chain reaction (qPCR) to quantitatively assess mRNA levels for the differentiation markers SP-A and SP-B and for genes of the GC and Shh signaling pathways in whole lungs of animals treated from P1 to P8 with SAG at the high dose (Fig. 4C). SAG did not significantly change the expression of the Shh targets Gli1 and Gli2 and the lung differentiation markers SP-B and SP-A [analysis of variance (ANOVA): not significantly different]. In contrast to its effects in the cerebellum, SAG treatment produced no significant changes in 11β-HSD1 or 11β-HSD2 (ANOVA: not significantly different). Finally, we tested the effects of simultaneous treatment of human fetal lung cultures with GC and SAG. As shown (Fig. 4D), SAG did not inhibit GC-induced up-regulation of the differentiation marker SP-B in human fetal lung cultures.
SAG protects against neurotoxic effects of prednisolone
We next investigated whether SAG can antagonize the neurotoxic effects of GCs on CGNPs in vivo (Fig. 5A). We have previously shown that daily treatment with prednisolone from P0 to P7 results in significant inhibition of CGNP proliferation (20), an effect that is mediated by the GC receptor because mouse CGNPs do not express mineralocorticoid receptor (20). As shown (Fig. 5, B and C), administration of SAG (20 μg/g) with prednisolone (0.67 μg/g) prevented such inhibition of proliferation within the external granule layer (EGL), which comprises exclusively CGNP. SAG treatment alone did not cause a significant increase in the baseline proliferation of CGNPs, or thickening of the EGL, characteristic of the precancerous state in Math1-cre, SmoM2 transgenic mice (18), nor was medulloblastoma observed at P21 (Fig. 5F) or at longer survival time points (Fig. 3A and Table 1).
Fig. 5.

SAG protects against neurotoxic effects of prednisolone. (A) Scheme for administration of vehicle, prednisolone, or prednisolone + SAG according to “daily (P0-P7)” or “acute (P7 only)” schedule and histological analysis at P7 or P21. (B to F) Immunocytochemistry of the mitotic marker pH3, apoptosis marker cleaved caspase 3 (Casp3), CGNP markers Zic1 and Pax6, and Purkinje cell marker calbindin (Calb) at P7 and P21. (C) At P7, daily SAG treatment prevented the significant antiproliferative (prednisolone versus prednisolone + SAG: P < 0.02, ANOVA with Tukey’s post hoc, n = 3) and (E) acute SAG treatment proapoptotic (prednisolone versus prednisolone + SAG: P < 0.003, ANOVA with Tukey’s post hoc, n = 3) effects of prednisolone on neonatal CGNPs of the EGL. The prednisolone group is significantly different from all other groups (vehicle, prednisolone + SAG, and SAG). Only the P values of the Tukey’s post hoc test between the prednisolone and the prednisolone + SAG groups are given. (F and G) Significant reduction in the volume of the IGL at P21 induced by prednisolone treatment (P < 0.02, ANOVA with Tukey’s post hoc, n = 3), which was prevented by SAG. Images of lobe VII of the cerebellum shown in (F) are representative of the results in all lobes of the cerebellum. Asterisks indicate significant changes with the Tukey’s post hoc test. Scale bars, 100 μm [(B) and (D)] and 300 μm (F).
Acute treatment with GCs between P4 and P7 in the mouse causes apoptosis of CGNPs (20, 33). SAG treatment prevented apoptosis of CGNPs in prednisolone-treated animals administered a one-time dose at P7 (Fig. 5, D and E). Treatment from P0 to P7 with GCs also results in permanent deficits in the volume of granule neurons, but not the number of Purkinje cells (20). However, as shown (Fig. 5, F and G), these effects of daily injection for 1 week on granule neurons are also effectively assuaged by SAG treatment. As expected, Purkinje cell populations, as measured by the density of calbindin-positive cells in the cerebellar Purkinje cell layer, were not affected by SAG (Fig. 5F). These findings indicate that SAG at the treatment dose effectively prevented GC-induced neonatal cerebellar developmental abnormalities.
DISCUSSION
Rates of cerebral palsy in the United States are rising because of increased rates of survival of extremely low birth weight preterm infants coupled with their susceptibility to neurological damage from intrinsic or iatrogenic causes (34). Here, we have focused on postnatal GC treatment of preterm neonates, which is associated with cognitive impairment, cerebral palsy, and cerebellar hypoplasia (35–37). Yet, it remains necessary to use GCs for life-threatening conditions such as severe chronic lung disease (also known as bronchopulmonary dysplasia), airway emergencies (for example, subglottic stenosis), intractable hypotension, and inflammation associated with cardiac bypass. Indeed, the most recent recommendations of the American Academy of Pediatrics continue to support the use of lower-dose GCs [for example, dexamethasone (0.15 mg/kg per day)] in premature neonates with chronic lung disease.
We used a 1-week course of GCs given to neonatal mice at a dose equivalent to what has been used to treat human neonates to prevent severe chronic lung disease (dexamethasone, 0.1 mg/kg per day). As shown in this and other studies (20, 33), even 11β-HSD2–sensitive GCs such as prednisolone and hydrocortisone can have neurotoxic effects on CGNPs in neonatal animals. Clinical studies also show impaired cerebellar morphogenesis in infants treated postnatally with dexamethasone (38) or hydrocortisone (39). In previous work aimed at investigating approaches to protect against the toxic effects of GCs, we showed that transgenic mice with forced expression of activated SmoM2 in CGNPs were protected against the neurotoxic effects of prednisolone through up-regulation of 11β-HSD2 in vivo at P7 (20). Here, we have extended these findings and demonstrated the efficacy of a small-molecule agonist of Shh-Smo signaling (SAG) in protecting against GC-induced cerebellar abnormalities in a mouse preclinical model of GC-induced cerebellar neurotoxicity.
In our previous work in which animals that were engineered to express activated SmoM2 in CGNPs were protected against GC-induced cerebellar damage, the animals later succumbed to medulloblastoma (18, 20), raising the question of whether a systemic therapy could provide neuroprotection without causing tumor formation. Our data show that SAG, given systemically, can cross the blood-brain barrier in neonatal mice to activate Shh target genes in the developing cerebellum. SAG treatment resulted in up-regulation of the Gli-luciferase reporter transgene (29), as well as the endogenous Shh target genes Gli1, Gli2 (10), and N-myc (16), which are implicated in cerebellar tumori-genesis, a serious concern for translation of SAG to humans. Cancers closely associated with the Shh pathway include basal cell carcinoma and medulloblastoma. However, unlike the well-established Ptc+/− mouse medulloblastoma model (40) that develops tumors at 6 months or later, mice that were used in this study do not carry any oncogenic predisposition. In Ptc+/− mice, precancerous rests of cerebellar cells can be identified between 3 and 8 weeks of age, resembling persistent EGL, and some of these go on to form tumors when the Shh–N-myc pathway is activated (32). Such findings suggest that Shh signaling must be maintained for prolonged periods for medulloblastoma initiation and maintenance. Indeed, in inhibitor studies using Ptc+/−, p53−/− mice (41), even interrupting Shh signaling in established medulloblastoma was sufficient to curtail tumor growth or eliminate the tumor. In our study, we found no evidence of medulloblastoma or precancerous EGL rests in animals studied at 1, 4, or 6 months, even when we used SAG at a high dose, seven times the amount sufficient for Shh target gene activation in the EGL. We did not observe cancers of blood or solid organs in animals studied at ages greater than 1 month. Thus, on the basis of these findings, and because effective SAG treatment only requires a transient dose, the risk of medulloblastoma formation after SAG treatment may be low.
Two side effects of SAG were observed only in the high-dose group of animals studied. First, lung histology showed hyperplasia at P9 after daily administration of SAG from P1 to P8. This was not, however, associated with any clinical respiratory symptoms or lack of pulmonary epithelial differentiation. Second, these animals showed reduced overall growth. Pathological inspection of the intestines of some SAG-treated animals showed hyperplasia, with the smooth muscle layer thickness being increased and the mucosa showing signs of hyperproliferation, raising the possibility that the poor growth may be a result of abnormal intestinal absorption. Such side effects were not observed when SAG was given transiently at the treatment dose for 1 week (20 μg/g per day from P0 to P7). Despite these reassuring findings, additional preclinical toxicity studies are warranted. In particular, studies of longer-term SAG administration are needed to determine whether such prolonged exposure would result in neoplastic transformation.
Our studies with SAG at treatment dose indicate that it is effective in mice at preventing the neurotoxic effects of prednisolone in the neonatal cerebellum, confirming and extending previous work with transgenic mice that show that Shh signaling is antagonistic to GC signaling in CGNPs through an 11β-HSD2–dependent mechanism (20). Specifically, SAG co-administration prevented prednisolone-induced CGNP apoptosis and prednisolone inhibition of CGNP proliferation in the neonatal cerebellum, and preserved the normal volume of the cerebellar granule neuron populations.
GCs are primarily given to human neonates for their beneficial effects on lung development and function. Thus, it was important to determine whether SAG inhibited this beneficial GC activity. Analysis of murine lung in vivo indicated that SAG does not prevent normal pulmonary epithelial maturation. Additionally, SAG did not inhibit GC-induced differentiation of human fetal lung cultures. Therefore, if SAG were to be used in the clinic as an adjuvant therapy with GCs to prevent neurotoxicity, our data suggest that it would not interfere with the beneficial effects of GCs in the developing lung.
In summary, our data indicate that SAG crosses the blood-brain barrier to activate Shh-Smo gene targets in the cerebellum, protecting vulnerable neuronal precursors from GC-induced growth inhibition, and that such systemic treatment does not induce medulloblastoma or other types of cancer. However, it will be important to confirm these findings with additional toxicity studies to determine the optimal dose schedule and further establish safety parameters (side effects, tumor development) in rodents and other animal systems (nonhuman primate) before clinical trials in humans. SAG can prevent the growth-inhibitory effects of GCs in the cerebellum without antagonizing the beneficial effects of GCs in the lung. These findings suggest that adjuvant therapy with SAG could be a practical way to prevent certain neurotoxic side effects of GCs given to pre-term neonates for life-threatening conditions. Testing of SAG in additional models of human neonatal brain injury may also be warranted. Because Shh-Smo signaling is the major driver of cerebellar growth, this signaling axis may be affected in cerebellar injury because of hemorrhage or hypoxia (2). If so, then small-molecule agonists of Shh signaling like SAG might help to prevent these newborn neurological injuries.
MATERIALS AND METHODS
Preparation of SAG
Synthesis of SAG has been described (27). The chlorobenzo[b]thiophene derivative SAG (molecular weight, 490.06) was dissolved in dimethyl sulfoxide (DMSO) to 5 mM and further diluted with normal saline or culture medium. These experiments used SAG prepared as a trifluoroacetic acid salt or a free-base form. Vehicle controls comprised saline containing an equivalent concentration of DMSO.
Animals
All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee of University of California, San Francisco (UCSF). The SW129/J and C57BL6J mouse lines were obtained from The Jackson Laboratory. The Gli-luciferase transgenic mouse line, which drives expression of luciferase through Gli-responsive cis-acting DNA regulatory sequences (29), was provided by E. Holland.
Systemic administration of prednisolone and SAG
On P0 or P1, C57BL6J or Swiss Webster B pups received daily intra-peritoneal injections of prednisolone (0.67 μg/g, Sigma-Aldrich), SAG (20 μg/g), or prednisolone in combination with SAG, or vehicle, for 7 days. For acute treatment, a one-time dose of prednisolone alone or prednisolone + SAG was given at P7, and the cerebellum was harvested for analysis 6 hours later.
CGNP primary cell culture, immunohistochemistry, Western blot, and quantitative reverse transcription–PCR
All the experiments were done as described (20). Quantitative reverse transcription–PCR (qRT-PCR) was performed with SYBR Green master mix (Roche) in LightCycler 480 (Roche). β-Actin was used as a reference gene to calculate 2−ΔΔCt, and experiments were performed in duplicate for each sample. For all experiments, at least two independent samples were used per genotype and age that we examined (n values are indicated in the figure legends).
Human fetal lung explant culture
Human fetal lung tissue from a 20-week gestation abortus was obtained from Advanced Bioscience Resources under an Institutional Review Board–approved protocol of the Children’s Hospital of Philadelphia. The tissue was minced and placed in culture, as previously described (42). Explants were cultured in serum-free Waymouth medium on a rocking platform with an atmosphere of 95% air/5% CO2 for 24 hours and then cultured for 72 hours in Waymouth medium (control) or in medium containing dexamethasone (10 nM) alone or DCI [10 nM dexamethasone, 0.1 mM 8-bromo-cAMP (8-bromo-3′,5′-adenosine monophosphate), and 0.1 mM isobutylmethylxanthine together] in the presence or absence of SAG (120 nM). SP-B induction in explants was calculated by qRT-PCR using human-specific primers.
Sample size and quantification
Surface area of the entire cerebellar EGL and IGL (internal granule layer) was calculated as in (20). Results of primary CGNP culture are representative of experiments repeated with pups from more than five independent litters. Total numbers of cells in 10 microscopic fields (magnification, ×20) were counted for the markers pH3 and cleaved caspase 3 for statistical comparison.
Statistics
Statistical analysis was performed with an ANOVA (single factor). If the ANOVA test gave a significant difference (P < 0.05), a Tukey’s post hoc test was performed.
Acknowledgments
We thank A. M. Barette, S. Kaing, and M. Wong for expert technical assistance; J. Otero, E. Huang, B. Hann, and the UCSF Comprehensive Cancer Center pathology core for blood and whole-body solid tumor screening; L. Gonzales for fetal lung cultures; and E. Holland for providing Gli-luciferase mice.
Funding: V.M.H. thanks the Netherlands Organization for Scientific Research for a TALENT stipend. A.G. is grateful for support by a fellowship from the American Brain Tumor Association. This work was supported by grants from the March of Dimes Foundation (to D.H.R.) and NIH (R01 CA136574 to J.K.C.; R01 NS047527 to D.H.R.; RO1 HL086323 and PO1 HL024075 to P.L.B.). D.H.R. is a Howard Hughes Medical Institute Investigator.
Footnotes
Author contributions: V.M.H., C.C., and A.G. acquired and analyzed the data. V.M.H., J.K.C., and D.H.R. conceived the study. All authors designed the experiments and wrote the manuscript.
Competing interests: The authors declare that they have no competing interests. The SAG compound was synthesized at Stanford University and is available under a materials transfer agreement.
REFERENCES AND NOTES
- 1.Allen MC. Neurodevelopmental outcomes of preterm infants. Curr Opin Neurol. 2008;21:123–128. doi: 10.1097/WCO.0b013e3282f88bb4. [DOI] [PubMed] [Google Scholar]
- 2.Doyle LW, Anderson PJ. Adult outcome of extremely preterm infants. Pediatrics. 2010;126:342–351. doi: 10.1542/peds.2010-0710. [DOI] [PubMed] [Google Scholar]
- 3.Yanney M, Marlow N. Paediatric consequences of fetal growth restriction. Semin Fetal Neonatal Med. 2004;9:411–418. doi: 10.1016/j.siny.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Allin MP, Salaria S, Nosarti C, Wyatt J, Rifkin L, Murray RM. Vermis and lateral lobes of the cerebellum in adolescents born very preterm. Neuroreport. 2005;16:1821–1824. doi: 10.1097/01.wnr.0000185014.36939.84. [DOI] [PubMed] [Google Scholar]
- 5.Bodensteiner JB, Johnsen SD. Cerebellar injury in the extremely premature infant: Newly recognized but relatively common outcome. J Child Neurol. 2005;20:139–142. doi: 10.1177/08830738050200021101. [DOI] [PubMed] [Google Scholar]
- 6.Limperopoulos C, Soul JS, Gauvreau K, Huppi PS, Warfield SK, Bassan H, Robertson RL, Volpe JJ, du Plessis AJ. Late gestation cerebellar growth is rapid and impeded by premature birth. Pediatrics. 2005;115:688–695. doi: 10.1542/peds.2004-1169. [DOI] [PubMed] [Google Scholar]
- 7.Volpe JJ. Cerebellum of the premature infant: Rapidly developing, vulnerable, clinically important. J Child Neurol. 2009;24:1085–1104. doi: 10.1177/0883073809338067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tavano A, Grasso R, Gagliardi C, Triulzi F, Bresolin N, Fabbro F, Borgatti R. Disorders of cognitive and affective development in cerebellar malformations. Brain. 2007;130:2646–2660. doi: 10.1093/brain/awm201. [DOI] [PubMed] [Google Scholar]
- 9.Kenney AM, Widlund HR, Rowitch DH. Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development. 2004;131:217–228. doi: 10.1242/dev.00891. [DOI] [PubMed] [Google Scholar]
- 10.Corrales JD, Blaess S, Mahoney EM, Joyner AL. The level of sonic hedgehog signaling regulates the complexity of cerebellar foliation. Development. 2006;133:1811–1821. doi: 10.1242/dev.02351. [DOI] [PubMed] [Google Scholar]
- 11.Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 12.Rakic P, Sidman RL. Histogenesis of cortical layers in human cerebellum, particularly the lamina dissecans. J Comp Neurol. 1970;139:473–500. doi: 10.1002/cne.901390407. [DOI] [PubMed] [Google Scholar]
- 13.Adams RD, Victor M, Ropper AH. Principles of Neurology. McGraw-Hill; New York: 1997. p. 1618. [Google Scholar]
- 14.Lewis PM, Gritli-Linde A, Smeyne R, Kottmann A, McMahon AP. Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Dev Biol. 2004;270:393–410. doi: 10.1016/j.ydbio.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 15.Kenney AM, Rowitch DH. Sonic hedgehog promotes G1 cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol. 2000;20:9055–9067. doi: 10.1128/mcb.20.23.9055-9067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. 2003;130:15–28. doi: 10.1242/dev.00182. [DOI] [PubMed] [Google Scholar]
- 17.Dubuc AM, Northcott PA, Mack S, Witt H, Pfister S, Taylor MD. The genetics of pediatric brain tumors. Curr Neurol Neurosci Rep. 2010;10:215–223. doi: 10.1007/s11910-010-0103-9. [DOI] [PubMed] [Google Scholar]
- 18.Schüller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, Ma Q, Alvarez-Buylla A, McMahon AP, Rowitch DH, Ligon KL. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beers MF, Shuman H, Liley HG, Floros J, Gonzales LW, Yue N, Ballard PL. Surfactant protein B in human fetal lung: Developmental and glucocorticoid regulation. Pediatr Res. 1995;38:668–675. doi: 10.1203/00006450-199511000-00007. [DOI] [PubMed] [Google Scholar]
- 20.Heine VM, Rowitch DH. Hedgehog signaling has a protective effect in glucocorticoid-induced mouse neonatal brain injury through an 11βHSD2-dependent mechanism. J Clin Invest. 2009;119:267–277. doi: 10.1172/JCI36376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edwards CR, Benediktsson R, Lindsay RS, Seckl JR. 11β-Hydroxysteroid dehydrogenases: Key enzymes in determining tissue-specific glucocorticoid effects. Steroids. 1996;61:263–269. doi: 10.1016/0039-128x(96)00033-5. [DOI] [PubMed] [Google Scholar]
- 22.Holmes MC, Sangra M, French KL, Whittle IR, Paterson J, Mullins JJ, Seckl JR. 11β-Hydroxysteroid dehydrogenase type 2 protects the neonatal cerebellum from deleterious effects of glucocorticoids. Neuroscience. 2006;137:865–873. doi: 10.1016/j.neuroscience.2005.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frey FJ, Escher G, Frey BM. Pharmacology of 11β-hydroxysteroid dehydrogenase. Steroids. 1994;59:74–79. doi: 10.1016/0039-128x(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 24.Diaz R, Brown RW, Seckl JR. Distinct ontogeny of glucocorticoid and mineralocorticoid receptor and 11β-hydroxysteroid dehydrogenase types I and II mRNAs in the fetal rat brain suggest a complex control of glucocorticoid actions. J Neurosci. 1998;18:2570–2580. doi: 10.1523/JNEUROSCI.18-07-02570.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beato M, Sánchez-Pacheco A. Interaction of steroid hormone receptors with the transcription initiation complex. Endocr Rev. 1996;17:587–609. doi: 10.1210/edrv-17-6-587. [DOI] [PubMed] [Google Scholar]
- 26.Datson NA, Morsink MC, Meijer OC, de Kloet ER. Central corticosteroid actions: Search for gene targets. Eur J Pharmacol. 2008;583:272–289. doi: 10.1016/j.ejphar.2007.11.070. [DOI] [PubMed] [Google Scholar]
- 27.Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of Smoothened activity. Proc Natl Acad Sci USA. 2002;99:14071–14076. doi: 10.1073/pnas.182542899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frank-Kamenetsky M, Zhang XM, Bottega S, Guicherit O, Wichterle H, Dudek H, Bumcrot D, Wang FY, Jones S, Shulok J, Rubin LL, Porter JA. Small-molecule modulators of Hedgehog signaling: Identification and characterization of Smoothened agonists and antagonists. J Biol. 2002;1:10. doi: 10.1186/1475-4924-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Becher OJ, Hambardzumyan D, Fomchenko EI, Momota H, Mainwaring L, Bleau AM, Katz AM, Edgar M, Kenney AM, Cordon-Cardo C, Blasberg RG, Holland EC. Gli activity correlates with tumor grade in platelet-derived growth factor–induced gliomas. Cancer Res. 2008;68:2241–2249. doi: 10.1158/0008-5472.CAN-07-6350. [DOI] [PubMed] [Google Scholar]
- 30.Lum L, Beachy PA. The Hedgehog response network: Sensors, switches, and routers. Science. 2004;304:1755–1759. doi: 10.1126/science.1098020. [DOI] [PubMed] [Google Scholar]
- 31.Masuya H, Inoue M, Wada Y, Shimizu A, Nagano J, Kawai A, Inoue A, Kagami T, Hirayama T, Yamaga A, Kaneda H, Kobayashi K, Minowa O, Miura I, Gondo Y, Noda T, Wakana S, Shiroishi T. Implementation of the modified-SHIRPA protocol for screening of dominant phenotypes in a large-scale ENU mutagenesis program. Mamm Genome. 2005;16:829–837. doi: 10.1007/s00335-005-2430-8. [DOI] [PubMed] [Google Scholar]
- 32.Kessler JD, Hasegawa H, Brun SN, Emmenegger BA, Yang ZJ, Dutton JW, Wang F, Wechsler-Reya RJ. N-myc alters the fate of preneoplastic cells in a mouse model of medulloblastoma. Genes Dev. 2009;23:157–170. doi: 10.1101/gad.1759909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Noguchi KK, Walls KC, Wozniak DF, Olney JW, Roth KA, Farber NB. Acute neonatal glucocorticoid exposure produces selective and rapid cerebellar neural progenitor cell apoptotic death. Cell Death Differ. 2008;15:1582–1592. doi: 10.1038/cdd.2008.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hintz SR, Kendrick DE, Wilson-Costello DE, Das A, Bell EF, Vohr BR, Higgins RD. NICHD Neonatal Research Network, Early-childhood neurodevelopmental outcomes are not improving for infants born at <25 weeks’ gestational age. Pediatrics. 2011;127:62–70. doi: 10.1542/peds.2010-1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dalziel SR, Lim VK, Lambert A, McCarthy D, Parag V, Rodgers A, Harding JE. Antenatal exposure to betamethasone: Psychological functioning and health related quality of life 31 years after inclusion in randomised controlled trial. BMJ. 2005;331:665. doi: 10.1136/bmj.38576.494363.E0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liggins GC, Howie RN. A controlled trial of antepartum glucocorticoid treatment for prevention of the respiratory distress syndrome in premature infants. Pediatrics. 1972;50:515–525. [PubMed] [Google Scholar]
- 37.Wilson-Costello D, Walsh MC, Langer JC, Guillet R, Laptook AR, Stoll BJ, Shankaran S, Finer NN, Van Meurs KP, Engle WA, Das A. Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network, Impact of postnatal corticosteroid use on neurodevelopment at 18 to 22 months’ adjusted age: Effects of dose, timing, and risk of bronchopulmonary dysplasia in extremely low birth weight infants. Pediatrics. 2009;123:e430–e437. doi: 10.1542/peds.2008-1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yeh TF, Lin YJ, Lin HC, Huang CC, Hsieh WS, Lin CH, Tsai CH. Outcomes at school age after postnatal dexamethasone therapy for lung disease of prematurity. N Engl J Med. 2004;350:1304–1313. doi: 10.1056/NEJMoa032089. [DOI] [PubMed] [Google Scholar]
- 39.Tam EWY, Chau V, Ferriero DM, Barkovich AJ, Poskitt KJ, Studholme C, Fok EDY, Grunau RE, Glidden DV, Miller SP. Preterm cerebellar growth impairment after post-natal exposure to glucocorticoids. Sci Transl Med. 2011;3:105ra105. doi: 10.1126/scitranslmed.3002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goodrich LV, Milenković L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 41.Romer JT, Curran T. Medulloblastoma and retinoblastoma: Oncology recapitulates ontogeny. Cell Cycle. 2004;3:917–919. [PubMed] [Google Scholar]
- 42.Gonzales LW, Ballard PL, Ertsey R, Williams MC. Glucocorticoids and thyroid hormones stimulate biochemical and morphological differentiation of human fetal lung in organ culture. J Clin Endocrinol Metab. 1986;62:678–691. doi: 10.1210/jcem-62-4-678. [DOI] [PubMed] [Google Scholar]
- 43.Venkatesh VC, Iannuzzi DM, Ertsey R, Ballard PL. Differential glucocorticoid regulation of the pulmonary hydrophobic surfactant proteins SP-B and SP-C. Am J Respir Cell Mol Biol. 1993;8:222–228. doi: 10.1165/ajrcmb/8.2.222. [DOI] [PubMed] [Google Scholar]