

Abstract
Chronological age is a well established risk factor for the development of cardiovascular diseases. The changes that accumulate in the vasculature with age, though, are highly variable. It is now increasingly recognized that indices of vascular health are more reliable than age per se in predicting adverse cardiovascular outcomes. The variation in the accrual of these age-related vascular changes is a function of multiple genetic and environmental factors. In this review, we highlight some of the pathophysiological mechanisms that characterize the vascular aging phenotype. Furthermore, we provide an overview of the key outcome studies that address the value of these vascular health indices in general and discuss potential effects on perioperative cardiovascular outcomes.
“You are as old as your arteries”
William Osler, MD
The Johns Hopkins Hospital, 1894
INTRODUCTION
Cardiovascular disease remains the leading cause of morbidity and mortality in the United States and other industrialized countries.1 The incidence of cardiovascular disease is predicted to increase as the population ages despite effective treatments for several established cardiovascular risk factors such as hypertension and hypercholesterolemia. This apparent incongruity results from the fact that age itself is associated with both morphological and functional changes in the vasculature. Moreover, the rate at which these changes develop exhibits considerable heterogeneity across individuals.2 The pathophysiological vascular changes that are most recognized by clinicians are those that are initiated in intima, i.e., atherosclerosis. However, other vascular abnormalities that lead to remodeling and stiffening of the media and adventitia occur with aging even in the absence of atherosclerosis.3-5 The latter contribution to cardiovascular disease is generally under-recognized and may explain in part why the predictive value of some conventional risk factors for cardiac disease, such as hypertension, decrease with age.6 Together, these observations have led to the emerging concept that “vascular age” rather than chronological age is the best predictor of cardiovascular events and mortality. Based on recent data showing that markers of vascular age are predictive of adverse cardiac, cerebrovascular, and renal outcomes in the general population, this concept may be applied to patients undergoing surgery.7-9 In this review, we provide a background for the pathophysiological basis of “vascular aging” and its implications for the perioperative care of an aging surgical population.
Morphological Changes with Arterial Aging
Aging is associated with morphological changes in all layers of the vascular tree. These changes include central aortic dilatation3 and thickening of the arterial wall even in the absence of atherosclerotic disease.10 The latter results mainly from intimal thickening but is also caused by sclerotic changes in the media and adventitia (Fig. 1).2 These changes result in increased vascular stiffness,11,12 central blood pressure augmentation,13 increased systolic and pulse pressures with a decrease in diastolic blood pressure,14 and a higher central blood pressure for any given peripheral (e.g., brachial) blood pressure.15
Figure 1.
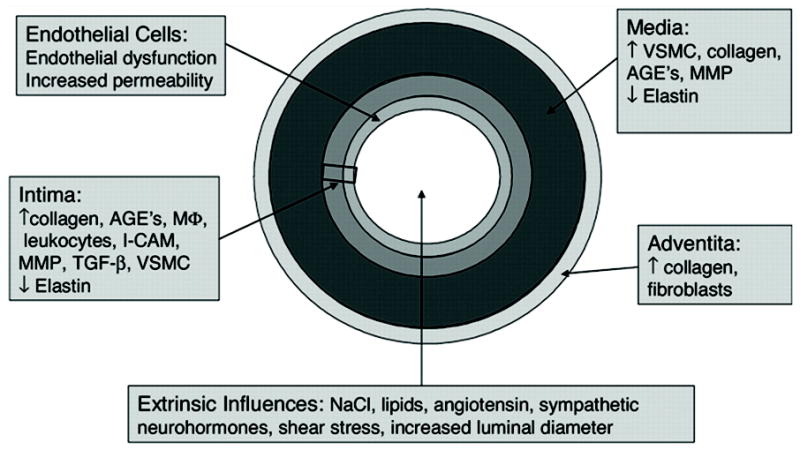
Summary of the multiple causes and locations of arterial stiffness. AGEs, advanced glycation end-products; I-CAM, Inter-Cellular Adhesion Molecule; MMP, matrix metalloproteinase; VSMC, vascular smooth muscle cells. (Arterioscler Thromb Vasc Biol 2005;25:932, reprinted with permission from the publisher, Wolters Kluwer Health).
Age-associated arterial wall thickening results from multiple pathophysiological processes. Many of these changes are not unique to aging per se, but also occur in patients with conventional risk factors for cardiovascular disease, such as hypertension, diabetes, renal insufficiency, and tobacco smoking. In general, arterial stiffness is related to the relative amounts of elastin and collagen in the vessel wall. The proximal parts of the aorta are more elastic than the distal vasculature and contain a relatively larger proportion of elastin compared to collagen.16 Age-associated collagen accumulation and fragmentation of elastin in the aortic wall, along with excessive intramural build-up of other proteins, such as integrins, fibronectin, and desmin, promote increased vascular stiffness.17,18 The increased accumulation of matrix metalloproteinases and fibronectin in the media likely contributes to matrix protein degradation.19-21 Overall, there is a tendency for vascular smooth muscle cells (VSMC) to change from cells characterized by contraction to cells characterized by synthesis/proliferation and migration. Although the mechanisms underlying these phenotype changes are complex, elastin fragments, which bind to elastin-laminin receptors found on the surface of VSMC (and other cells, including endothelial cells), are thought to be important contributors.22 Not only are VSMC phenotypically altered in aged vessels (increased VSMC size, decreased VSMC number), but they also migrate to the intima and contribute to increased intimal thickening.23,24
Age- or disease-related activation of inflammation also contributes to vascular stiffening. Increased activity of inflammatory cytokines, such as monocyte chemoattractant protein-1 and transforming growth factor β1, promotes deposition of matrix proteins and collagen as well as VSMC proliferation (discussed above), each of which contributes to intimal thickening.24 With age, vascular endothelial cells exhibit not only functional changes, but also morphological changes, including increased nuclear polyploidy and changes in the cytoskeleton.
Another contributor to age-related vascular stiffening is an increase in cross-link formation of extracellular matrix proteins. At least two separate mechanisms have been proposed to cause high levels of cross-link formation: advanced glycation end-products (AGE products) and the increased activity of the enzyme transglutaminase 2. AGE products (of which HgbA1C is an example) are formed by a non-enzymatic reaction between reducing sugars, such as glucose, and proteins, including collagen.25 This process is similar to that exploited with caramelization of sugars during cooking (e.g., hard topping of crème du lait). AGE-related cross-link formation of extracellular matrix proteins contributes to the stiffening of the arterial wall. The process is increasingly recognized to be important in many senescence-related changes in the cardiovascular system, such as endothelial dysfunction, atherosclerosis, myocardial dysfunction, and hypertension. Importantly, recent laboratory experiments have indicated that these changes can be pharmacologically modified, suggesting potential treatments for vascular stiffness. With age, excessive collagen deposition occurs in all connective tissues. Transglutaminase is responsible for irreversible cross-link formation between structural proteins.26 Increased transglutaminase activity in aged vessels could also contribute to increased vascular stiffness.
Data from the Framingham Heart Study have shown that aortic stiffness is a heritable trait.27 Genetic polymorphisms for the angiotensin receptor, metalloproteinases, fibrillin-1, and the endothelin pathway, among others, have been reported to influence central vascular stiffness,28-31 most likely by altering the expression or function of these vascular-related genes. Thus, age-related vascular stiffening is likely the net result of both genetic and environmental factors.
Normal Physiology of Vasculature
Central vascular stiffness is a function of the aorta and its main branches, which are embryologically, structurally, and physiologically distinguishable from distal arteries and arterioles.32-37 Central vessels function mainly to cushion and dampen the pressure oscillations produced by ventricular ejection and also to transfer energy and mechanical stress in the form of pulse waves along the vascular tree. Clinically, vascular stiffness reflects the cushion effect of the central vasculature tree and can be assessed by measuring pulse wave velocity, pulse pressure, or augmentation index (see below). The peripheral pulse results from a pressure wave that is generated by left ventricular (LV) ejection and acute distension of the aorta. A pressure wave is propagated antegrade during systole until it reaches branch points or arteries with abrupt lumen diameter changes, at which point the wave is reflected back to the central circulation. The final pressure wave in the central circulation is the sum of the forward and reflected waves,32 as shown in Figure 2. Both antegrade/incident and retrograde/reflected waves contribute to the actual pressure waveform. The propagation speed of the pulse wave is measured by the pulse wave velocity and increases with age in parallel with vascular stiffness (waves travel faster in a stiffer medium). The contribution of a reflected wave to the net pulse wave in the central circulation depends on the magnitude and the time when the reflected wave reaches the central arteries. The magnitude of wave reflection depends mainly on the geometry and stiffness of the peripheral muscular arteries/arterioles at the major reflecting sites.38 The contribution of pulse wave velocity to the waveform contour in the central circulation is critical. The net result is that in a compliant, non-stiff, usually young vasculature with elastic arteries, most reflected waves return to central circulation during early diastole and augment coronary blood flow. In contrast, in a stiff, usually elderly vasculature with noncompliant arteries, most reflected waves return to the central circulation sooner, during late systole, thereby increasing cardiac workload and decreasing coronary perfusion by decreasing diastolic pressure. Increased amplitude of reflected waves leading to central systolic deregulation is a major contributor to the development of LV hypertrophy in elderly patients.38
Figure 2.
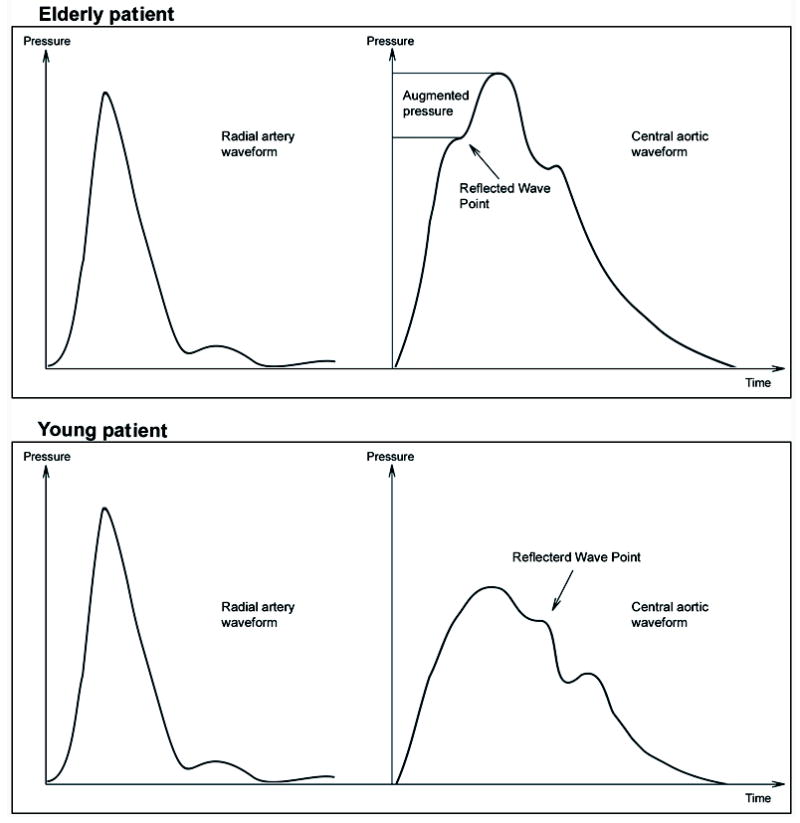
Illustration of the influence of increased vascular stiffness on peripheral (radial) and central (aortic) derived pressures. Note the similarity of peripheral radial pressures in individuals with normal (lower left panel) and increased (upper left panel) vascular stiffness. In young individuals with normal vascular stiffness, central aortic pressures are lower than radial pressures (lower panels). In contrast, in older individuals with increased vascular stiffness, central aortic pressures are increased and can approach or equal peripheral pressures as a result of wave reflection and central wave augmentation during systole (top panels).
The architecture of a normal arterial tree is elegant for its conduit and cushioning functions. The central aorta receives blood in pulses of stroke volume from the heart. The mean pressure decreases by only 1–2 mm Hg between the ascending aorta and a peripheral artery, indicating an excellent conduit function.39 The cushioning effect stores the energy of incoming blood during systole and gives it back to promote forward flow in diastole. The cushioning mechanism is so efficient that it requires only 10% more energy than if the heart output were continuous and nonpulsitile.40 The kinetic energy of each stroke volume contributes only 5% to the total blood flow. Ninety-five percent of the energy is potential energy stored in the distended conduit arteries. Because of the cushioning effect, most of the energy stored in blood pressure pulsations is transformed and dissipated in the major arteries, such that eventually, blood flow is almost laminar and steady through peripheral arterioles and capillaries. However, this is not the case for the renal, brain, and coronary beds. These vascular beds are similar to the central circulation in that they are exposed to pulsations and the associated energy as a result of the high flow into these organs. O’Rourke and Safar41 point out that the kidney and brain are unique because they are pulsating with blood flow. This condition occurs because they are continually and passively perfused at high-volume flow throughout both systole and diastole because their vascular resistance is very low, similar to that observed in other vascular beds during vasodilatation. In addition, wave reflections from kidney and brain are very low. As a result of high volume, low resistance, and wave reflections, the pulsations of pressure and flow extend well into the arterial network of these organs, and pulsations can be recorded even in their venous efflux. The small vessels in other organs are protected from intense pulsations by relatively intense vasoconstriction upstream. The brain and kidney are accustomed to such flow conditions under normal pulse pressures; however, they are susceptible to increased damage from high blood pressure fluctuations upstream as occurs during vascular aging and stiffening.
Blood pressure waveform changes as the pulse wave propagates along the vascular tree. In a young healthy vasculature, systolic blood pressure increases with distance from the heart, whereas diastolic blood pressure and mean arterial pressure decrease slightly (1–2 mmHg). These changes cause pulse pressure amplification of 10–15 mmHg between the central aorta and large peripheral arteries (e.g., radial artery). In reality, pulse pressure amplification is a protective mechanism because the LV needs to eject against a lower systolic blood pressure and hence against lower afterload. With age and development of vascular stiffness, pulse pressure amplification is attenuated and its protective role is lost, exposing the LV to augmented central pulse.42
Heart rate has been proposed to play an important role in central pressure waveform generation, such that reduced heart rate leads to an increase in central pressures with minimal effects on the peripheral pulse. One explanation for this phenomenon is related to the basic fact that cardiac output is the product of stroke volume and heart rate. For the same systemic vascular resistance (SVR, see below), a reduction in heart rate leads to an increase in stroke volume to maintain cardiac output and mean arterial blood pressure. When the central conduit arteries are compliant, such as in the young, they can accommodate the increased stroke volume and dampen central aortic pressure despite the higher volume of blood. This ability to accommodate is lacking in noncompliant or “stiff” central vasculature. In the latter, the increased stroke volume associated with slow heart rate is ejected into a much less distensible proximal aorta. Loss of distensibility and inability to accommodate increased incoming volume cause increases in central aortic systolic and pulse pressure even in the presence of unchanged SVR.
A second explanation for the observation that central aortic pressure increases with decreased heart rate is simply that decreasing heart rate prolongs the duration of cardiac ejection. In addition, a decreased contraction rate (dP/dt), as observed with negative inotropic drugs, will prolong systole. This prolongation will delay the time to peak of the outgoing incident pulse wave and cause the reflected wave to return in late systole for any given arterial stiffness and corresponding pulse wave velocity.43
Vascular Stiffness
Stiffening of major conduit arteries is referred to as arteriosclerosis (in distinction from atherosclerosis) and is the basis for the main manifestations of vascular aging: increased systolic blood pressure, decreased diastolic pressure, increased pulse pressure, and increased pulse wave velocity (Figs. 2 and 3).6,11,14,44-49 It is now generally accepted that increased arterial stiffness is a sensitive, early, noninvasive measure of vascular disease. Although arteriosclerosis and increased vascular stiffness frequently develop in aged/aging vessels in the absence of atherosclerosis, increased vascular stiffness is an inevitable development during atherosclerosis.50-52
Figure 3.
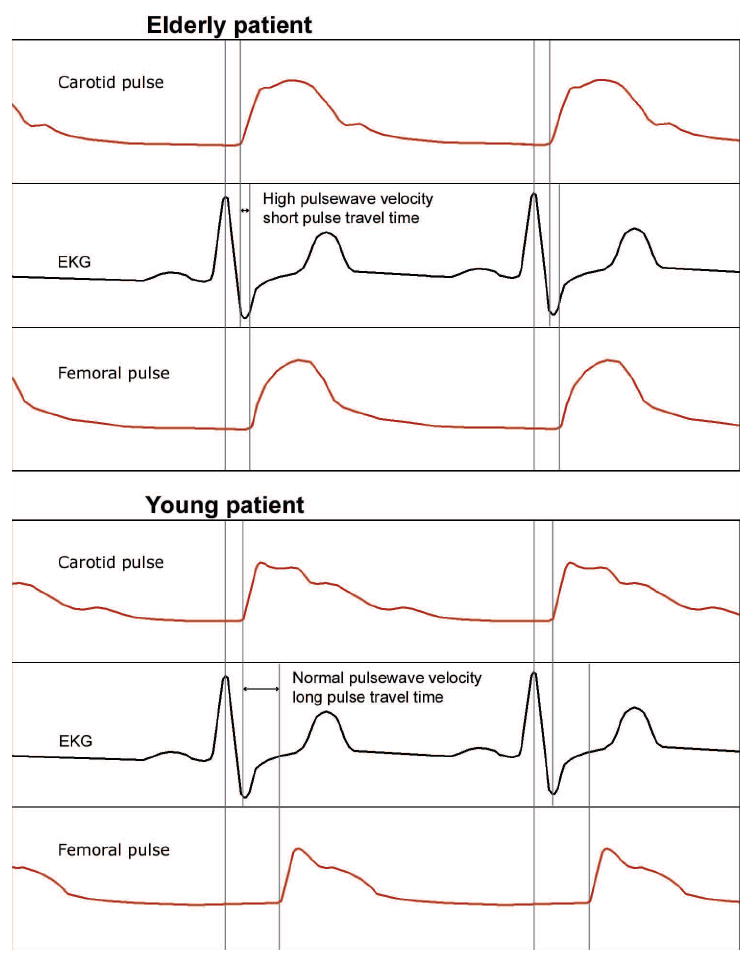
Example of pulse wave velocity measurements in two individuals, one with profoundly increased vascular stiffness (top panel) and the other with high normal vascular stiffness (lower panel). Pulse wave velocity is calculated as the distance (femoral—carotid) divided by time (derived from the electrocardiogram).
The measurement of pulse wave velocity has been successfully used to quantify aortic stiffness. Pulse wave velocity can increase two-fold between ages 20 and 80 years, a change that corresponds to a four-fold decrease in distensibility of the aorta.18 Thus, the early return of reflected arterial waves from the peripheral arterioles has the potential to increase the afterload on the LV four-fold (Fig. 3).53 The LV afterload is doubled by the decrease in aortic wall distensibility and doubled again by the early return of wave reflection in a heart-rate-dependent manner.53 Peripheral blood pressure also increases with age; however, the magnitude of this increase is less than that in the central circulation.
Blood pressure ultimately is determined by input into the central vasculature from the LV and outflow into the resistance arterioles. The outflow is continuous and clinically defined as SVR. The inflow into the central aorta, however, is pulsatile. Hence, LV afterload comprises two components: a static component composed of SVR, and a dynamic pulsatile component. Together they constitute arterial impedance or the true resistance against which the LV ejects. SVR, or arterial resistance (the static component), is the main determinant of LV arterial afterload under normal physiological circumstances in a healthy compliant vasculature. The pulsatile component contributes significantly to total LV afterload in stiff vessels.54 It consists of two phasic elements; one is compliance-related forward pulse wave and the second is reflected pulse wave. An increase in either the static or dynamic component may result in hypertension. Importantly, healthy aging is not associated with increased SVR. In contrast, central vascular stiffness, which may increase with aging, leads to an increased dynamic resistance and thus increased total impedance or LV afterload, systolic, and pulse pressures. As a result, isolated systolic hypertension, with normal diastolic blood pressure and increased pulse pressure, is the most common form of hypertension in the elderly. Systolic blood pressure increases progressively with age along with arterial stiffness, whereas diastolic blood pressure increases until middle age and then usually begins decreasing at approximately 45 years of age.14 Even in patients with diastolic hypertension (which is mainly due to increase in SVR), diastolic blood pressure stabilizes or even declines with age. The vascular impedance or the total afterload on the heart depends also on the pulse frequency and therefore heart rate. The relationship between pulse frequency (heart rate) and arterial impedance (afterload for the LV) is nonlinear; O’Rourke and Safar showed that decreasing the heart rate increases the arterial impedance.53 Sunugawa et al.55-57 suggested that total arterial impedance could be quantified by calculating the ratio of the pressure generated to the stroke volume ejected. This approach characterizes the total arterial load as an “effective elastance.”
As described above, vascular stiffness causes arterial waves to return to the central aorta at end-systole rather than during diastole; this change increases systolic and decreases diastolic blood pressure, thus increasing pulse pressure. Importantly, this phenomenon imparts increased loading conditions on the LV. Furthermore, the normal diastolic pulse augmentation is lost, which leads to reduced myocardial diastolic perfusion and a predisposition to myocardial ischemia.58-60 The main contributing factor to the increased afterload, LV hypertrophy, and increased LV mass is the increased amplitude of the reflected waves when they arrive at the central circulation.61
VASCULAR STIFFNESS AND CARDIOVASCULAR DISEASE
In a compliant vasculature, energy is dissipated in the central circulation by viscous dampening of reflected waves.32 Loss of this protective mechanism with vascular stiffening exposes organs to high pulsatile pressure that leads to arterial remodeling and microcirculatory damage in the brain and kidney.53,62-65 Vascular stiffening is associated with cerebral small vessel changes, including small lacunar infarcts and white matter lesions that are common in individuals with cognitive impairment and dementia.66-68 Vascular stiffness is also associated with endothelial dysfunction that can lead to neuronal injury by compromising cerebral blood flow and the blood-brain barrier.53,62-66,69,70 Hence, it is plausible that chronic small vessel changes associated with vascular stiffness might predispose patients to impaired cerebral and renal blood flow and end-organ damage.
The heart also undergoes changes with age. Increased vascular stiffness causes increased afterload and leads to the development of myocardial hypertrophy, diastolic dysfunction, and heart failure. In addition, the myocardium undergoes age-related changes that are mechanistically related to those in the vasculature. Ultimately, increased vascular stiffness and the resultant increase in myocardial loading conditions lead to a coupled increase in both systolic and diastolic myocardial stiffness (Fig. 4). As arterial stiffness increases, LV systolic stiffness increases in parallel. That is, the changes in the vasculature and LV are coupled. Eventually, however, beyond moderate increases in arterial stiffness, the processes uncouple because the central vasculature loses its ability to accommodate the stroke volume; the result is less efficient LV ejection. In contrast to indices of stiffness, indices of contractility do not correlate with age, suggesting that arterial stiffness, and by inference structural elements in the myocardium, may be important factors in age-related changes. Moreover, it has been clearly demonstrated that indices of vascular stiffness (increased pulse pressure, decreased arterial elastance, increased pulse wave velocity) and myocardial systolic stiffness increase with age.71,72 However, SVR does not increase with age. Vascular-ventricular coupling of stiffer vessels with a stiffer heart has an overall deleterious effect on pressure and flow regulation (including coronary blood flow), resulting in decreased cardiovascular reserve and a lower threshold for the development of cardiovascular disease.71,72
Figure 4.
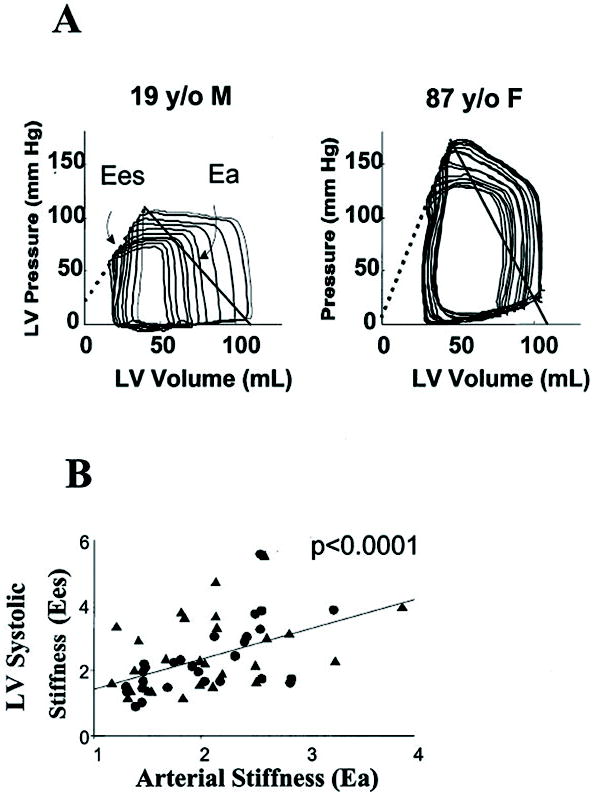
A, Example pressure-volume loops with ventricular systolic (Ees) and effective arterial elastance (Ea) relations from a young and old patient. Ees defines chamber systolic stiffness, and the intersection point of the Ea and Ees lines is the ventricular-arterial equilibrium set point. The ratio of Ea/Ees is used to index relative coupling between the heart and vascular systems. In both instances, the Ea and Ees values are similar to each other, coupling with a relative ratio near 1.0. However, in the elderly subject, both variables are increased, consistent with both vascular stiffening and ventricular systolic stiffening. Note the relatively small changes in left ventricular (LV) pressure with changes in LV volume in the young patient. In contrast, the elderly patient exhibits increases in LV diastolic pressure with similar increases in LV volume. This indication of diastolic dysfunction portends marked fluctuations in blood pressure with changes in LV preload as may occur with drug-induced venodilation. B, Group data from 57 patients. There is a significant relation between an increase in effective arterial stiffness reflected by Ea and ventricular systolic stiffness as reflected by Ees. Tandem increases in both characterize aging (Heart Failure Reviews 7:51-62, 2002, reprinted with permission by the publisher, Springer).
There is a clear link between increased vascular stiffness and development of diastolic dysfunction. This pathological LV hypertrophy in aged myocardium is due not only to cardiac myocyte hypertrophy but also to the significant increase in connective tissue content that leads to pathological remodeling and heart fibrosis. The sclerotic changes in the heart cause LV stiffness, which leads to reduced LV compliance—a decreased ability of the LV to accommodate diastolic blood volume without high pressures. Decreased LV compliance manifests mainly during diastole; LV diastolic filling becomes impaired, and higher filling pressures are required to achieve the same LV diastolic volume. LV filling from the left atrium is impeded, leading to an increase in left atrial pressure. This high pressure is transmitted backwards to pulmonary veins and capillaries, and the resulting interstitial fluid accumulation is manifested as shortness of breath and even pulmonary edema.
The combination of LV diastolic dysfunction and vascular stiffness might have particular relevance for acute hemodynamic perturbations as may occur perioperatively or in critically ill patients. Arteriolar remodeling may necessitate higher arterial pressure for brain and kidney perfusion. At the same time, preload dependence associated with diastolic dysfunction may predispose patients to marked fluctuations in blood pressure that result from alterations in intravascular blood volume. As illustrated in Figure 4, older patients operate on the steeper portion of the diastolic pressure-volume relationship compared with younger individuals. The clinical consequence is that small changes in the preload (defined as end-diastolic LV volume) will cause relatively large changes in systemic blood pressure. Other mechanisms may also contribute to end-organ (brain, kidney) damage in the setting of increased vascular stiffness. Autoregulation in the brain and kidney may become impaired or lower autoregulatory limits may develop in patients with increased vascular stiffness. Organ hypoperfusion may result when such individuals are exposed to what otherwise might be considered an “acceptable” blood pressure.73
Clinical Measurements of Vascular Stiffness
Increased arterial stiffness is a manifestation of vascular aging and is accompanied by increased central aortic pressure. However, peripheral blood pressure measurements do not fully reflect the central aortic pressure profile. Thus, the reliance on peripheral blood pressure measurements as markers of vascular alteration can underestimate vascular aging even in asymptomatic individuals. Several indices of vascular stiffness have been used in population-based studies, including brachial artery pulse pressure, central aorta augmentation index, pulse wave velocity, aortic distensibility, and the magnitude of reflected wave. These have been shown to be robust predictive indices of adverse cardiovascular outcomes in population-based studies and notably are independent of conventional risk factors, including age itself (Table 1).6,11,48,74
Table 1.
Pivotal studies linking vascular stiffness and adverse cardiovascular events in community-dwelling and cardiac surgical patients.
| Authors | Study Population | N | Main findings (verbatim) |
|---|---|---|---|
| Laurent et al.44 | Adult patients with essential hypertension | 1980 | After a mean follow-up of 112 ± 53 months, PWV was significantly associated with all-cause and cardiovascular mortality independent of previous cardiovascular diseases, age, and diabetes. |
| Sutton-Tyrrell et al.48 | The Health Aging and Body Composition (ABC) Study | 2488 | Arterial PWV was associated with both total mortality and cardiovascular mortality. Arterial PWV was also significantly associated with coronary heart disease (P=0.007) and stroke (P=0.001) after adjustment for age, gender, race, systolic blood pressure, known cardiovascular disease, and other variables related to events. |
| Mattace-Raso et al.6 | The Rotterdam Study | 2835 | PWV was an independent predictor of coronary heart disease and stroke after adjustment for age, gender, MAP, and heart rate. The aortic PWV index provided additional predictive value above cardiovascular risk factors, measures of atherosclerosis, and PP. |
| Hansen, et al.11 | Monica | 1678 | After 9.4 years of follow-up, PWV was significantly (P<0.05) related to cardiovascular mortality and fatal and nonfatal coronary heart disease after adjusting for sex, age, body mass index, MAP measured in the office, PP, ambulatory BP monitoring (24-h PP), smoking, and alcohol intake. For each 1-SD increment in PWV (3.4 m/s), the risk of an event increased by 16% to 20%. |
| Kingwell, et al.98 | Patients with CAD who underwent a treadmill exercise test | 96 | Within a patient group with moderate CAD, large-artery stiffness was a major determinant of myocardial ischemic threshold. |
| Guerin AP.74 | Patients with ESRD were followed for 51 months | 150 | In patients with ESRD, the insensitivity of PWV to decreased BP was an independent predictor of mortality; use of ACE inhibitors had a favorable effect on survival that was independent of BP changes. |
| Hansen, et al.11,99 | Population study in Europeans 40–70 years old with mean follow-up of 9.4 years | 1829 (1678) | In a randomly recruited European population, the AASI was a strong predictor of stroke beyond traditional cardiovascular risk factors, including MAP and PP. |
| Dolan et al.100 | Dublin Study | 11,291 | Over a median follow-up of 5.3 years AASI derived from ambulatory BP readings and PP predicted total cardiovascular mortality, but AASI was a stronger predictor than PP for stroke with the opposite trend for cardiac mortality. In subjects with normal daytime ambulatory BP (<135/<85 mm Hg), AASI was more predictive than PP of cardiovascular mortality and of stroke mortality, whereas neither independently predicted cardiac mortality. |
| Williams et al.88 | CAFE study | 2199 | Despite similar reductions in brachial systolic BPs between treatments, amlodipine±perindopril led to reductions in central aortic pressure compared with an atenolol±thiazide regimen. Based on Cox proportional-hazards modeling, central PP was significantly associated with the composite outcome of total cardiovascular events/procedures and development of renal impairment. |
| Aronson et al.7 | McSPI database of CABG surgery patients | 4801 | PP was an independent predictor of postoperative renal dysfunction and/or failure such that for every additional 20-mm Hg increment in PP >40 mm Hg, there was an OR of 1.49 (CI, 1.17–1.89; P=0.001). Patients with PP hypertension >80 mm Hg were 3 times more likely to die from a renal-related death than those without (3.7% vs. 1.1%). |
| Benjo et al.8 | CABG surgery patients at the Johns Hopkins Hospital | 703 | PP was higher in patients with stroke than in non-stroke patients (81.2 mm Hg vs. 64.5 mm Hg; P=0.0006). PP was an independent predictor of stroke based on adjusted Cox hazard modeling (hazard ratio: 2.62; 95% CI: 1.49–4.60, respectively; P=0.001 for every 10-mm Hg increase in PP. |
| Fontes et al.9 | McSPI database of CABG surgery patients | 5436 patients | PP increments of 10 mm Hg (above a threshold of 40 mm Hg) were associated with an increased risk of cerebral events. The incidence of a cerebral event and/or death from neurologic complications nearly doubled for patients with PP > 80 mm Hg versus ≤80 mm Hg (5.5% vs. 2.8%; P = 0.004). PP > 80 mm Hg was associated with congestive heart failure (P=0.003) and death from cardiac cause (P=0.006). |
AASI, ambulatory arterial stiffness index; ACE, angiotensin converting enzyme; BP, blood pressure; CABG, coronary artery bypass graft; CAD, coronary artery disease; ESRD, end-stage renal disease; MAP, mean arterial blood pressure; OR, odds ratio; PP, pulse pressure; PWV, pulse wave velocity
Historically, the “gold standard” for arterial stiffness assessment is the measurement of pulse wave velocity (Fig. 3). Velocity is defined as change in distance over change in time. For pulse wave velocity, distance is measured at two separate points along the arterial tree, usually the carotid and femoral arteries or the carotid and radial arteries. The time interval is determined by measuring the time between the electrocardiogram R wave and the start or peak of the pulse wave at each of the arterial sites of measurement to derive the “time elapsed” as shown in Figure 3. This measurement is obtained by timing the arrival of the pulse to peripheral sites (e.g., radial and femoral pulses) compared with more central pulses (e.g., the carotid pulse) in relation to a fixed time point, the electrocardiogram QRS complex. The pulse wave velocity in a young person is approximately 6 m/s; it increases to 10 m/s by the age of 65 years and continues to increase with advancing age.
Carotid-femoral pulse wave velocity constitutes a useful, safe, reproducible, and noninvasive method for assessing arterial stiffness.75 Increased carotid-femoral pulse wave velocity has been shown to be an independent predictor of cardiovascular events in the general population,6,11 in the elderly,48 and in patients with hypertension,44,64 diabetes mellitus,76 and end-stage renal disease.77 Recent meta-analyses have confirmed that measures of arterial stiffness (e.g., pulse wave velocity) independently predict adverse cardiovascular outcomes and all-cause mortality.78,79 The pooled relative risk for cardiovascular events (myocardial infarction, stroke, revascularization) was 2.26 (95% confidence interval [CI]: 1.89 to 2.70; 14 studies) and the pooled relative risk for cardiovascular mortality was 2.02 (95% CI: 1.68 to 2.42; 10 studies) for subjects with high versus low aortic pulse wave velocity. As expected, the relative risk was higher in patients with coronary artery disease, renal disease, and hypertension compared with the general population. An increase in aortic pulse wave velocity by 1 m/s corresponded to an age-, sex-, and risk factor-adjusted risk increase of 15% for adverse events and mortality. An increase in aortic pulse wave velocity by 1 standard deviation was associated with a relative risk increase of 47% for these events.79 Vascular stiffness can be assessed noninvasively, and thus, can be incorporated into routine clinical assessements.80 Noninvasive assessment of arterial stiffness usually falls into one of three categories: measurement of pulse wave velocity, determination of arterial distensibility, or assessment of central arterial pressure augmentation (e.g., augmentation index) and pulse pressure. Pulse wave velocity measurements have been suggested as a means of assessing subclinical target organ damage.81
Indices of vascular stiffness all increase with advancing age; however, the age-related changes in augmentation index and aortic pulse wave velocity are nonlinear. Some investigators have suggested that augmentation index might be a more sensitive marker of arterial stiffening and cardiovascular risk in younger individuals because this measure increases at a younger age compared to other measurements. In contrast, aortic pulse wave velocity is thought to be a better measure of vascular risk in older individuals, because prominent changes in pulse wave velocity have been observed only in older individuals.82 Indeed, arterial pulse wave velocity, but not augmentation index, has been shown to be associated with the extent of coronary artery disease.83 The potential explanation for the lower predictive value of augmented pressure and augmentation index in predicting adverse cardiovascular events compared to pulse wave velocity may be that the former measurement depends on the timing of arrival of a reflected wave to the central circulation. Pulse wave velocity and magnitude of the reflected waves are time-independent measures of arterial stiffness that predict long-term cardiovascular mortality independent of conventional risk factors and other measures of arterial stiffness84. The magnitude of the reflected wave is derived from pulse wave analysis via a mathematical algorithm.84
Recently, MRI has been suggested as another noninvasive modality for assessing central arterial compliance/stiffness.85 Aortic arch distensibility as assessed by MRI was found to be the most sensitive marker of arterial aging in individuals <50 years of age, whereas aortic arch MRI-derived pulse wave velocity was more sensitive in individuals >50 years of age. This study found a dramatic decrease in aortic arch distensibility by the third decade of life in individuals otherwise free of overt cardiovascular disease. Although the authors reported that the relationship between age and aortic stiffness was nonlinear, these results suggest that MRI might be useful for detecting early and subclinical vascular disease.
Several devices to measure arterial stiffness are commercially available (Table 2). Most derive their measurements from peripherally acquired waveforms by using tonometry. Central pressure waveforms are then extrapolated by mathematical modeling based on transfer function of waveforms. These methods have been validated against central aortic measurements.86 Measurements can be made easily with good inter- and intraoperator reproducibility.87
Table 2.
Commercial devices that measure central arterial stiffness
| Device | Manufacturer | Measure | Pros | Cons |
|---|---|---|---|---|
| SphygmoCor | AtCor Medical (www.atcormedical.com) | PWV via applanation tonometry | Measures central PWV and AIx | Limited by technical difficulty in obtaining measurements |
| Colin VP-1000 | Omron Healthcare (www.omronhealthcare.com) | PWV, AIx, ABI | Measures central AIx | Does not measure central PWV |
| HDI/PulseWave CR-2000 and CVProfilor DO-2020 | Hypertension Diagnostics (www.hdi-pulsewave.com) | Large- and small-vessel elasticity (compliance) | Distinguishes stiffness of small and large peripheral arteries | Measures compliance expressed as elasticity, which is limited by variations in compliance throughout the arterial tree largely as a result of change in vessel size |
| Complior | Artech Medical (www.artech-medical.com) | PWV | Obtains central PWV, taking simultaneous measures at carotid and femoral sites | Digitized waveforms create difficulty in discerning “foot” (arrival time) of the wave |
| PulseTrace PWV and PCA 2 | Micro Medical (www.micromedical.co.uk) | PWV via photoplethysmography | Measures central PWV | SI and RI lack reproducibility, outcome data |
ABI, ankle-brachial index; AIx, augmentation index; PWV, pulse wave velocity; RI, reflection index; SBP, systolic blood pressure; SI, stiffness index; SVR, systemic vascular resistance.
From DeLoach SS, Townsend RR. Vascular stiffness: its measurement and significance for epidemiologicand outcome studies. Clin J Am Soc Nephrol 2008;3:184-92.
The predictive value of vascular stiffness in surgical patients has become increasingly appreciated.7-9 Pulse pressure was shown to be an independent predictor of renal,7 neurological,8 and cardiovascular events9 in patients undergoing cardiac surgery. Thus, there is emerging evidence that measurements of vascular stiffness provide unique prognostic information for future cardiovascular events in nonsurgical and surgical settings. These findings provide not only a means for improving current risk stratification models but also the potential for interventions to improve patient outcomes.
Pharmacologic Modification of Vascular Stiffness
Data from studies in nonsurgical patients suggest that therapy to reduce central arterial stiffness may improve patient outcomes independent of blood pressure reduction. In the REASON study, angiotensin blockade was shown to reduce arterial stiffness, wave reflections, and central pulse pressure independent of mean arterial blood pressure reduction after 1 year of treatment. In addition to decreasing SVR, blockade of angiotensin II restores the remodeling of large arteries and decreases the thickness of resistance arteries; the resulting changes in arterial morphology, in turn, lead to a reduction in pressure wave reflections and augmentation index 43.
The Conduit Artery Function Evaluation (CAFE) Study88 demonstrated that an antihypertensive regimen consisting of an angiotensin converting enzyme inhibitor and a calcium antagonist preferentially reduced central vascular pressures (and presumably central vascular stiffness) and decreased the long-term cardiovascular event rates compared with a regimen of β-blockers and diuretics, even though both regimens had similar effects on peripheral blood pressure (Fig. 5). The CAFE study suggested that β-blockers may have been less beneficial because they are less effective at decreasing central aortic pressure. Subsequent analysis of the CAFE study’s heart rate data showed that heart rate reduction was the main reason that β-blocker–based therapies were less effective than the others at reducing central pressure.89 In addition, it has been shown that augmentation index is inversely related to heart rate, such that slow heart rate leads to increased augmentation index and high central aortic pressures, but only minimal increases in peripheral systolic blood pressure.90,91 The authors of the CAFE heart rate study found that the main impact of heart rate reduction was on the augmentation of the reflected wave, which increased by 3 mm Hg per 10 beats/min reduction in heart rate, with minimal effect on the incident outgoing pressure waves. Similarly, after adjustment for the brachial blood pressure, heart rate was the major determinant of central systolic and pulse pressure, pressure wave reflections, and pulse pressure amplification. Even more importantly, after the authors adjusted for heart rate, the differences in central systolic and pulse pressures between treatment arms were no longer significant, and the differences in indices of central blood pressure augmentation were minimal. The authors suggested that reduction in heart rate was the major reason that β-blocker–based therapy has been less effective at reducing cardiovascular events, especially stroke, when compared with other treatments. In addition, the decrease in pulse pressure observed with nitrates, calcium channel blocking drugs, and angiotensin converting enzyme inhibitors has a marked benefit on microvascular function and may explain their ability to protect brain and kidney function.92
Figure 5.
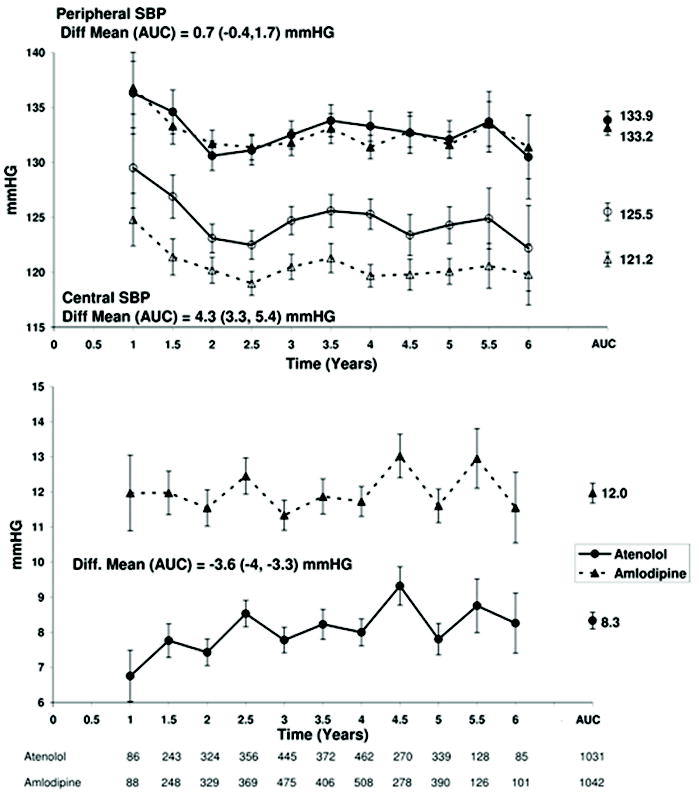
Top: Changes in brachial (solid symbols) and derived central aortic (open symbols) pulse pressure (PP) over time (mean, 95% CI) for patients randomized to receive atenolol±thiazide- or amlodipine±perinodopril-based therapy. Bottom, Changes in PP difference (brachial – derived central aortic; mean, 95% CI) over time. For calculation of area under the curve (AUC), see the data supplement numbers below the abscissa, which represent the number of patients seen at each time point. Time represents the period from randomization to patient follow-up visit at which tonometry measurement was made in the CAFE study (Circulation 2006;113:1213, reprinted with permission from the publisher, Wolters Kluwer Health).88
The suggestive protective effect of calcium channel blockers was illustrated in a study by Aronson et al.93 that assessed the efficacy and safety of the novel, ultra-short-acting dihydropyridine calcium channel blocker clevidipine. The efficacy of treating acute hypertension with clevidipine, nicardipine, nitroglycerin, and sodium nitroprusside was assessed by analyzing the duration and extent of blood pressure excursions beyond predetermined upper and lower limits. Notably, sodium nitroprusside and nitroglycerine were associated with longer time periods outside the predefined blood pressure limits, and this was associated with a tendency toward increased mortality, stroke, and renal dysfunction. However the authors measured only peripheral blood pressure; no data regarding central aortic pressure were reported. Given the known cardioprotective role of statin therapy (e.g., ASCOT trial), the CAFE-LLA study was designed to investigate whether the beneficial effects of atorvastatin were at least partially due to a decrease in central aortic pressures. CAFE-LLA demonstrated no effects of atorvastatin on either central aortic pressures or hemodynamic indices.94
All conventional strategies with well-established benefits in the prevention of cardiovascular events improve endothelial function. Such strategies include physical exercise training, avoidance of stress, and smoking cessation. The decreased cardiovascular risk is associated with a reduction in vascular stiffness. Specifically, regular physical exercise has been reported to slow the increases in arterial stiffness with age and improve arterial compliance in healthy subjects and in patients with chronic inflammatory diseases.95-97
Finally, it is important to emphasize that two different patients with the same peripheral blood pressure-decreasing effect from antihypertensive therapy may have completely different central aortic pressure responses, depending on vascular stiffness. Thus, indices of vascular stiffness may not only improve the precision of risk stratification models for patients undergoing surgery, but may also allow select patients to benefit from treatment paradigms targeting patient-specific hemodynamic end points.
Despite the importance of, growing interest in, and number of studies focusing on indices of vascular stiffness and central aortic pressure in the medical literature, only a few studies have been performed in a surgical population. Given the importance of pulsatile phenomena for the cardiovascular system and its dependence on vascular age, we strongly believe that measurements of vascular stiffness and central aortic pressure should be used in the perioperative period as predictors of adverse outcomes and to set goals for hemodynamic management. Future studies will explore the perioperative implications of vascular aging and provide evidence-based guidance.
CONCLUSIONS
Cardiovascular disease attributable to aging remains the leading cause of mortality in Western countries despite a growing number of effective treatments and preventative therapies. Increased arterial stiffness is a direct manifestation of early vascular aging and is accompanied by increased central aortic pressure. Current data suggest that aortic pulse wave velocity and augmentation index are the best available noninvasive estimates of arterial stiffness in patients above and below 50 years, respectively. Furthermore, these values are more sensitive than conventional risk factors at predicting cardiovascular outcomes. Emerging data have linked increased pulse pressure with stroke, renal failure, and mortality after cardiac surgery, thus providing evidence that measures of vascular stiffness may provide important risk-stratifying information. Data show that blood pressure-decreasing therapies specifically aimed at reducing central aortic pressure may provide additional therapeutic advantages in everyday clinical practice beyond their essential blood pressure- decreasing effects. Whether therapies aimed at decreasing central aortic pressure can modify risk for patients undergoing cardiac and noncardiac surgery remains to be explored.
Acknowledgments
Funding: Supported in part by a grant from the NIH (R01) AG021523 to (DEB).
Footnotes
The authors declare no conflicts of interest.
Contributor Information
Viachaslau M. Barodka, The Department of Anesthesiology/Critical Care Medicine, The Johns Hopkins Medical Institutions, Baltimore, Maryland.
Brijen L. Joshi, The Department of Anesthesiology/Critical Care Medicine, The Johns Hopkins Medical Institutions, Baltimore, Maryland.
Dan E. Berkowitz, The Department of Anesthesiology/Critical Care Medicine, The Johns Hopkins Medical Institutions, Baltimore, Maryland.
Charles W. Hogue, Jr, The Department of Anesthesiology/Critical Care Medicine, The Johns Hopkins Medical Institutions, Baltimore, Maryland.
Daniel Nyhan, The Department of Anesthesiology/Critical Care Medicine, The Johns Hopkins Medical Institutions, Baltimore, Maryland.
References
- 1.American Heart Association. Association AH. Dallas, TX: 2009. Heart Disease and Stroke Statistics — 2009 Update. [Google Scholar]
- 2.Farb A, Burke AP, Tang AL, Liang TY, Mannan P, Smialek J, Virmani R. Coronary plaque erosion without rupture into a lipid core. A frequent cause of coronary thrombosis in sudden coronary death. Circulation. 1996;93:1354–63. doi: 10.1161/01.cir.93.7.1354. [DOI] [PubMed] [Google Scholar]
- 3.Lakatta EG. Cardiovascular regulatory mechanisms in advanced age. Physiol Rev. 1993;73:413–67. doi: 10.1152/physrev.1993.73.2.413. [DOI] [PubMed] [Google Scholar]
- 4.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular sisease enterprises: Part I: Aging arteries: a “set up” for vascular disease. Circulation. 2003;107:139–46. doi: 10.1161/01.cir.0000048892.83521.58. [DOI] [PubMed] [Google Scholar]
- 5.Najjar SS, Scuteri A, Lakatta EG. Arterial aging: is it an immutable cardiovascular risk factor? Hypertension. 2005;46:454–62. doi: 10.1161/01.HYP.0000177474.06749.98. [DOI] [PubMed] [Google Scholar]
- 6.Mattace-Raso FS, van der Cammen TJ, Hofman A, van Popele NM, Box ML, Schalekamp MA, Asmar R, Reneman RS, Hoeks AP, Breteler MM, Witteman JC. Arterial stiffness and risk of coronary heart disease and stroke. The Rotterdam study. Circulation. 2006;113:657–3. doi: 10.1161/CIRCULATIONAHA.105.555235. [DOI] [PubMed] [Google Scholar]
- 7.Aronson S, Fontes ML, Miao Y, Mangano DT Investigators of the Multicenter Study of Perioperative Ischemia Research Group, Ischemia Research and Education Foundation. Risk index for perioperative renal dysfunction/failure: critical dependence on pulse pressure hypertension. Circulation. 2007;115:733–42. doi: 10.1161/CIRCULATIONAHA.106.623538. [DOI] [PubMed] [Google Scholar]
- 8.Benjo A, Thompson RE, Fine D, Hogue CW, Alejo D, Kaw A, Gerstenblith G, Shah A, Berkowitz DE, Nyhan D. Pulse pressure is an age-independent predictor of stroke development after cardiac surgery. Hypertension. 2007;50:630–5. doi: 10.1161/HYPERTENSIONAHA.107.095513. [DOI] [PubMed] [Google Scholar]
- 9.Fontes ML, Aronson S, Mathew JP, Miao Y, Drenger B, Barash PG, Mangano DT Multicenter Study of Perioperative Ischemia (McSPI) Research Group, Ischemia Research and Education Foundation (IREF) Investigators. Pulse pressure and risk of adverse outcome in coronary bypass surgery. Anesth Analg. 2008;107:1122–9. doi: 10.1213/ane.0b013e31816ba404. [DOI] [PubMed] [Google Scholar]
- 10.Nagai Y, Metter EJ, Earley CJ, Kemper MK, Becker LC, Lakatta EG, Fleg JL. Increased carotid artery intimal-medial thickness in asymptomatic older subjects with exercise-induced myocardial ischemia. Circulation. 1998;98:1504–9. doi: 10.1161/01.cir.98.15.1504. [DOI] [PubMed] [Google Scholar]
- 11.Hansen TW, Staessen JA, Torp-Pedersen C, Rasmussen S, Thijs L, Ibsen H, Jeppesen J. Prognostic value of aortic pulse wave velocity as index of arterial stiffness in the general population. Circulation. 2006;113:664–70. doi: 10.1161/CIRCULATIONAHA.105.579342. [DOI] [PubMed] [Google Scholar]
- 12.Vaikevicius PV, Fleg JL, Engel JH, O’Connor FC, Wright JG, Lakatta LE, Yin FC, Lakatta EG. Effects of age and aerobic capacity on arterial stiffness in healthy adults. Circulation. 1993;88:1456–62. doi: 10.1161/01.cir.88.4.1456. [DOI] [PubMed] [Google Scholar]
- 13.Nichols WW. Clinical measurement of arterial stiffness obtained from noninvasive pressure waveforms. Am J Hypertens. 2005;18:3S–10S. doi: 10.1016/j.amjhyper.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 14.Franklin SS, Gustin WI, Wong ND, Larson MG, Weber MA, Kannel WB, Levy D. Hemodynamic patterns of age-related changes in blood pressure: The Framingham Heart Study. Circulation. 1997;96:308–15. doi: 10.1161/01.cir.96.1.308. [DOI] [PubMed] [Google Scholar]
- 15.Wilkinson IB, Franklin SS, Hall IR, Tyrrell S, Cockcroft JR. Pressure amplification explains why pulse pressure is unrelated to risk in young subjects. Hypertension. 2001;38:1461–6. doi: 10.1161/hy1201.097723. [DOI] [PubMed] [Google Scholar]
- 16.Harkness ML, Harkness RD, McDonald DA. The collagen and elastin content of the arterial wall in the dog. Proc R Soc Lond B Biol Sci. 1957;146:541–51. doi: 10.1098/rspb.1957.0029. [DOI] [PubMed] [Google Scholar]
- 17.Cattan V, Kakou A, Louis H, Lacolley P. Pathophysiology, genetic, and therapy of arterial stiffness. Biomed Mater Eng. 2006;16:S155–61. [PubMed] [Google Scholar]
- 18.O’Rourke MF, Nichols WW. Aortic diameter, aortic stiffness, and wave deflection increase with age and isolated systolic hypertension. Hypertension. 2005;45:652–8. doi: 10.1161/01.HYP.0000153793.84859.b8. [DOI] [PubMed] [Google Scholar]
- 19.Pauly RR, Passaniti A, Crow M, Kinsella JL, Papadopoulos N, Monticone R, Lakatta EG, Martin GR. Experimental models that mimic the differentiation and dedifferentiation of vascular cells. Circulation. 1992;86:III68–III73. [PubMed] [Google Scholar]
- 20.Wang M, Takagi G, Asai K, Resuello RG, Natividad FF, Vatner DE, Vatner SF, Lakatta EG. Aging Increases Aortic MMP-2 Activity and Angiotensin II in Nonhuman Primates. Hypertension. 2003;41:1308–16. doi: 10.1161/01.HYP.0000073843.56046.45. [DOI] [PubMed] [Google Scholar]
- 21.Wang M, Lakatta EG. Altered regulation of matrix metalloproteinase-2 in aortic remodeling during aging. Hypertension. 2002;39:865–73. doi: 10.1161/01.hyp.0000014506.13322.66. [DOI] [PubMed] [Google Scholar]
- 22.Mathew JP, Fontes ML, Tudor IC, Ramsay J, Duke P, Mazer CD, Barash PG, Hsu PH, Mangano DT Investigators of the Ischemia Research and Education Foundation; Multicenter Study of Perioperative Ischemia Research Group. A multicenter risk index for atrial fibrillation after cardiac surgery. J Am Med Assoc. 2004;291:1720–9. doi: 10.1001/jama.291.14.1720. [DOI] [PubMed] [Google Scholar]
- 23.Li Z, Cheng H, Lederer WJ, Froehlich J, Lakatta EG. Enhanced Proliferation and Migration and Altered Cytoskeletal Proteins in Early Passage Smooth Muscle Cells from Young and Old Rat Aortic Explants. Experimental and Molecular Pathology. 1997;64:1–11. doi: 10.1006/exmp.1997.2204. [DOI] [PubMed] [Google Scholar]
- 24.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–7. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 25.Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol. 2005;25:932–43. doi: 10.1161/01.ATV.0000160548.78317.29. [DOI] [PubMed] [Google Scholar]
- 26.Santhanam L, Tuday EC, Webb AK, Dowzicky P, Kim JH, Oh YJ, Sikka G, Kuo M, Halushka MK, Macgregor AM, Dunn J, Gutbrod S, Yin D, Shoukas A, Nyhan D, Flavahan N, Belkin AM, Berkowitz DE. Decreased S-Nitrosylation of Tissue Transglutaminase Contributes to Age-Related Increases in Vascular Stiffness. Circ Res. 2010;107:117–25. doi: 10.1161/CIRCRESAHA.109.215228. [DOI] [PubMed] [Google Scholar]
- 27.Mitchell GF, DeStefano AL, Larson MG, Benjamin EJ, Chen MH, Vasan RS, Vita JA, Levy D. Heritability and a genome-wide linkage scan for arterial stiffness, wave reflection, and mean arterial pressure: the Framingham Heart Study. Circulation. 2005;112:194–9. doi: 10.1161/CIRCULATIONAHA.104.530675. [DOI] [PubMed] [Google Scholar]
- 28.Benetos A, Gautier S, Ricard S, Topouchian J, Asmar R, Poirier O, Larosa E, Guize L, Safar M, Soubrier F, Cambien F. Influence of angiotensin-converting enzyme and angiotensin II type 1 receptor gene polymorphisms on aortic stiffness in normotensive and hypertensive patients. Circulation. 1996;94:698–703. doi: 10.1161/01.cir.94.4.698. [DOI] [PubMed] [Google Scholar]
- 29.Medley TL, Cole TJ, Gatzka CD, Wang WY, Dart AM, Kingwell BA. Fibrillin-1 genotype is associated with aortic stiffness and disease severity in patients with coronary artery disease. Circulation. 2002;105:810–5. doi: 10.1161/hc0702.104129. [DOI] [PubMed] [Google Scholar]
- 30.Medley TL, Kingwell BA, Gatzka CD, Pillay P, Cole TJ. Matrix metalloproteinase-3 genotype contributes to age-related aortic stiffening through modulation of gene and protein expression. Circ Res. 2003;92:1254–61. doi: 10.1161/01.RES.0000076891.24317.CA. [DOI] [PubMed] [Google Scholar]
- 31.Lajemi M, Gautier S, Poirier O, Baguet JP, Mimran A, Gosse P, Hanon O, Labat C, Cambien F, Benetos A. Endothelin gene variants and aortic and cardiac structure in never-treated hypertensives. Am J Hypertens. 2001;14:755–60. doi: 10.1016/s0895-7061(01)02162-8. [DOI] [PubMed] [Google Scholar]
- 32.Safar M, Levy B, Struijker-Boudier H. Current perspectives on arterial stiffness and pulse pressure in hypertension and cardiovascular diseases. Circulation. 2003;107:2864–9. doi: 10.1161/01.CIR.0000069826.36125.B4. [DOI] [PubMed] [Google Scholar]
- 33.Davidson JM, Hill KE, Mason ML, Giro MG. Longitudinal gradients of collagen and elastin gene expression in the porcine aorta. J Biol Chem. 1985;260:1901–8. [PubMed] [Google Scholar]
- 34.Dingemans KP, Teeling P, Lagendijk JH, Becker AE. Extracellular matrix of the human aortic media: an ultrastructureal histochemical and immunohistochemical study of the adult aortic media. Anat Rec. 2000;258:1–14. doi: 10.1002/(SICI)1097-0185(20000101)258:1<1::AID-AR1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 35.Rosenquist TH, Beall AC, Modis L, Fishman R. Impaired elastic matrix development in the great arteries after ablation of the cardiac neural crest. Anat Rec. 1990;226:347–59. doi: 10.1002/ar.1092260312. [DOI] [PubMed] [Google Scholar]
- 36.Bevan JA, Bevan RD, Hwa JJ, Owen MP, Tayo FM. Calcium regulation in vascular smooth muscle: is there a pattern to its variability within the arterial tree? J Cardiovasc Pharmacol. 1986;8:S71–S5. [PubMed] [Google Scholar]
- 37.Waldo KL, Kumiski DH, Kirby ML. Development of the great arteries. Boston: Birkenhauser; 1998. [Google Scholar]
- 38.Safar ME. Mechanism(s) of systolic blood pressure reduction and drug therapy in hypertension. Hypertension. 2007;50:167–71. doi: 10.1161/HYPERTENSIONAHA.107.088799. [DOI] [PubMed] [Google Scholar]
- 39.Pauca AL, Wallenhaupt SL, Kon ND, Tucker WY. Does radial artery pressure accurately reflect aortic pressure? Chest. 1992;102:1193–8. doi: 10.1378/chest.102.4.1193. [DOI] [PubMed] [Google Scholar]
- 40.O’Rourke MF. Steady and pulsatile energy losses in the systemic circulation under normal conditions and in simulated arterial disease. Cardiovasc Res. 1967;1:313–26. doi: 10.1093/cvr/1.4.313. [DOI] [PubMed] [Google Scholar]
- 41.O’Rourke MF, Safar ME. Relationship between aortic stiffening and microvascular disease in brain and kidney: cause and logic of therapy. Hypertension. 2005;46:200–4. doi: 10.1161/01.HYP.0000168052.00426.65. [DOI] [PubMed] [Google Scholar]
- 42.Avolio AP, Van Bortel LM, Boutouyrie P, Cockcroft JR, McEniery CM, Protogerou AD, Roman MJ, Safar ME, Segers P, Smulyan H. Role of pulse pressure amplification in arterial hypertension: experts’ opinion and review of the data. Hypertension. 2009;54:375–83. doi: 10.1161/HYPERTENSIONAHA.109.134379. [DOI] [PubMed] [Google Scholar]
- 43.London GM, Asmar RG, O’Rourke MF, Safar ME. Mechanism(s) of selective systolic blood pressure reduction after a low-dose combination of perindopril/indapamide in hypertensive subjects: comparison with atenolol. J Am Coll Cardiol. 2004;43:92–9. doi: 10.1016/j.jacc.2003.07.039. [DOI] [PubMed] [Google Scholar]
- 44.Laurent S, Boutouyrie P, Asmar R, Gautier I, Laloux B, Guize L, Ducimetiere P, Benetos A. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension. 2001;37:1236–41. doi: 10.1161/01.hyp.37.5.1236. [DOI] [PubMed] [Google Scholar]
- 45.Smulyan H, Asmar RG, Rudnicki A, London GM, Safar ME. Comparative effects of aging in men and women on theproperties of the arterial tree. J Am Coll Card. 2001;37:1374–80. doi: 10.1016/s0735-1097(01)01166-4. [DOI] [PubMed] [Google Scholar]
- 46.Mitchell GR, Parise H, Benjamin EJ, Larson MG, Keyes MJ, Vita JA, Vasan RS, Levy D. Changes in arterial stiffness and wave reflection with advancing age in healthy men and wome: the Framingham Heart Study. Hypertension. 2004;43:1239–45. doi: 10.1161/01.HYP.0000128420.01881.aa. [DOI] [PubMed] [Google Scholar]
- 47.Izzo JLJ, Shykoff BE. Arterial stiffness: clinical relevance, measurement, and treatment. Rev Cardiovasc Med. 2001;2:29–34. [PubMed] [Google Scholar]
- 48.Sutton-Tyrrell K, Najjar SS, Boudreau RM, Venkitachalam L, Kupelian V, Simonsick EM, Havlik R, Lakatta EG, Spurgeon H, Kritchevsky S, Pahor M, Bauer D, Newman A for the Health ABC Study. Elevated aortic pulse wave velocity, a marker of arterial stiffness, predicts cardiovascular events in well-functioning older adults. Circulation. 2005;111:3384–90. doi: 10.1161/CIRCULATIONAHA.104.483628. [DOI] [PubMed] [Google Scholar]
- 49.McEniery CM, Yasmin, Wallace S, Maki-Petaja K, McDonnell B, Sharman JE, Retallick C, Franklin SS, Brown MJ, Lloyd RC, Cockcroft JR, Wilkinson IB on behalf of the ENIGMA Study Investigators. Increased stroke volume and aortic stiffness contribute to isolated systolic hypertension in young adults. Hypertension. 2005;46:221–6. doi: 10.1161/01.HYP.0000165310.84801.e0. [DOI] [PubMed] [Google Scholar]
- 50.Cohn JN, Duprez DA, Grandits GA. Arterial elasticity as part of a comprehensive assessment of cardiovascular risk and drug treatment. Hypertension. 2005;46:217–20. doi: 10.1161/01.HYP.0000165686.50890.c3. [DOI] [PubMed] [Google Scholar]
- 51.Grey E, Bratteli C, Glasser SP, Alinder C, Finkelstein SM, Lindgren BR, Cohn JN. Reduced small artery but not large artery elasticity is an independent risk marker for cardiovascular events. Amer J Hyperten. 2003;16:265–9. doi: 10.1016/s0895-7061(02)03271-5. [DOI] [PubMed] [Google Scholar]
- 52.Cohn JN. Arterial stiffness, vascular disease, and risk of cardiovascular events. Circulation. 2006;113:601–3. doi: 10.1161/CIRCULATIONAHA.105.600866. [DOI] [PubMed] [Google Scholar]
- 53.O’Rourke MF, Safar ME. Relationship between aortic stiffening and microvascular disease in brain and kidney. Hypertension. 2005;46:200–4. doi: 10.1161/01.HYP.0000168052.00426.65. [DOI] [PubMed] [Google Scholar]
- 54.Hayward CD, Kelly RP. Gender-related differences in teh central arterial pressure waveform. J Am Coll Card. 1997;30:1863–71. doi: 10.1016/s0735-1097(97)00378-1. [DOI] [PubMed] [Google Scholar]
- 55.Sunagawa K, Maughan WL, Burkhoff D. Left ventricular interaction with arterial load studied in isolated canine ventricle. Am J Physiol. 1983;245:H773–H80. doi: 10.1152/ajpheart.1983.245.5.H773. [DOI] [PubMed] [Google Scholar]
- 56.Sunagawa K, Sagawa K, Maughan WL. Ventricular interaction with the vascular system in terms of pressure-volume relationships. In: Yin FCP, editor. Ventriculo-vascular coupling: clinical, physiologic, and engineering aspects. New York: Springer Verlag; 1987. pp. 210–39. [Google Scholar]
- 57.Sunagawa K, Maughan WL, Sagawa K. Optimal arterial resistance for hte maximal stroke work studied in isolated canine left ventricle. Circ Res. 1985;56:586–95. doi: 10.1161/01.res.56.4.586. [DOI] [PubMed] [Google Scholar]
- 58.Lartaud-Idjouadiene I, Lompre A-M, Kieffer P, Colas T, Atkinson J. Cardiac consequences of prolonged exposure to an isolated increase in aortic stiffness. Hypertension. 1999;34:63–9. doi: 10.1161/01.hyp.34.1.63. [DOI] [PubMed] [Google Scholar]
- 59.Roman MJ, Ganau A, Saba PS, Pini R, Pickering TG, Devereux RB. Impact of arterial stiffening on left ventricular structure. Hypertension. 2000;36:489–94. doi: 10.1161/01.hyp.36.4.489. [DOI] [PubMed] [Google Scholar]
- 60.Kass DA. Ventricular arterial stiffening: integrating the pathophysiology. Hypertension. 2005;46:185–93. doi: 10.1161/01.HYP.0000168053.34306.d4. [DOI] [PubMed] [Google Scholar]
- 61.Hashimoto J, Westerhof BE, Westerhof N, Imai Y, O’Rourke MF. Different role of wave reflection magnitude and timing on left ventricular mass reduction during antihypertensive treatment. J Hypertens. 2008;26:1017–24. doi: 10.1097/HJH.0b013e3282f62a9b. [DOI] [PubMed] [Google Scholar]
- 62.Rizzoni D, Porteri E, Boari GE, De Ciuceis C, Sleiman I, Muiesan ML, Castellano M, Miclini M, Agabiti-Rosei E. Prognostic significance of small-artery structure in hypertension. Circulation. 2003;108:2230–5. doi: 10.1161/01.CIR.0000095031.51492.C5. [DOI] [PubMed] [Google Scholar]
- 63.Liao D, Cooper L, Cai J, Toole J, Bryan N, Burke G, Shahar E, Nieto J, Mosley T, Heiss G. The prevalence and severity of white matter lesions, their relationship with age, ethnicity, gender, and cardiovascular disease risk factors: the ARIC Study. Neuroepidemiology. 1997;16:149–62. doi: 10.1159/000368814. [DOI] [PubMed] [Google Scholar]
- 64.Laurent S, Katsahian S, Fassot C, Tropeano A-I, Gautier I, Laloux B, Boutouyrie P. Aortic stiffness is an independent predictor of fatal stroke in essential hypertension. Stroke. 2003;34:1203–6. doi: 10.1161/01.STR.0000065428.03209.64. [DOI] [PubMed] [Google Scholar]
- 65.Boutouyrie P, Bussy C, Lacolley P, Girerd X, Laloux B, Laurent A. Association between local pulse pressure, mean blood pressure and large artery remodeling. Circulation. 1999;100:1087–93. doi: 10.1161/01.cir.100.13.1387. [DOI] [PubMed] [Google Scholar]
- 66.Hanon O, Haulon S, Lenoir H, Seux ML, Rigaud AS, Safar M, Girerd X, Forette F. Relationship between arterial stiffness and cognitive function in elderly subjects wit complaints of memory loss. Stroke. 2005;36:2193–7. doi: 10.1161/01.STR.0000181771.82518.1c. [DOI] [PubMed] [Google Scholar]
- 67.Pasquier F, Leys D. Why are stroke patients prone to develop dementia? J Neurol. 1997;244:135–42. doi: 10.1007/s004150050064. [DOI] [PubMed] [Google Scholar]
- 68.Breteleler MM, van Swieten JC, Bots ML, Grobbee DE, Claus JJ, van den Hout JH, van Harskamp F, Tanghe HL, de Jon PT, van Gijn J. Cerebral white matter lesions, vascualar risks factors, and cognitive function in a populatino-based study: the Rotterdam study. Neurology. 1994;44:1246–52. doi: 10.1212/wnl.44.7.1246. [DOI] [PubMed] [Google Scholar]
- 69.Hardy JA, Mann DM, Wester P, Winbland B. An integrative hypothesis concerning the pahtogenesis and progression of Alzheimer’s disease. Neurobiol Aging. 1986;7:489–502. doi: 10.1016/0197-4580(86)90086-2. [DOI] [PubMed] [Google Scholar]
- 70.Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. Beta-amyloid-mediate vascoactiveity and vascualr endothelial damage. Nature. 1996;380:168–71. doi: 10.1038/380168a0. [DOI] [PubMed] [Google Scholar]
- 71.Kass DA. Age-related changes in ventricular-arterial coupling: pathophysiologic implications. Heart Fail Rev. 2002;7:51–62. doi: 10.1023/a:1013749806227. [DOI] [PubMed] [Google Scholar]
- 72.Kass DA, Saeki A, Tunin RS. Adverse influence of systemic vascular stiffening on cardiac dysfunction and adaptation to acute coronary occlusion. Circulation. 1996;93:1533–41. doi: 10.1161/01.cir.93.8.1533. [DOI] [PubMed] [Google Scholar]
- 73.Nyhan D, Berkowitz DE. Perioperative blood pressure management: does central vascular stiffness matter? Anesth Analg. 2008;107:1103–6. doi: 10.1213/ane.0b013e318182d864. [DOI] [PubMed] [Google Scholar]
- 74.Guerin AP, Blacher J, Pannier B, Marchais SJ, Safar ME, London GM. Impact of aortic stiffness attenuation on survival of patients in end-stage renal failure. Circulation. 2001;103:987–92. doi: 10.1161/01.cir.103.7.987. [DOI] [PubMed] [Google Scholar]
- 75.Asmar R, Benetos A, Topouchian J, Laurent P, Pannier B, Brisac AM, Target R, Levy BI. Assessment of arterial distensibility by automatic pulse wave velocity measurement. Validation and clinical application studies. Hypertension. 1995;26:485–90. doi: 10.1161/01.hyp.26.3.485. [DOI] [PubMed] [Google Scholar]
- 76.Cruickshank K, Riste L, Anderson SG, Wright JS, Dunn G, Gosling RG. Aortic pulse-wave velocity and its relationship to mortality in diabetes and glucose intolerance: an integrated index of vascular function? Circulation. 2002;106:2085–90. doi: 10.1161/01.cir.0000033824.02722.f7. [DOI] [PubMed] [Google Scholar]
- 77.Blacher J, Guerin AP, Pannier B, Marchais SJ, Safar ME, London GM. Impact of aortic stiffness on survival in end-stage renal disease. Circulation. 1999;99:2434–9. doi: 10.1161/01.cir.99.18.2434. [DOI] [PubMed] [Google Scholar]
- 78.Vlachopoulos C, Aznaouridis K, O’Rourke MF, Safar ME, Baou K, Stefanadis C. Prediction of cardiovascular events and all-cause mortality with central haemodynamics: a systematic review and meta-analysis. Eur Heart J. 2010;31:1819–22. doi: 10.1093/eurheartj/ehq024. [DOI] [PubMed] [Google Scholar]
- 79.Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of cardiovascular events and all-cause mortality with arterial stiffness: a systematic review and meta-analysis. J Am Coll Cardiol. 2010;55:1318–27. doi: 10.1016/j.jacc.2009.10.061. [DOI] [PubMed] [Google Scholar]
- 80.DeLoach SS, Townsend RR. Vascular stiffness: its measurement and significance for epidemiologic and outcome studies. Clin J Am Soc Nephrol. 2008;3:184–92. doi: 10.2215/CJN.03340807. [DOI] [PubMed] [Google Scholar]
- 81.Mancia G, De Backer G, Dominiczak A, Cifkova R, Fagard R, Germano G, Grassi G, Heagerty AM, Kjeldsen SE, Laurent S, Narkiewicz K, Ruilope L, Rynkiewicz A, Schmieder RE, Boudier HA, Zanchetti A, Vahanian A, Camm J, De Caterina R, Dean V, Dickstein K, Filippatos G, Funck-Brentano C, Hellemans I, Kristensen SD, McGregor K, Sechtem U, Silber S, Tendera M, Widimsky P, Zamorano JL, Erdine S, Kiowski W, Agabiti-Rosei E, Ambrosioni E, Lindholm LH, Viigimaa M, Adamopoulos S, Bertomeu V, Clement D, Farsang C, Gaita D, Lip G, Mallion JM, Manolis AJ, Nilsson PM, O’Brien E, Ponikowski P, Redon J, Ruschitzka F, Tamargo J, van Zwieten P, Waeber B, Williams B. 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC) J Hypertens. 2007;25:1105–87. doi: 10.1097/HJH.0b013e3281fc975a. [DOI] [PubMed] [Google Scholar]
- 82.McEniery CM, Yasmin, Hall IR, Qasem A, Wilkinson IB, Cockcroft JR. Normal vascular aging: differential effects on wave reflection and aortic pulse wave velocity: the Anglo-Cardiff Collaborative Trial (ACCT) J Am Coll Cardiol. 2005;46:1753–60. doi: 10.1016/j.jacc.2005.07.037. [DOI] [PubMed] [Google Scholar]
- 83.Hope SA, Antonis P, Adam D, Cameron JD, Meredith IT. Arterial pulse wave velocity but not augmentation index is associated with coronary artery disease extent and severity: implications for arterial transfer function applicability. J Hypertens. 2007;25:2105–9. doi: 10.1097/HJH.0b013e3282a9be41. [DOI] [PubMed] [Google Scholar]
- 84.Wang KL, Cheng HM, Sung SH, Chuang SY, Li CH, Spurgeon HA, Ting CT, Najjar SS, Lakatta EG, Yin FC, Chou P, Chen CH. Wave reflection and arterial stiffness in the prediction of 15-year all-cause and cardiovascular mortalities: a community-based study. Hypertension. 2010;55:799–805. doi: 10.1161/HYPERTENSIONAHA.109.139964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Redheuil A, Yu WC, Wu CO, Mousseaux E, de Cesare A, Yan R, Kachenoura N, Bluemke D, Lima JA. Reduced ascending aortic strain and distensibility: earliest manifestations of vascular aging in humans. Hypertension. 2010;55:319–26. doi: 10.1161/HYPERTENSIONAHA.109.141275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen CH, Nevo E, Fetics B, Pak PH, Yin FC, Maughan WL, Kass DA. Estimation of central aortic pressure waveform by mathematical transformation of radial tonometry pressure. Validation of generalized transfer function. Circulation. 1997;95:1827–36. doi: 10.1161/01.cir.95.7.1827. [DOI] [PubMed] [Google Scholar]
- 87.Wilkinson IB, Fuchs SA, Jansen IM, Spratt JC, Murray GD, Cockcroft JR, Webb DJ. Reproducibility of pulse wave velocity and augmentation index measured by pulse wave analysis. J Hypertens. 1998;16:2079–84. doi: 10.1097/00004872-199816121-00033. [DOI] [PubMed] [Google Scholar]
- 88.Williams B, Lacy P, Thom SM, Cruickshank K, Stanton A, Collier D, Hughes AD, Thurston H. Differential impact of blood pressure-lowering drugs on central aortic pressure and clinical outcomes. Circulation. 2006;113:1213–25. doi: 10.1161/CIRCULATIONAHA.105.595496. [DOI] [PubMed] [Google Scholar]
- 89.Williams B, Lacy PS. Impact of heart rate on central aortic pressures and hemodynamics: analysis from the CAFE (Conduit Artery Function Evaluation) study: CAFE-Heart Rate. J Am Coll Cardiol. 2009;54:705–13. doi: 10.1016/j.jacc.2009.02.088. [DOI] [PubMed] [Google Scholar]
- 90.Wilkinson IB, MacCallum H, Flint L, Cockcroft JR, Newby DE, Webb DJ. The influence of heart rate on augmentation index and central arterial pressure in humans. J Physiol. 2000;525(Pt 1):263–70. doi: 10.1111/j.1469-7793.2000.t01-1-00263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wilkinson IB, Mohammad NH, Tyrrell S, Hall IR, Webb DJ, Paul VE, Levy T, Cockcroft JR. Heart rate dependency of pulse pressure amplification and arterial stiffness. Am J Hypertens. 2002;15:24–30. doi: 10.1016/s0895-7061(01)02252-x. [DOI] [PubMed] [Google Scholar]
- 92.Safar ME. Peripheral pulse pressure, large arteries, and microvessels. Hypertension. 2004;44:121–2. doi: 10.1161/01.HYP.0000135448.73199.75. [DOI] [PubMed] [Google Scholar]
- 93.Aronson S, Dyke CM, Stierer KA, Levy JH, Cheung AT, Lumb PD, Kereiakes DJ, Newman MF. The ECLIPSE trials: comparative studies of clevidipine to nitroglycerin, sodium nitroprusside, and nicardipine for acute hypertension treatment in cardiac surgery patients. Anesth Analg. 2008;107:1110–21. doi: 10.1213/ane.0b013e31818240db. [DOI] [PubMed] [Google Scholar]
- 94.Williams B, Lacy PS, Cruickshank JK, Collier D, Hughes AD, Stanton A, Thom S, Thurston H. Impact of statin therapy on central aortic pressures and hemodynamics: principal results of the Conduit Artery Function Evaluation-Lipid-Lowering Arm (CAFE-LLA) Study. Circulation. 2009;119:53–61. doi: 10.1161/CIRCULATIONAHA.108.785915. [DOI] [PubMed] [Google Scholar]
- 95.Tanaka H, Dinenno FA, Monahan KD, Clevenger CM, DeSouza CA, Seals DR. Aging, habitual exercise, and dynamic arterial compliance. Circulation. 2000;102:1270–5. doi: 10.1161/01.cir.102.11.1270. [DOI] [PubMed] [Google Scholar]
- 96.Vivodtzev I, Minet C, Wuyam B, Borel JC, Vottero G, Monneret D, Baguet JP, Levy P, Pepin JL. Significant improvement in arterial stiffness after endurance training in patients with COPD. Chest. 2010;137:585–92. doi: 10.1378/chest.09-1437. [DOI] [PubMed] [Google Scholar]
- 97.Sharman JE, McEniery CM, Dhakam ZR, Coombes JS, Wilkinson IB, Cockcroft JR. Pulse pressure amplification during exercise is significantly reduced with age and hypercholesterolemia. J Hypertens. 2007;25:1249–54. doi: 10.1097/HJH.0b013e3280be5911. [DOI] [PubMed] [Google Scholar]
- 98.Kingwell BA, Waddell TK, Medley TL, Cameron JD, Dart AM. Large artery stiffness predicts ischemic threshold in patients with coronary artery disease. J Am Coll Cardiol. 2002;40:773–9. doi: 10.1016/s0735-1097(02)02009-0. [DOI] [PubMed] [Google Scholar]
- 99.Hansen TW, Staessen JA, Torp-Pedersen C, Rasmussen S, Li Y, Dolan E, Thijs L, Wang JG, O’Brien E, Ibsen H, Jeppesen J. Ambulatory arterial stiffness index predicts stroke in a general population. J Hypertens. 2006;24:2247–53. doi: 10.1097/01.hjh.0000249703.57478.78. [DOI] [PubMed] [Google Scholar]
- 100.Dolan E, Thijs L, Li Y, Adams N, McCormack P, McClory S, O’Brien E, Staessen J, Stanton A. Ambulatory arterial stiffness index as a predictor of cardiovascular mortality in the dublin outcome study. Hypertension. 2006;47:365–70. doi: 10.1161/01.HYP.0000200699.74641.c5. [DOI] [PubMed] [Google Scholar]