

Abstract
Mesoporous silica nanoparticles have the capacity to load and deliver therapeutic cargo and incorporate imaging modalities, making them prominent candidates for theranostic devices. One of the most widespread imaging agents utilized in this and other theranostic platforms is nanoscale superparamagnetic iron oxide. Although several core-shell magnetic mesoporous silica nanoparticles presented in the literature have provided high T2 contrast in vitro and in vivo, there is ambiguity surrounding which parameters lead to enhanced contrast. Additionally, there is a need to understand the behavior of these imaging agents over time in biologically relevant environments. Herein, we present a systematic analysis of how the transverse relaxivity (r2) of magnetic mesoporous silica nanoparticles is influenced by nanoparticle diameter, iron oxide nanoparticle core synthesis, and the use of a hydrothermal treatment. This work demonstrates that samples which did not undergo a hydrothermal treatment experienced a drop in r2 (75% of original r2 within 8 days of water storage), while samples with hydrothermal treatment maintained roughly the same r2 for over 30 days in water. Our results suggest that iron oxide oxidation is the cause of the r2 loss, and this oxidation can be prevented both during synthesis and storage by the use of deoxygenated conditions during nanoparticle synthesis. The hydrothermal treatment also provides colloidal stability, even in acidic and highly salted solutions, and a resistance against acid degradation of the iron oxide nanoparticle core. The results of this study show the promise of multifunctional mesoporous silica nanoparticles but will also likely inspire further investigation into multiples types of theranostic devices, taking into consideration their behavior over time and in relevant biological environments.
Keywords: transverse relaxivity, mesoporous silica, iron oxide, nanoparticles, hydrothermal treatment
INTRODUCTION
Theranostics, devices which enable diagnosis, therapy, and monitoring of therapeutic efficacy, are becoming increasingly important tools for treating diseases such as cancer. The ability to monitor treatment easily can inform physicians regarding choices in dosage or even the type of drug used. Nanoparticle (NP) platforms are well suited for incorporation of imaging and therapy functionalities for this type of personalized medicine.1 In particular, mesoporous silica (MS) NPs have emerged as prominent candidates for theranostics.2,3 Thus far, various types of imaging agents, including fluorescent dyes,4 gold nanorods,5 radionuclides (64Cu),6 superparamagnetic iron oxide NPs (SPIONs, Fe3O4 and γ-Fe2O3)7,8 and gadolinium complexes,9 have been incorporated into MS NPs while maintaining a high drug loading capacity in the same NP. These materials have enabled MS NP use in various types of bio-imaging, including fluorescence, computed tomography (CT), positron emission tomography (PET), and magnetic resonance imaging (MRI).4–9 Among these multifunctional MS NPs, SPION-containing MS NPs are one of the most prominent in the literature because they can be used both as separation tools and as T2 MRI contrast agents. The quality of an MRI contrast agent is generally measured via the relaxivity, ri (i=1 or 2), which describes the ability of a contrast agent to shorten the T1 or T2 relaxation time of water. For SPION-based MRI contrast agents, the signal enhancement ability (r1 or r2), is determined by several factors: (1) NP size, (2) NP composition, (3) NP surface coating, and (4) synergistic magnetism effects from multiple SPIONs.10–22
To incorporate SPIONs into MS NPs, some groups have attached SPIONs to the exterior of MS NPs, creating stimuli-sensitive ‘capped’ MS NPs for controlled drug release applications.23,24 Others have synthesized magnetic NPs inside the pores of MS NPs.25 Alternative syntheses are based on a core-shell architecture, which is the focus of the present investigation. Many groups have developed syntheses of core-shell magnetic MS NPs (mMS NPs),7,8,26,27 which display a range of conflicting r2 values. These differences may be attributable to core differences; for example, Kim et al. achieved an r2 of 245 mM−1s−1 with 15-nm diameter SPION cores synthesized via decomposition of iron oleate,8 but Lin et al. found an r2 of 153 mM−1s−1 with 10-nm diameter cores synthesized by decomposition of iron acetylacetonate.7 To put this difference in perspective, a 20 % greater dose of the lower r2 mMS NPs would be required to obtain the same contrast as the other mMS NPs on a MR image. Despite the fact that such large discrepancies exist in literature-reported systems, there is little understanding behind the causes of such differences. Based on these literature precedents and others, it is clear that SPIONs prepared using different methods lead to size and crystallinity differences that may influence the r2 values; however, it is not immediately clear whether size, crystallinity, or other variables are the major contributors to r2 differences in mMS NPs. To compare the SPION synthetic effects on r2, one must prepare SPIONs with a similar size from two different synthetic methods and then incorporate a MS shell. Aside from differences in core character, discrepancies in r2 between mMS NP syntheses could be derived from the character of the silica shell. In recent work, solid silica shell thickness13,14 and polyethylene glycol (PEG) coating15,16 effects on the r2 values of SPION cores have been thoroughly considered, but there have not been systematic studies for MS-coated SPIONs. These studies are critical based on the much higher drug loading capacity and much lower unintentional cytotoxicity of MS NPs compared to their solid silica counterparts. To characterize the effect of a MS coating on SPION r2 values, it is essential to synthesize SPION-incorporated MS NPs having various sizes, using the same synthetic route to avoid other possible influencing factors such as particle stability, variation in extent of silica condensation, or extent of oxidation of SPIONs, which would likely change r2.
In addition to determining NP synthetic effects on r2, this work also explores practical concerns for long-term mMS NP storage and biological use. Most r2 measurements in the literature are performed at only one time point and in pure deionized (DI) water. While this initial value is important, care must be taken to further examine r2 in multiple scenarios which are relevant before and during theranostic use. Recent studies on other core-shell magnetic NPs have revealed relaxivity or magnetic property changes over time, depending on the storage conditions or suspension environment.28–30 For example, Lee et al. found that the T1 relaxation time of MnO-SiO2 core-shell NPs significantly changed over time at pH 5 due to dissolution of MnO to Mn2+ ions.29 Meade and co-workers found that mixed ferrite NPs coated with silica underwent significant T1 and T2 changes upon exposure to conditions as gentle as neutral water during dialysis.30 Should mMS NPs be used clinically, they will likely be stored in aqueous suspensions for days, weeks, or months. Stability following introduction into the body is also critical, both in mMS NP structure and in relaxivity. To the best of our knowledge, none of the literature studies explore long-term T2 relaxivity stability of silica-coated SPIONs in aqueous suspensions.
It is well-known that the degree of aggregation of SPIONs significantly influences r2.17–19 These results indicate that particle stability of the silica-coated SPIONs is a critical consideration for long-term relaxivity studies. Unless controlled, unmodified mMS NPs tend to aggregate during the synthesis or template removal processes.31 To enable long-term relaxivity measurements, mMS NPs having long-term particle stability in various media are required. Our group has recently utilized a hydrothermal treatment to improve colloidal stability of PEG-modified MS NPs without added SPION cores.31,32 This treatment significantly improved the PEG modification efficiency and reduced the free silanol groups on the MS NPs, resulting in a great stability improvement for PEGylated MS NPs in biological media compared to those without the hydrothermal treatment. Herein, this hydrothermal treatment can be simply applied to the synthesis of PEGylated mMS NPs to promote long-term colloidal stability.
Personalized medicine will require high-quality imaging hours or days after imaging agent administration. To this end, mMS NPs must retain particle stability in biological environments, and the effectiveness of the contrast agent must be maintained. Herein, we further examine the protective effect of the MS coating on SPIONs within acidic environments by monitoring T2 in mild (pH 5.5) and highly acidic (pH 0) suspensions over time. In addition, we correlate these results with percent iron dissolution at pH 0. Lastly, the protective effect is examined in biologically relevant media such as acetate buffer (pH 5) and phosphate buffered saline (PBS, pH 7.4).
To the best of our knowledge, there is no report which examines the effects of synthetic variables on the r2 of mMS NPs. In the interest of advancing mMS NPs toward clinical trials and beyond, herein we systematically investigate the effects of multiple synthetic variables on r2 to understand the major factors which affect contrast quality on multiple time scales. In this work, MS shell thickness, SPION core synthesis, and the inclusion of a hydrothermal treatment are all investigated in relevant environments. The results of this study will better inform synthetic choices regarding clinical mMS NPs, ultimately leading to higher image contrast and better personalized patient care.
EXPERIMENTAL SECTION
Chemicals
All chemicals were used as received. n-cetyltrimethylammonium bromide (CTAB), tetraethyl orthosilicate (TEOS), polyvinyl-pyrrolidone (PVP10, average MW10,000), iron(III)acetylacetonate (Fe(acac)3), benzyl ether, 1,2-dodecandiol (90%), oleyl amine (70%) and 10X PBS were purchased from Sigma Aldrich (Milwaukee, WI). 2-[Methoxy(polyethyleneoxy)propyl] trimethoxysilane, (PEG-silane, MW 596–725 g/mol, 9-12EO) was obtained from Gelest (Morrisville, PA). Ammonium nitrate (NH4NO3), chloroform (99.8%), iron(III) chloride hexahydrate (FeCl3·6H2O, >99%) and ammonium hydroxide (NH4OH, 28–30 wt% as NH3) were obtained from Mallinckrodt (Phillipsburg, NJ). Iron(II) chloride tetrahydrate (FeCl2·4H2O), 99–102%), and oleic acid were obtained from Fisher Scientific (Rockford, IL). Hydrochloric acid (HCl) was obtained from BDH (West Chester, PA). Absolute anhydrous 99.5% ethanol and 95% ethanol were purchased from Pharmco-Aaper (Brookfield, CT). The DI water was generated using a Millipore Milli-Q system (Billerica, MA). Heat-inactivated fetal bovine serum was purchased from Hyclone (Logan, UT). Powder DMEM without phenol red and sodium pyruvate was purchased from SAFC Biosciences (Lenexa, KS).
Synthesis of Hydrophobic Fe3O4 NPs from Co-Precipitation and Thermal Decomposition Methods
Syntheses of Fe3O4 NPs were performed according to published procedures.33,34 The detailed synthesis of Fe3O4 NPs prepared by a thermal decomposition method is described in the Supporting Information.
Preparation of PEGylated mMS NPs and Hydrothermally Treated mMS NPs with Various Sizes
Synthesis of PEGylated mMS NPs with various diameters and purification schemes were performed according to our previous work.26 First, 0.29 g CTAB and 0.20 g PVP-10 were dissolved in 5 mL DI water. A total of 8.2 mg (for 62-nm-diameter mMS NP synthesis), 32.7 mg (for 43-nm-diameter mMS NP synthesis), and 65.4 mg (for 35-nm-diameter mMS NP synthesis) Fe3O4 NPs suspended in chloroform were added to the reaction mixture. The mixture was sonicated and heated at 40 °C for 1 h to evaporate the solvent and allow aqueous suspension of organically-coated Fe3O4 NPs. Next, the Fe3O4 NP suspension was added to 150 mL of a 0.256 M NH4OH solution and heated at 50 °C with stirring (300 rpm) for 1 h. The reaction flask was covered with parafilm for the first hour to prevent evaporation of ammonia and subsequent pH changes. The parafilm was removed, stirring was increased to 600 rpm, and 3 mL 0.88 M ethanolic TEOS were added. The suspension became lighter in color and more opaque and was then heated at 50 °C for 1 h. Next, 540 μL of PEG-silane was added. The suspension was stirred for 30 more min, and then allowed to age at 50 °C for 20 h without stirring. The suspension was passed through a 0.45 μm filter and diluted to 50 mL with DI water. If hydrothermal treatment was applied, the as-synthesized NP suspension was heated in a sealed container at 90 °C for 24 h. Deoxygenated hydrothermal treatment conditions were attained by vacuum sonication of the as-synthesized NP suspension for 15 minutes, then backfilling the container headspace with nitrogen. Surfactants were removed from the pores by several centrifugation steps. The as-synthesized suspension was centrifuged (66,226 g for 30 min), then redispersed in 75 mM ethanolic NH4NO3 and heated at 60 °C for 1 h with stirring (300 rpm). The suspension was centrifuged and redispersed in 95% ethanol and 0.012 M HCl, consecutively. Once redispersed in HCl, the suspension was heated and stirred at 60 °C for 2 h, then centrifuged and redispersed in 95% ethanol and 99% ethanol, consecutively. Finally, the suspension was centrifuged once more, redispersed in ~10 mL of 99% ethanol, and stored at room temperature (RT) for further use. It should be noted that this synthesis has been performed previously and has demonstrated low toxicity toward various types of mammalian cells.26
Characterization
N2 adsorption-desorption, powder X-ray diffraction (XRD), and magnetic curve measurements
The N2 adsorption-desorption isotherms were measured on a Micromeritics ASAP 2020 (Norcross, GA) at 77K. Prior to measurements, samples were degassed at 120 °C for 18 h. The surface area and pore size of samples were determined by the BET and BJH methods, respectively. The powder XRD patterns were generated on a Siemens Bruker-AXS D-5005 X-ray diffractometer (Karlsruhe, Germany) using filtered Cu Kα radiation (λ = 1.5406 Å) at 45 kV and 40 mA. The magnetic curves were measured by a Quantum Designs MPMS-5S cryogenic susceptometer at RT.
T2-relaxivity measurements
Relaxometry measurements were performed on a Bruker Minispec mq60 NMR Analyzer (Billerica, MA) at 60 MHz. T2 relaxation values were obtained by performing a Carr-Purcell-Meiboom-Gill sequence on 400 μL sample solutions and are reported as the average of three measurements. r2 values were obtained by plotting 1/T2 vs. [Fe] for five dilutions of each sample and fitting to the following equation:
T2 measurements were obtained from mMS NP samples dispersed in differing media, including 1 M aqueous HCl, 10 μM aqueous HCl, PBS, and acetic acid/sodium acetate buffer (pH 5). To transfer purified mMS NPs from ethanol suspensions, the samples were first quantified by drying 1 mL of ethanol suspension by rotary evaporation and weighing the powder. Next, 2 mL of each ethanol stock suspension was centrifuged (66,226 g, 30 min) and redispersed by sonication in DI H2O twice. The final aqueous sample was diluted to 2 mg/mL with DI H2O, and then to 1 mg/mL with 2× the desired concentration of the intended medium. For example, a 2 mg/mL mMS NP aqueous suspension would be diluted with 20 μM aqueous HCl for a final concentration of 1 mg/mL mMS NP in 10 μM aqueous HCl.
ICP-OES for iron quantitation
ICP-OES quantitation of iron was performed on a Thermo Scientific iCAP 6500 dual view ICP-OES (West Palm Beach, FL) at a power of 1150 W. For accurate iron quantitation, the MS shells had to be completely dissolved before the SPION could be digested by nitric acid. Samples at 1 mg/mL were diluted 10 fold with a 10% solution of HBF4 (prepared by reacting 358 ml of conc. HF with 125 g of boric acid). The solutions were allowed to sit until they become clear and colorless. They were then diluted another 10 fold with 0.2 M HNO3 for iron digestion, and yittrium was added as an internal standard.
T2-weighted MR phantom imaging
Phantoms were prepared by suspending mMS NPs in 2 mL water at a concentration of 0.15 mM Fe (as determined by ICP-OES). Next, enough suspension was transferred to a 2 cm long NMR tube to completely fill the tube, taking care to avoid bubbles. Five samples could be inserted into the MRI instrument at once by placement in a 50 mL centrifuge tube with plastic supports to keep the NMR tubes stationary. The centrifuge tube was filled with DI water for all measurements. Samples were scanned in a 31 cm bore 9.4 T scanner (Oxford Magnet/Agilent DirectDrive Console). All images were acquired with a volume transmit/receive coil having an inner diameter of 3 cm (Agilent Technologies, Santa Clara, CA). To measure T2, fast spin-echo sequences with spin-echo preparation for single slices were used with following parameters: repetition time (TR) = 4000 ms, echo train length (ETL) = 4, echo time (TE) of spin-echo preparation = 9.86, 15.6, 24.8, 39.4, 62.5, 99.3, 158 and 250 ms, field of view (FOV) = 40 mm × 40 mm, slice thickness = 1.5 mm, and acquisition matrix = 128×128. The T2 value was calculated in MATLAB by fitting the image intensities and TE times into a mono-exponential curve.
RESULTS AND DISCUSSION
Synthesis and Characterization of mMS NPs
To determine the effects of various synthetic parameters on r2, PEGylated mMS NPs were synthesized using our previously developed method26 with several different conditions. First, the synthesis of the SPION cores (Fe3O4 NPs) was performed via two routes, a co-precipitation reaction of iron salts33 and a decomposition of iron acetylacetonate, Fe(acac)3.34 Samples of varying overall mMS NP size (35-, 43-, and 62-nm diameters, Figure 1, Figure S1, and Table 1) were synthesized by seeding different amounts of SPION cores into the reaction.26 Lastly, a subset of samples underwent a hydrothermal treatment wherein they were aged in water at 90 °C for 24 h. All mMS NPs demonstrate a consistent core diameter of 9–10 nm. The different core syntheses alone do not appear to have a significant effect on mMS NP size, shape, or number of incorporated cores. In all conditions, the core is located inhomogenously in the MS matrix, not necessarily at the center of the mMS NPs. This inhomogeneity makes it inappropriate to compare the effects of silica shell thickness on r2. Instead, r2 is monitored as a function of total mMS NP diameter. The morphological effects of the hydrothermal treatment can be seen in Figure 1b, d, f, and h (Enlarged TEM images are shown in Figure S1). The increased PEG modification on/in the pores and more complete silica condensation contribute to some pore structure disorder compared to the NPs that did not undergo hydrothermal treatment. This effect of hydrothermal treatment was expected based on results of our previous work.31,32
Figure 1.
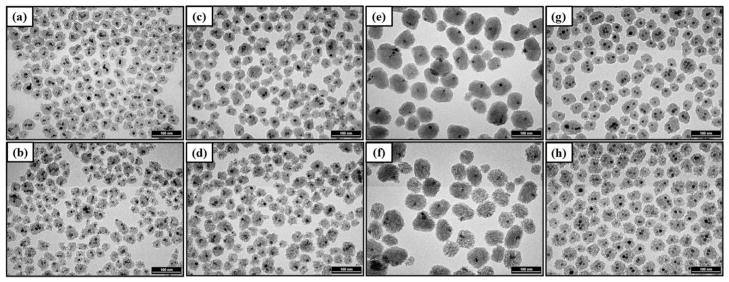
Representative TEM images showing mMS NPs of varying sizes, SPION cores, and hydrothermal treatment. (a) mcMS35-no hy, (b) mcMS35-hy, (c) mcMS43-no hy, (d) mcMS43-hy, (e) mcMS62-no hy, (f) mcMS62-hy, (g) mdMS43-no hy, and (h) mdMS43-hy. All scale bars represent 100 nm.
Table 1.
TEM diameter and SPION core analysis of various mMS NPs. “n” designates the number of individual NPs measured during image analysis.
| Sample | mMS NP Diameter (nm) | Number of SPION Cores | SPION Diameter (nm) |
|---|---|---|---|
| mcMS35-no hy | 35 ± 7 (n=900) | 1.6 ± 0.8 | 9.2 ± 2.0 (n=1459) |
| mcMS35-hy | 34 ± 7 (n=900) | 1.5 ± 0.8 | 9.0 ± 2.0 (n=1357) |
| mcMS43-no hy | 43 ± 8 (n=900) | 1.5 ± 0.8 | 9.0 ± 2.0 (n=1332) |
| mcMS43-hy | 46 ± 9 (n=900) | 1.7 ± 0.9 | 9.0 ± 2.0 (n=1522) |
| mcMS62-no hy | 63 ± 12 (n=900) | 1.3 ± 0.9 | 9.1 ± 2.2 (n=1166) |
| mcMS62-hy | 62 ± 13 (n=900) | 1.2 ± 0.9 | 9.3 ± 2.1 (n=1058) |
| mcMS43-no hy | 44 ± 8 (n=900) | 1.7 ± 1.0 | 9.9 ± 1.3 (n=1547) |
| mcMS43-hy | 46 ± 8 (n=900) | 1.7 ± 0.8 | 10.0 ± 1.3 (n=1507) |
Further mMS NP characterization data including XRD and nitrogen adsorption-desorption measurements are shown in Figure S2 and S3. XRD spectra at low angles show peaks indicative of 2D hexagonal order, representing the ordering of the MS pores within the NP. The small peaks at (110) and (200) indicate long range order, which is more apparent in larger diameter mMS NPs and in samples that did not undergo hydrothermal treatment (Figure S2). The broader and lower intensity peaks for hydrothermally treated samples are indicative of pore disorder in agreement with TEM (Figure 1). N2 adsorption-desorption isotherms (Figure S3), surface area, and pore size (Table 2) of all the mMS samples are shown to be dependent on overall NP diameter and hydrothermal treatment. Hydrothermal treatment results in a loss of surface area and a decrease in pore size, likely because the small PEG-silane moieties diffuse into pores and modify the inner pore surface or incorporate into the MS framework during the heating process. These results are consistent with our previously reported work.31,32
Table 2.
Surface area and primary pore diameter of PEGylated mMS-no hy and mMS-hy NPs with varied sizes and SPION cores.
| Sample | BET surface area (m2/g) | BJH pore size (Å) |
|---|---|---|
| mcMS35-no hy | 603 ± 21 | 24.0 ± 0.3 |
| mcMS35-hy | 435 ± 17 | 20.0 ± 0.8 |
| mcMS43-no hy | 685 ± 6 | 23.3 ± 0.7 |
| mcMS43-hy | 492 ± 11 | 19.9 ± 0.4 |
| mcMS62-no hy | 804 ± 21 | 23.3 ± 0.6 |
| mcMS62-hy | 576 ± 13 | 19.3 ± 0.3 |
| mdMS43-no hy | 672 ± 23 | 23.7 ± 0.9 |
| mdMS43-hy | 503 ± 17 | 19.7 ± 0.4 |
The hydrodynamic sizes of mMS NPs in various media, including DI water, PBS, cell culture media (Dulbecco’s Modified Eagle’s Medium with 10% fetal bovine serum, DMEM+10% FBS) and simulated body fluid (SBF) are summarized in Table 3. While hydrothermally treated mMS NPs demonstrate consistent hydrodynamic sizes in all media, those samples without hydrothermal treatment experience a significant increase in size, indicative of aggregation or the formation of a protein corona. In addition, Figure 2 further shows the hydrothermal treatment effect on long-term particle stability (hydrodynamic diameter over time) of mdMS43 NPs in SBF at 37 °C. Compared to mdMS43-hy, the mdMS43-no hy sample aggregated and settled out of suspension after one day of aging in SBF at 37 °C (inset of Figure 2). These results further confirm our previous work that the hydrothermal treatment supports MS NP colloidal stability in biologically relevant environments.31,32 Herein, we show that addition of the SPION core does not change the stability behavior. Subsequent T2 and r2 measurements were only performed on samples which displayed colloidal stability in relevant media. For example, samples with hydrothermal treatment could be measured over time in acetate buffer or PBS, but those without hydrothermal treatment were not measured to avoid the complications associated with aggregation effects.
Table 3.
Hydrodynamic diameters of various mMS NPs in biologically relevant media.
| Sample | H2O (nm) | PBS (nm) | SBF (nm) | DMEM+10 % FBS (nm) |
|---|---|---|---|---|
| mcMS35-no hy | 57 ± 5 | 76 ± 9 | 88 ± 10 | 72 ± 2 |
| mcMS35-hy | 52 ± 7 | 54 ± 5 | 53 ± 6 | 54 ± 5 |
| mcMS43-no hy | 68 ± 3 | 82 ± 5 | 88 ± 12 | 77 ± 2 |
| mcMS43-hy | 68 ± 3 | 67 ± 2 | 67 ± 1 | 67 ± 4 |
| mcMS62-no hy | 88 ± 4 | 275 ± 51 | 440 ± 78 | 98 ± 7 |
| mcMS62-hy | 82 ± 4 | 80 ± 6 | 80 ± 7 | 78 ± 7 |
Figure 2.
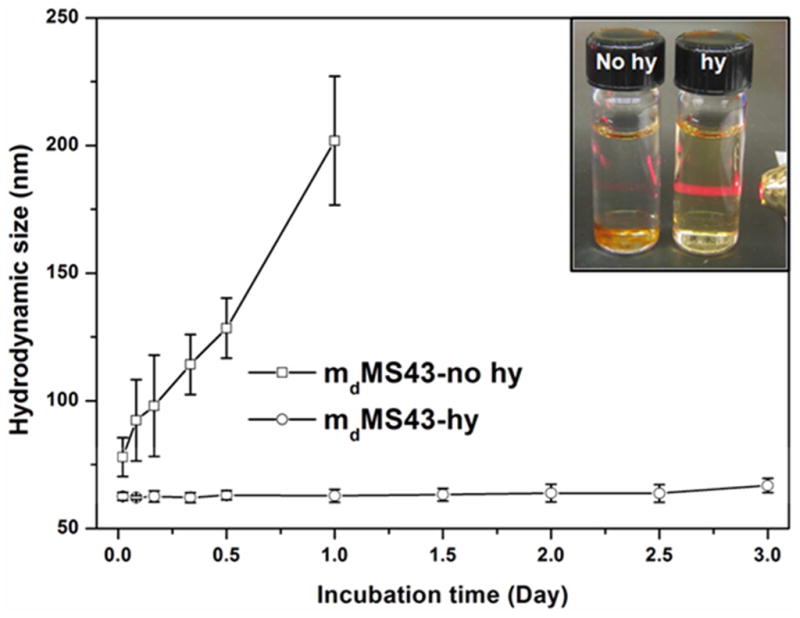
Long-term particle stability of mdMS43-no hy and mdMS43-hy in SBF at 37 °C. Inset: A photograph of mdMS43-no hy and mdMS43-hy suspensions after 3-day aging in SBF at 37 °C.
Effects of mMS NP Diameter on T2 Relaxivity
The r2 values of mMS NPs of varying diameters with and without hydrothermal treatment upon exposure to DI water are shown in Figure 3a and b. No significant r2 difference exists between any of the tested diameters (either in hydrothermally treated or non-hydrothermally treated samples), indicating a lack of r2 dependence on NP diameter for at least the range of sizes considered herein. Indeed, dextran-coated SPIONs (no silica shell) formed by co-precipitation display similar r2 values to those seen here.35 This result is encouraging, as it suggests that MS diameter (up to 62 nm) has no critical influence on r2. Researchers will be free, then, to change NP size for practical reasons without worrying about r2 losses due to different diameters from MS coating. In contrast, an increase of 26 nm to the diameter of γ-Fe2O3 NPs coated with nonporous silica resulted in an r2 decrease of more than 50 % (compare r2 of 47 mM−1s−1 for 40 nm diameter to r2 of 23 mM−1s−1 for 66 nm diameter).13 The r2 of SPIONs is largely dictated by an outer sphere relaxation mechanism wherein water molecules diffuse in and out of the inhomogenous magnetic field generated by the SPIONs.36 The drop in r2 caused by nonporous silica coatings is presumably due to the exclusion of water from the SPION environment.13,14 In practical applications, the separation of water molecules from a contrast agent is critically important as the strength of a magnetic field surrounding a magnetic material is proportional to the inverse of the distance cubed. In the case of mMS NPs, however, we do not observe an appreciable diameter effect on r2 of mMS NPs. It is likely that water molecules are able to diffuse through the mesopores toward the SPIONs located inside the MS NPs, fulfilling the outer sphere relaxation condition.
Figure 3.
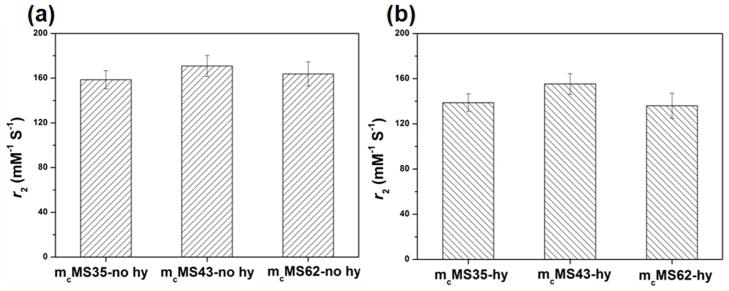
r2 values of mcMS NPs having varied sizes (a) without and (b) with hydrothermal treatment. There is no significant difference among these conditions.
Effects of SPION Core Synthesis and Hydrothermal Treatment on T2 Relaxivity
In addition to investigating the size effects of MS coating on SPIONs, we also compared the effects of different SPION syntheses and hydrothermal treatments on the r2 of mMS NPs. TEM images of the as-synthesized SPION cores prepared from the co-precipitation and thermal decomposition methods are shown in Figure 4a and b, respectively. It is clear that the SPIONs prepared from the thermal decomposition method are more uniform in size and shape compared to SPIONs made from the co-precipitation method. Figure 4c shows the comparison of measured r2 values for mMS NPs with an overall 43 nm diameter prepared with the different SPION cores. A significant difference in r2 is observed between mcMS43 and mdMS43 NPs. Both the mdMS43-no hy and mdMS43-hy NPs show a clear enhancement of r2 (~28–35 % greater r2) compared to mcMS NPs regardless of hydrothermal treatment. This result is likely due to a lower saturation magnetization (Ms) of co-precipitation SPION cores compared to thermal decomposition SPIONs. It has been shown that large size distribution and polycrystallinity contribute to the lower Ms of co-precipitation SPIONs.10,22 The clear imaging benefit of the thermal decomposition SPIONS will have to be balanced in practical use, as they require higher temperatures, more expensive precursors, and more stringent synthetic conditions than co-precipitated SPION cores.
Figure 4.

TEM images of (a) co-precipitation Fe3O4 NPs (mc) and (b) thermal decomposition Fe3O4 NPs (md), scale bar = 50 nm. Despite differences in diameter distributions, the SPIONs present in mMS NPs have diameters which center on 9–10 nm and allow a reasonable comparison between the two (see Table 1). (c) Effect of core synthesis on r2 of mMS43-no hy and mM43-hy NPs. (d) r2 value comparison for mcMS and mdMS NPs with and without hydrothermal treatment. * represents p < 0.05.
Both mcMS and mdMS NPs display higher r2 values without the hydrothermal treatment, although the difference is only significant between mdMS43-no hy and mdMS43-hy (Figure 4d). This lower r2 value for hydrothermally treated samples is likely due to a partial oxidation of Fe3O4 (magnetite) to γ-Fe2O3 (maghemite) that occurs during the hydrothermal treatment process, causing a decrease of Ms in hydrothermally treated mMS NPs. The decrease of Ms in mdMS43-hy NPs is expected based on recent work by Rebodos and Vikesland37 and has been confirmed by superconducting quantum interference device magnetometer (SQUID) measurements (Figure 5). Fe3O4 NPs have been shown to easily oxidize to γ-Fe2O3 by heating at 100 °C for 30 min under aeration in the literature,38 and this oxidation can be simply observed by a color change in the mMS NP solution after hydrothermal treatment. As seen in Figure 7a, the hydrothermally treated mdMS NPs display a more orange color than samples without hydrothermal treatment. Despite the advantage of higher r2 values, the poor particle stability of mMS NPs without hydrothermal treatment limits their practical use.3,31
Figure 5.

Room temperature magnetization curves of mdMS43-no hy and mdMS43-hy.
Figure 7.

Evidence for oxidation during hydrothermal treatment and water storage. (a) mdMS62 samples before and after hydrothermal treatment. (b) Fe edge EELS spectra from a mdMS62-no hy sample after zero and 14 days of storage in DI water. (c) mdMS62 sample before and after hydrothermal treatment in a deoxygenated environment. (d) Percent original r2 of mdMS samples with no hydrothermal treatment and hydrothermal treatment with or without oxygen over time in DI water.
Long-Term R2 Stability and Dissolution Resistance Based on Hydrothermal Treatment
Several reports on MRI contrast agents have shown that NP relaxivity values may change over time depending on the environment of the NP.28–30 Therefore, r2 values of mMS NPs with differing cores and hydrothermal treatments should be monitored over time in various media. Initial measurements were made on mMS NPs suspended in DI water. DLS data (not shown) indicate that these mMS NPs exhibit colloidal stability in DI water, suggesting that r2 changes are not being caused by aggregation. The r2 values of all mMS NPs over 32 day aging in DI water are shown in Figure 6. Both mcMS and mdMS NPs exhibited similar behavior, with hydrothermally treated samples retaining the original r2 values while samples without hydrothermal treatment dropped to 80% of original r2 values in just eight days of storage in DI water at RT. Though all the initial r2 values of non-hydrothermally treated mMS NPs are higher than their hydrothermally treated counterparts, the values drop to become comparable or even lower than those of hydrothermally treated mMS NPs after eight days of storage. This result is important as it suggests that, without hydrothermal treatment or other modifications, mMS NPs will have a short shelf life at maximum r2. We considered two possible causes for the drop in r2. First, the SPION cores could be oxidizing during storage. The decrease of saturation magnetization of mcMS43-no hy after 28-day storage confirmed some oxidation of SPION cores, which would result in a decrease of r2 (Figure S4). Samples with hydrothermal treatment may experience a less substantial effect because the Fe3O4 core oxidizes to the more stable γ-Fe2O3 phase during hydrothermal treatment, a ‘pre-oxidation.’ Second, more complete PEG coating and silica condensation during hydrothermal treatment may lead to a protective layer surrounding the SPION cores, keeping them from oxidizing longer than in the case of the samples without hydrothermal treatment. To test these hypotheses, we performed a number of experiments, the results of which are shown in Figure 7. The oxidation of iron oxide is well known to be associated with a distinct color change from dark brown/black to a lighter orange or yellow.38 Samples that underwent hydrothermal treatment displayed this color change (Figure 7a). To obtain more rigorous evidence, electron energy loss spectroscopy (EELS) was performed on a mdMS62-no hy sample after zero and 14 days of storage in water. The iron edge spectra after background subtraction are shown in Figure 7b. Literature precedent has shown that oxidation from Fe3O4 to γ-Fe2O3 will result in two spectral changes at the iron edge. First, the ratio of the L3 edge to the L2 edge intensities will increase with oxidation. Second, The L3 edge will shift to higher energies with increasing amounts of oxidation.39,40 The L3/L2 ratio did increase from 3.37 to 3.9 between days zero and 14 (see Table S1). Any shift in the L3 peak was smaller than the resolution limit of the EELS spectrometer and should not be taken as a statistically significant difference. Finally, a mdMS62 sample was hydrothermally treated in a deoxygenated environment (degassed water, nitrogen headspace). As seen in Figure 7c, the sample color remained brown black, indicating that the presence of oxygen was necessary for the previously seen color change and further confirming the oxidation argument. When this deoxy-hy sample was monitored for r2 stability in water, it maintained ~ 90% for up to 24 days, similar to other hy samples. The lack of color change and long term stability in water suggest that not only was oxidation prevented by a deoxygenated process during hydrothermal treatment – it was prevented during storage as well, perhaps by the presence of a thin, condensed shell of silica surrounding the SPION core.
Figure 6.

r2 behavior and of mMS-no hy and mMS-hy NPs in DI water over 32 days at RT. Figures a–d are plotted as absolute r2 value vs. time. Figures e–f are plotted as percent original r2 vs. time.
The presence of this shell could be further elucidated by observing the behavior of mMS NPs in dilute or concentrated solutions of acids, which are also relevant for study due to the fact that NPs interacting with cells are likely to be taken up into acidic compartments41–43 and SPIONs are easily dissolved in acid.44,45 First, the relaxation times of 1 mg/mL of mdMS43-no hy and mdMS43-hy samples in 10 μM aqueous HCl (pH ~5.5) were monitored over time. Because degradation behavior is a concentration-dependent process, contrast agent quality was determined by monitoring T2 as opposed to r2. Changes in T2 represented as a percentage of initial T2 are shown in Figure 8a (larger T2 values correspond to smaller r2 values). Samples without hydrothermal treatment experienced longer T2 values over time, potentially indicative of slow acid dissolution of the SPION core, while hydrothermally treated samples maintained the original T2 values. It has been shown that the mechanism of iron oxide dissolution in acidic conditions proceeds through a number of steps.46 If the starting material is Fe3O4, it is first oxidized to γ-Fe2O3 before undergoing further dissolution. Additionally, Fe3O4 has a faster rate of dissolution than γ-Fe2O3 under identical conditions.46,47 Therefore, we suspect that the long term acid resistance of the hydrothermally treated mMS NPs is due to the oxidation that occurs during the hydrothermal treatment. Either the exterior shell or the entirety of the SPION is oxidized to γ-Fe2O3 during the treatment, imparting mMS-hy samples with slower dissolution rates. This same reasoning helps to explain why mdMS samples lost r2 more quickly than mcMS samples in DI water. Beyond the oxidation effect, however, we suspect that the more complete condensation of the silica shell around the SPION core provides some protection against acid entry. To further investigate this hypothesis, 1 mg/mL suspensions of samples with and without hydrothermal treatment were placed in 1 M aqueous HCl (pH 0), and SPION dissolution was measured for 24 h. After 24 h acid aging, the NPs were collected by centrifugation, and dissolution was monitored based on the color of the NP pellet. Figure 8c clearly shows that a colorless pellet was obtained from mdMS43-no hy NPs, indicating complete dissolution of incorporated SPIONs. In addition, iron dissolution data (analyzed by ICP-OES) show that 99% of SPIONs in mdMS43-no hy samples were dissolved after 24 h incubation in 1 M HCl at RT but only 13% of SPIONs dissolved from mdMS43-hy samples (Figure 8b). The TEM images of mdMS43-no hy and mdMS43-hy after acid aging (Figure 8d and e) clearly show that, for hydrothermally treated mdMS43 NPs, most of the magnetic cores remain intact after one day RT aging in pH 0 at a NP concentration of 1 mg/mL, while the cores of non-hydrothermally treated samples are completely absent. This result further confirms the presence of a more highly condensed silica shell surrounding the SPION core. It is likely that this shell will provide protection for multiple phases of iron oxide, including Fe3O4 or γ-Fe2O3 (both phases are commonly used as starting core materials) as both are subject to oxidation and acid dissolution when unprotected.
Figure 8.

Behavior of mdMS43 NPs with and without hydrothermal treatment in varying acidic conditions. (a) T2 behavior over time in 10 cM HCl. (b) percent iron dissolution of mdMS43 NPs in 1 M HCl. (c) A photograph and TEM images of (d) mdMS43-no hy and (e) mdMS43-hy NPs after 24-h aging in 1 M HCl.
To examine the T2 behavior over time in biologically relevant conditions, hydrothermally treated mdMS NPs were aged in acetate buffer (pH 5) and PBS (pH 7). Samples without hydrothermal treatment aggregated and visibly settled out of suspension within 4 h of exposure to acetate buffer (Figure 9 inset), and samples without hydrothermal treatment are known to aggregate in PBS.31 Figure 9 shows that hydrothermally treated samples maintained their original T2 value in acetate for over 72 h, while samples in PBS did experience a slight increase in T2 during that time. This increase for PBS suspensions is likely due to the presence of chloride (PBS contains NaCl and KCl). It has been shown that although Cl− ions are not necessary for oxidation and subsequent dissolution of Fe3O4 and γ-Fe2O3, the presence of Cl− ions increases the rates of reaction.47 Biological systems, including blood, lysosomes, and cytosol, all contain high concentrations of Cl− ions, so this result would likely be seen and possibly intensified in vivo (Cl− ions increase dissolution rates in a roughly exponential fashion).
Figure 9.

T2 behavior of mdMS43 NPs in biological media (hydrothermally treated samples only). Inset: samples without hydrothermal treatment aggregated and settled out of acetate buffer suspensions within 4 h. Previous results show aggregation in PBS over days for samples without hydrothermal treatment.31
To further demonstrate the effect of hydrothermal treatment on T2 stability over time, T2-weighted MR images were collected at one and 15 days after addition of mMS NPs to DI water. Two samples, mdMS43-hy and mdMS43-no hy, were suspended in DI water at a concentration of 0.15 mM Fe. The results of the MRI measurements are shown in Figure 10 and S5. The negative contrast of samples without hydrothermal treatment is visibly reduced over the course of 15 days in DI water while samples with hydrothermal treatment remain stable (Figure 10a and S5). The change in percent of original T2 (Figure 10b) for both samples is consistent with data collected by relaxometry. The T2 value of samples without hydrothermal treatment increased to 130 % of original T2 values in 15 days of storage in DI water at RT, consistent with the loss in contrast observed in Figure 10a.
Figure 10.
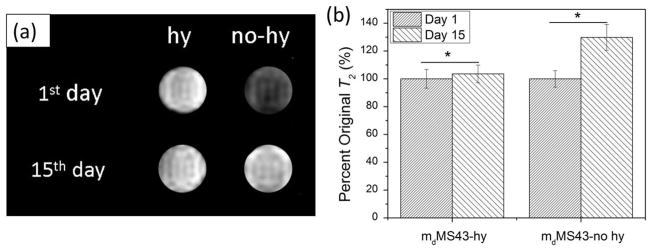
MRI data for mdMS43-no hy and mdMS43-hy samples over 15 days of aging in DI water. (a) T2 weighted images show an increase in T2 over time for samples without hydrothermal treatment while those with hydrothermal treatment remain stable. (b) Percent of original T2 values calculated from T2 maps (n=81 for each condition). The increase in T2 values for samples without hydrothermal treatment is consistent with measurements obtained by relaxometry. * indicates p<0.05.
CONCLUSIONS
In this work, we have investigated the effects of several synthetic parameters on the r2 values of mMS NPs for biomedical imaging applications. The mMS NP diameter appears to have no significant effect on r2 in the measured range, indicating a fundamental difference between porous and nonporous silica as a magnetic NP coating material. Successful mMS NP syntheses were performed by coating MS onto Fe3O4 NPs synthesized by two different routes. Fe3O4 NPs made via a decomposition reaction displayed 26–35% higher r2 values, but cost and scale up considerations will be important in choosing between the co-precipitation and thermal decomposition reactions. The hydrothermal treatment, found in previous work to support colloidal stability, has also demonstrated an effect on both r2 stability and acid resistance of mMS NPs. Samples without hydrothermal treatment lose 20 % of their r2 within just eight days of suspension in water, a likely storage medium. This r2 loss was determined to be a result of iron oxide oxidation, which was prevented in the case of hydrothermal treatment in deoxygenated water. The iron dissolution rate of mMS NPs with/without hydrothermal treatment was investigated, and it was found that the hydrothermal treatment engenders resistance to acidic etching of the magnetic core. Finally, mMS NPs were studied over time in biological suspensions, acetate buffer (pH 5) and PBS (pH 7.4). Hydrothermally treated samples were able to maintain T2 stability in acetate, but T2 increased slowly for samples in PBS due to high chloride concentrations. These results should be carefully considered for future design of SPION-based MRI contrast agents, in particular those involving mesoporous silica.
Supplementary Material
Acknowledgments
This research was supported by National Science Foundation (CHE-0645041), the Keck Foundation, and the NIH Biotechnology Research Center (BTRC) grant P41 RR008079 (NCRR) and grant P41 EB015894 (NIBIB). The authors wish to thank Professor M. Garwood for helpful discussions, expertise, and the use of MRI equipment at the CMRR. Additionally, the authors thank Professor V. C. Pierre and Dr. E. D. Smolensky for help with T2 relaxivity measurements and A. Nicol and R. Knurr for help with iron quantification. The TEM and XRD measurements were performed in the College of Science and Engineering Characterization Facility, University of Minnesota, which receives partial support from NSF through National Nanotechnology Infrastructure Network (www.mrfn.org) via the MRSEC program. SQUID measurements were performed in the University of Minnesota Institute for Rock Magnetism. K.R.H. and Y.-S.L. acknowledge financial support from a National Science Foundation Graduate Research Fellowship and a Taiwan Merit Scholarship (NSC-095-SAF-I-70 564-052-TMS), respectively.
Footnotes
The authors declare no competing financial interest.
Author Contributions
K.R.H. and Y.S.L. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Enlarged TEM images, particle size histograms, XRD spectra, N2 adsorption-desorption isotherms, and SQUID magnetic curves of mMS NPs; color-scale T2 map MRI images; detailed synthesis procedure of SPIONs prepared by the thermal decomposition method.
References
- 1.Wolf LK. Personalizing Nanomedicine. Chemical and Engineering News. 2011;89:29. [Google Scholar]
- 2.Lee JE, Lee N, Kim T, Kim J, Heyon T. Acc Chem Res. 2011;44:893. doi: 10.1021/ar2000259. [DOI] [PubMed] [Google Scholar]
- 3.Lin YS, Hurley KR, Haynes CL. J Phys Chem Lett. 2012;3:364. doi: 10.1021/jz2013837. [DOI] [PubMed] [Google Scholar]
- 4.Lee CH, Cheng SH, Wang YJ, Chen YC, Chen NT, Souris J, Chen CT, Mou CY, Yang CS, Lo LW. Adv Funct Mater. 2009;19:215. [Google Scholar]
- 5.Luo T, Huang P, Gao G, Shen G, Fu S, Cui D, Zhou C, Ren Q. Opt Express. 2011;19:17030. doi: 10.1364/OE.19.017030. [DOI] [PubMed] [Google Scholar]
- 6.Huang X, Zhang F, Lee S, Swierczewska M, Kiesewetter DO, Lang L, Zhang G, Zhu L, Gao H, Choi HS, Niu G, Chen X. Biomaterials. 2012;33:4370. doi: 10.1016/j.biomaterials.2012.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin YS, Wu SH, Hung Y, Chou YH, Chang C, Lin ML, Tsai CP, Mou CY. Chem Mater. 2006;18:5170. [Google Scholar]
- 8.Kim J, Kim HS, Lee N, Kim T, Kim H, Yu T, Song IC, Moon WK, Hyeon T. Angew Chem Int Ed. 2008;47:8438. doi: 10.1002/anie.200802469. [DOI] [PubMed] [Google Scholar]
- 9.Tsai CP, Hung Y, Chou YH, Huang DM, Hsiao JK, Chang C, Chen YC, Mou CY. Small. 2008;4:186. doi: 10.1002/smll.200700457. [DOI] [PubMed] [Google Scholar]
- 10.Stephen ZR, Kievit FM, Zhang M. Mater Today. 2011;14:330. doi: 10.1016/S1369-7021(11)70163-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoo D, Lee JH, Shin TH, Cheon J. Acc Chem Res. 2011;44:863. doi: 10.1021/ar200085c. [DOI] [PubMed] [Google Scholar]
- 12.Neoh KG, Kang ET. Soft Matter. 2012;8:2057. [Google Scholar]
- 13.Pinho SLC, Pereira GA, Voisin P, Kassem J, Bouchaud V, Etienne L, Peters JA, Carlos L, Mornet S, Geraldes CFGC, Rocha J, Delville MH. ACS Nano. 2010;4:5339. doi: 10.1021/nn101129r. [DOI] [PubMed] [Google Scholar]
- 14.Pinho SLC, Laurent S, Rocha J, Roch A, Delville MH, Mornet S, Carlos LD, Elst LV, Muller RN, Geraldes CFGC. J Phys Chem C. 2012;116:2285. [Google Scholar]
- 15.Hu F, MacRenaris KW, Waters EA, Liang T, Schultz-Sikma EA, Eckermann AL, Meade TJ. J Phys Chem C. 2009;113:20855. doi: 10.1021/jp907216g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tong S, Hou S, Zheng Z, Zhou J, Bao G. Nano Lett. 2010;10:4607. doi: 10.1021/nl102623x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perez JM, Josephson L, O’Loughlin T, Högemann D, Weissleder R. Nat Biotechnol. 2002;20:816. doi: 10.1038/nbt720. [DOI] [PubMed] [Google Scholar]
- 18.Larsen BA, Haag MA, Serkova NJ, Shroyer KR, Stoldt CR. Nanotechnology. 2008;19:265102. doi: 10.1088/0957-4484/19/26/265102. [DOI] [PubMed] [Google Scholar]
- 19.Carroll MR, Huffstetler PP, Miles WC, Goff JD, Davis RM, Riffle JS, House MJ, Woodwart RC, St Pierre TG. Nanotechnology. 2011;22:325702. doi: 10.1088/0957-4484/22/32/325702. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka K, Narita A, Kitamura N, Uchiyama W, Morita M, Inubshi T, Chujo Y. Langmuir. 2010;26:11759. doi: 10.1021/la1015077. [DOI] [PubMed] [Google Scholar]
- 21.Wu SH, Lin CY, Hung Y, Chen W, Chang C, Mou CY. J Biomed Mater Res. 2011;99B:81. doi: 10.1002/jbm.b.31874. [DOI] [PubMed] [Google Scholar]
- 22.Roca AG, Veintemillas-Verdaguer S, Port M, Robic C, Serna CJ, Morales MP. J Phys Chem B. 2009;113:7033. doi: 10.1021/jp807820s. [DOI] [PubMed] [Google Scholar]
- 23.Giri S, Trewyn BG, Stellmaker MP, Lin VSY. Angew Chem Int Ed. 2005;44:5038. doi: 10.1002/anie.200501819. [DOI] [PubMed] [Google Scholar]
- 24.Lee JE, Lee N, Kim H, Kim J, Choi SH, Kim JH, Kim T, Song IC, Park SP, Moon WK, Hyeon T. J Am Chem Soc. 2010;132:552. doi: 10.1021/ja905793q. [DOI] [PubMed] [Google Scholar]
- 25.Lee KR, Kim S, Kang DH, Lee JI, Lee YJ, Kim WS, Cho DH, Lim HB, Kim J, Hur NH. Chem Mater. 2008;20:6738. [Google Scholar]
- 26.Lin YS, Haynes CL. Chem Mater. 2009;21:3979. [Google Scholar]
- 27.Liong M, Lu J, Kovochich M, Xia T, Ruehm SG, Nel AE, Tamanoi F, Zink JI. ACS Nano. 2008;2:889. doi: 10.1021/nn800072t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoon TJ, Lee H, Shao H, Weissleder R. Angew Chem Int Ed. 2011;50:4663. doi: 10.1002/anie.201100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee YC, Chen DY, Dodd SJ, Bouraoud N, Koretsky AP, Krishnan KMT. Biomaterials. 2012;33:3560. doi: 10.1016/j.biomaterials.2012.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schultz-Sikma EA, Joshi HM, Ma Q, MacRenaris KW, Eckermann AL, Dravid VP, Meade TJ. Chem Mater. 2011;23:2657. doi: 10.1021/cm200509g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin YS, Abadeer N, Haynes CL. Chem Comm. 2011;47:532. doi: 10.1039/c0cc02923h. [DOI] [PubMed] [Google Scholar]
- 32.Lin YS, Abadeer N, Hurley KR, Haynes CL. J Am Chem Soc. 2011;133:20444. doi: 10.1021/ja208567v. [DOI] [PubMed] [Google Scholar]
- 33.Molday RS. U.S. Patent 4,452,773. 1984
- 34.Sun S, Zeng H, Robinson DB, Raoux S, Rice PM, Wang S, Li G. J Am Chem Soc. 2004;126:273. doi: 10.1021/ja0380852. [DOI] [PubMed] [Google Scholar]
- 35.Ma M, Zhang Y, Shen Y, Xia X, Zhang S, Liu Z. J Nanopart Res. 2011;13:3249. [Google Scholar]
- 36.Gillis P, Koenig SH. Magn Reson Med. 1987;5:323. doi: 10.1002/mrm.1910050404. [DOI] [PubMed] [Google Scholar]
- 37.Rebodos RL, Vikesland PJ. Langmuir. 2010;26:16745. doi: 10.1021/la102461z. [DOI] [PubMed] [Google Scholar]
- 38.Kang YS, Risbud S, Rabolt JF, Stroeve P. Chem Mater. 1996;8:2209. [Google Scholar]
- 39.Colliex C, Manoubi T, Ortiz C. Phys Rev B. 1991;44:11402. doi: 10.1103/physrevb.44.11402. [DOI] [PubMed] [Google Scholar]
- 40.van Aken PA, Liebscher B, Styrsa VJ. Phys Chem Miner. 1998;25:323. [Google Scholar]
- 41.Slowing I, Trewyn BG, Lin VSY. J Am Chem Soc. 2006;128:14792. doi: 10.1021/ja0645943. [DOI] [PubMed] [Google Scholar]
- 42.Chung TH, Wu SH, Yao M, Lu CW, Lin YS, Hung Y, Mou CY, Chen YC, Huang DM. Biomaterials. 2007;28:2959. doi: 10.1016/j.biomaterials.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 43.Davila-Ibanez AB, Salgueirino V, Martinez-Zorzano V, Marino-Fernandez R, Garcia-Lorenzo A, Maceira-Campos M, Munoz-Ubeda M, Junquera E, Aircart E, Rivas J, Rodriguez-Berrocal F, Legido JL. ACS Nano. 2012;6:747. doi: 10.1021/nn204231g. [DOI] [PubMed] [Google Scholar]
- 44.Brillet PY, Gazeau F, Luciani A, Bessoud B, Cuénod CA, Siauve N, Pons JN, Poupon J, Clément O. Eur Radiol. 2005;15:1369. doi: 10.1007/s00330-004-2586-8. [DOI] [PubMed] [Google Scholar]
- 45.Arbab AS, Wilson LB, Ashari P, Jordan EK, Lewis BK, Frank JA. NMR Biomed. 2005;18:383. doi: 10.1002/nbm.970. [DOI] [PubMed] [Google Scholar]
- 46.Frenier WW, Growcock FB. Corrosion. 1984;40:663. [Google Scholar]
- 47.Sidhu PS, Gilkes RJ, Cornell RM, Posner AM, Quirk JP. Clay Clay Miner. 1981;29:269. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.