

Abstract
The absolute stereostructures of the components of symplocin A (3), a new N,N-dimethyl-terminated peptide from the Bahamian cyanobacterium, Symploca sp., were assigned from spectroscopic analysis, including MS and 2D NMR and Marfey’s analysis. The complete absolute configuration of symplocin A, including the unexpected D-configurations of the terminal N,N-dimethylisoleucine and valic acid residues, were assigned by chiral-phase HPLC of the corresponding 2-naphthacyl esters, a highly sensitive, complementary strategy for assignment of N-blocked peptide residues where Marfey’s method is ineffectual, or other methods fall short. Symplocin A exhibited potent activity as an inhibitor of cathepsin E (IC50 300 pM).
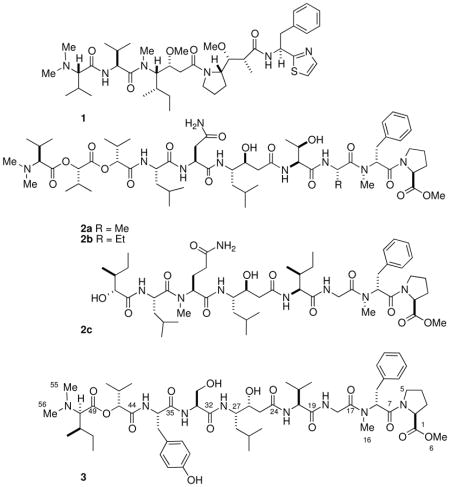
Linear peptides terminated with N,N-dimethyl amino acid residues (DMAAs) are a minority family of natural products among the larger array of cyclic peptides reported from cyanobacteria.i For example, the exceedingly potent anticancer compounds, dolastatin-15ii and - 10 (1)iii – which has completed phase I clinical trialsiv – were initially found in the sea hare Dolabella auricularia from the Indian Ocean, and later, in the cyanobacterium Symploca sp.1b collected in Palau. Grassystatins A and B (2a,b),1c two DMAA-terminated peptides from Lyngbya cf. confervoides and grassystatin C (2c), exhibit selective inhibition of the protease, cathepsin E. Gallinamide A,1a from a Panamanian Schizothrix sp., is moderately active against the malarial parasite, Plasmodium falciparum, and the cytotoxic belamide A,id from a Panamanian Symploca sp., shows tubulin destabilizing activity.
While analysis of many highly modified α-amino acids have been achievedv conveniently by application of Marfey’s analysis,3 the configuration of DMAA-containing peptides is complicated by lack of reactivity: the hindered N-terminal amino acid is inert in standard Marfey’s analysis and Edman degradation. A similar problem is encountered with analysis of 2-hydroxyacids that occasionally replace α-amino acid residues in DMAA-modified peptides or other cyclic peptides arising from non-ribosomal polypeptide synthetase (NRPS) pathways.vi Here, we report a new peptide, symplocin A (3) from a mixed cyanobacterial assemblage containing Symploca sp. collected in the Bahamas, and its complete stereoassignment aided by a new protocol for DMAA and other non-standard residues by analysis of their corresponding 2-naphthacyl esters. The sensitivity of the latter protocol is comparable to Marfey’s method and well-suited to DMAA, 2-hydroxy acids, and may find application to other N-terminal blocked peptides.
The molecular formula for 3, C56H86N8O14 was established from HRESIMS data (m/z 1094.6257, [M+H]+). The high N-content of 3 together with the presence of several C=O groups (HMBC) were suggestive of a peptide; this was confirmed by identification of peptide residue 1H and 13C spin systems through analysis of COSY, TOCSY, HSQC and HMBC spectra. Amino acid (aa) analysis of the hydrolysate of 3 (6M HCl, 110°) revealed the presence of the residues glycine (Gly), proline (Pro), serine (Ser), tyrosine (Tyr) and valine (Val). The 1H NMR spectrum of 3 (CD3CN, Table 1) showed broad N-H doublets coupled to α-CH protons of amino acid residues, the partial sequence of which was established by HMBC (Figure 1). A downfield AA′BB′ system (δ 6.73, d, J = 8.3 Hz; 7.09, d, J = 8.3 Hz) was assigned to the Tyr residue and supported by HMBC C-H correlations (Table 1). The remaining aromatic proton signals were suggestive of a phenylalanine (Phe) residue, however, Phe was not detected in standard analysis for standard proteinogenic amino acids. An HSQC spectrum showed the presence of eight C-substituted methyl groups indicating hydrophobic amino acid residues. In addition, three upfield N-Me signals (δ 2.92, s; 2.83, s, 6H, two overlapped Me) appeared in the 1H NMR spectrum of 3. The HMBC spectrum showed a correlation of the methyl group (δ 2.92, s) to a spin system consistent with N-MePhe.
Table 1.
1H (600 MHz) and 13C NMR (125 MHz) for 3 (CD3CN)
| Amino Acid | No. | δC, mult.a | δH, mult (J in Hz) | COSY | HMBCb | ROESY |
|---|---|---|---|---|---|---|
| O-Me-Pro | 1 | 173.4, C | ||||
| 2 | 59.1, CH | 4.26, dd (2.3, 5.9) | 3a, 3b | 1, 3, 4 | 3a, 3b | |
| 3a | 29.5, CH2 | 1.77, m | 2, 3b | 1, 2, 4, 5 | 2, 3b, 5 | |
| 3b | 2.12, m | 2, 3a | 1, 4, 5 | 2, 3a | ||
| 4a | 25.7, CH2 | 1.85, m | 3b, 5 | 2, 3, 5 | ||
| 4b | 1.78, m | 3a, 3b, 5 | 2, 5 | 3a, 3b, 5 | ||
| 5 | 47.4, CH2 | 3.31, m | 4a, 4b | 3, 4 | 3a | |
| 6 | 52.5, CH2 | 3.65, s | 1 | |||
| N-MePhe | 7 | 168.7, C | ||||
| 8 | 57.5, CH | 5.42, t (7.1) | 9a, 9b | 7, 9, 10, 16 | 5, 11 | |
| 9a | 35.3, CH2 | 2.75, m | 8, 9b | 8, 10, 11 | 7, 8, 9b, 11 | |
| 9b | 3.18, m | 8, 9a | 7, 8, 10, 11 | 8, 9b, 11 | ||
| 10 | 138.7, C | |||||
| 11/15 | 130.2, CH | 7.23, m | 9, 10, 13 | 8, 9a, 9b, 16 | ||
| 12/14 | 129.0, CH | 7.23, m | 10, 13 | 8, 9a, 9b, 16 | ||
| 13 | 127.2, CH | 7.18, m | 12/14 | |||
| 16 | 30.6, CH3 | 2.92, s | 8, 17 | |||
| Gly | 17 | 169.5, C | ||||
| 18a | 41.5, CH2 | 3.94, m | NH | 16 | ||
| 17, 19 | ||||||
| 18b | 4.02, m | NH | 17, 19 | 16 | ||
| NH | 7.45, m | 18b | ||||
| Val | 19 | 172.5, C | ||||
| 20 | 59.9, CH | 4.12c | NH, 21 | 21, 22, 23 | 21, 22, 25, 28a | |
| 21 | 30.9, CH | 2.08, m | 20, 23 | 20, 22, 23 | 20, 23 | |
| 22 | 18.4, CH3 | 0.91, s | 21 | 20, 21 | 21 | |
| 23 | 22.56, CH3 | 0.86, s | 21 | 20, 21 | 21 | |
| NH | 7.36, m | 20 | 21, 25, 30 | |||
| Sta | 24 | 172.7, C | ||||
| 25a | 41.1, CH2 | 2.36, m | 26 | 24, 26, 27 | 26,28, NH(Val), NH | |
| 25b | 2.42, m | 26 | 24, 26, 27 | |||
| 26 | 71.4, CH | 3.92, m | c | 25 | ||
| 27 | 52.5, CH | 3.83, m | c | |||
| 28a | 39.7, CH2 | 1.29, m | c | 27, 28b | ||
| 28b | 1.49, m | c | 28a | |||
| 29 | 25.4, CH | 1.57, m | 30 | 28, 30, 31 | 31 | |
| 30 | 23.6, CH3 | 0.88, s | 29 | |||
| 31 | 22.4, CH3 | 0.85, s | 29 | |||
| NH | 7.04b | 27 | ||||
| Ser | 32 | 171.8, C | ||||
| 33 | 56.9, CH | 4.31, m | 34, NH | 34 | 34, NH | |
| 34 | 62.6, CH2 | 3.77, m | 33 | 32 | ||
| Tyr | 35 | 172.5, C | ||||
| 36 | 56.1, CH | 4.58, m | 37a, 37b, NH | 37, 38, 32 | 37a,b, NH | |
| 37a | 36.9, CH2 | 2.81, m | 36, 37b | 35, 39/43 | 37b | |
| 37b | 3.14, m | 36, 37a | 35,36, 39/43 | 36, 37a, 39/43 | ||
| 38 | 129.0, C | |||||
| 39/43 | 131.0, CH | 7.09, d (8.3) | 40/42 | 37,38, 40/42, 41 | 37a,b, 36 | |
| 40/42 | 115.9, CH | 6.72, d (8.4) | 39/43 | 38, 41, 40/42, | ||
| 41 | 156.6, C | |||||
| NH | 7.94, m | 44 | 36, 37a, 46, 39/43 | |||
| Valic acid | 44 | |||||
| 45 | 81.2, CH | 4.87, d (4.1) | 46 | 46, 47, 48 | 46, 47, 48, NH (Tyr) | |
| 46 | 31.1, CH | 2.01, m | 45, 47, 48 | 45, 47, 48 | 45, NH (Tyr) | |
| 47 | 16.8, CH3 | 0.67, d (6.8) | 46 | 45, 46, 48 | 46, 45 | |
| 48 | 18.9, CH3 | 0.84d | 46 | 45, 46, 47 | 46 | |
| N,N-Me2-Ile | 49 | 168.0, C | ||||
| 50 | 71.8, CH3 | 3.94, d (4.5) | 51 | 51, 52, 53 | 51, 52, 53 | |
| 51 | 34.1, CH | 2.15, m | 50, 52 | 49, 50, 52, 53, 54 | 52 | |
| 52 | 14.0, CH3 | 0.95, s | 51 | 50, 51, 53 | ||
| 53a | 27.5, CH2 | 1.32, m | 50, 51, 52, 54 | 50, 51 | ||
| 53b | 1.42, m | 53a | 50, 53a | |||
| 54 | 12.0, CH3 | 0.96, s | 53a,b | 51, 52 | ||
| 55/56 | 43.0, CH3 | 2.83, s | 50 |
Determined from edited HSQC.
nJCH = 8 Hz.
significant overlap with isoleucine.
overlapped
Figure 1.
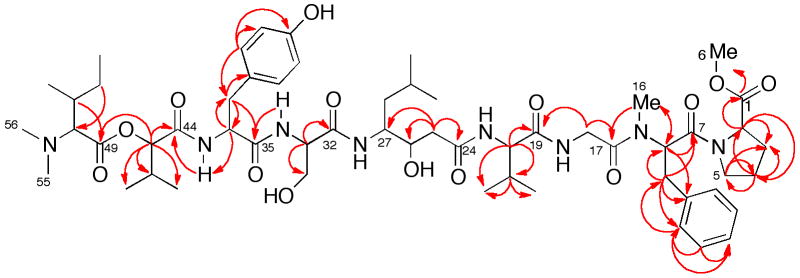
HMBC correlations in compound 3.
A TOCSY experiment readily traced the α-CH proton signal (δ 3.94, d) to a spin network belong to isoleucine (Ile). Integration of the δ 2.83 singlet in the 1H NMR spectrum showed that the presence of two equivalent methyl groups; thus 3 is a linear peptide terminated with an N,N-dimethylisoleucine residue. An O-CH3 group (δH 3.65 s) was linked to the terminal proline methyl ester (O-MePro) by HMBC correlations. The COSY and HMBC correlations from the downfield shifted CH (C-45 δ 81.2) were consistent with a 3-methyl-2-hydroxybutanoyl residue (2-valic acid). Finally, the remaining COSY and HMBC correlations of 3 revealed the presence of a γ-amino acid, statine (4-amino-3-hydroxy-6-methylheptanoic acid).
The configurations of the proteinogenic amino acid residues were determined, uneventfully, after hydrolysis of 3 (6M HCl, 110 °C) and derivatization-HPLC analysis using Marfey’s method.vii The α-amino acid residues Tyr, Ser, Val and O-MePro were found to be of the L-configuration, and the remaining residues were assigned either by Marfey’s method (N-MePhe, statine) or an alternative O-derivatization (2-naphthacyl esters; see below) after preparation of amino acid standards as follows.
N-Me-L-Phe was prepared (Scheme 2) by methylation of N-Boc-L-phenylalanine ethyl ester (freshly prepared Ag2O, CH3I) to obtain N-Boc-N-methyl-L-phenylalanine ethyl ester followed by acid-deprotection (3M HCl, 80 °C).viii Comparison of the L-Marfey’s derivative of the peptide hydrolysate of 3 with that of standard N-MePhe (both L-and D-FDAA reagents; tR = 33.35 and 44.65 min, respectively) revealed the presence of N-Me-D-Phe.
Scheme 2.

Synthesis of diastereomeric statine standards, (3S,4S)-5a and (3R,4S)-5b. See references ix and x.
Two diastereomers of statine were prepared by modifications of published procedures from Schmidtix and Galeottix (Scheme 2). (S)-N-Boc-Leu underwent intermolecular couplingcyclization with Meldrum’s acid (DCC, DMAP) to produce the corresponding pyrrolidin-2,4-dionexi which was reduced (NaBH4) diastereoselectively to provide the 4-hydroxypyrrolidinone (4S,5S)-4a (pyrrole numbering) as a single diastereomer. Ring opening of 4a (NaOMe, MeOH) followed by deprotection (TFA) gave pure (3S,4S)-statine (5a). Alternatively, the (3S,4R)-diasteromer 5b was obtained, along with 5a (2:3, 1H NMR), when borohydride reduction of the pyrrolidindione was carried out after removal of the N-Boc protecting group (TFA-CH2Cl2), followed by acid hydrolysis (6M HCl, 110 °C, 2.5 h).x,xii The diastereomeric ratio of the mixture of 5a:5b was the same as that of the 4-hydroxypyrrolidinone from which it derived, suggesting that no epimerization had taken place at C4.xiii HPLC-Marfey’s analysis of 3, as described above, revealed symplocin A contains the (3R,4S) diastereomer of statine.
Assignment of absolute configurations of the N,N-dimethylisoleucine and valic acid residues presented a larger obstacle: both are insufficiently nucleophilic to react with Marfey’s reagents (FDAAvii or FDLAxiv). As an alternative, α-bromoacetophenone has been extensively for derivatization of carboxylic acids to their corresponding phenacyl esters.xv We anticipated that free N,N-Ile and valic acid, present in the peptide hydrolysate of 3, would undergo O-alkylation exclusively at their carboxy groups at pH = 6, conditions under which the OH group in α-hydroxyacids and α-N,N-dimethylamino groups should be protonated and unreactive. Subsequent assignment of the configuration of the ester products should be feasible under chiral-phase HPLC conditions.
The four stereoisomers of N,N-dimethylisoleucine (6a-d) were prepared by separate reductive alkylation (CH2O, H2, Pd-C)xvi of stereoisomers of Ile. For reasons of sensitivity, we chose α-bromo-2-acetonaphthone (7) to prepare naphthacyl esters. The latter possesses two advantages over phenacyl esters: a stronger chromophore (λmax 248 nm, log10 ε 3.9413) that is inherently fluorescent. Compounds 6a-d were separately converted (Scheme 3) into the corresponding 2-naphthacyl esters 8a-d which were readily visualized and collected from preparative TLC plates under long-wavelength UV irradiation. Optimized conditions for chiral-phase HPLC separation of the four esters (Chiralcel OD-H, 2:98 i-PrOH + 0.1% TFA/hexane, 0.5 mL/min) gave baseline separations of 8a-d (tR = 28.60, 29.91, 27.42, 25.22, respectively). Derivatization of the acid hydrolysate of 3 and HPLC under identical conditions gave a compound that co-eluted with 8b; therefore, 3 contains N,N-Me2-D-Ile.
Scheme 3.
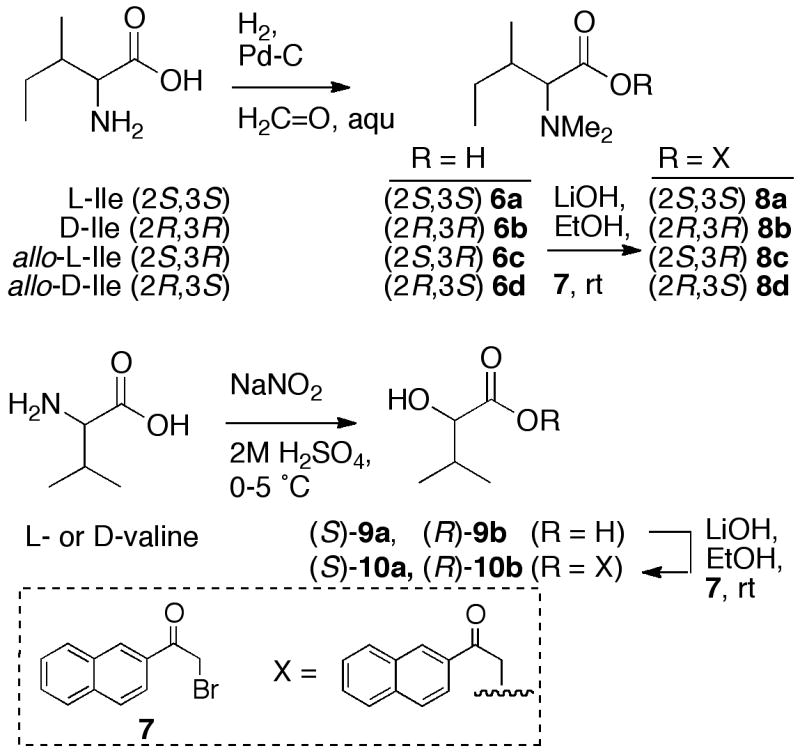
Synthesis of 2-naphthacyl esters of N,N-dimethylisoleucine stereoisomers and (S)- and (R)-valic acid.
The absolute configuration of valic acid was determined in a similar manner. Samples of L- and D-valic acid (9a,b), prepared by diazotization-hydrolysisxvii of L- and D-Val (Scheme 3), were converted to the corresponding 2-naphthacyl esters 10a,b under conditions similar to those described above. The two enantiomers were separated with baseline resolution on chiral-phase HPLC (tR = 30.20; 18.16, respectively). The EtOAc-soluble material, obtained by two-phase extraction of the acid hydrolysate of 3, was derivatized in the same manner. Chiral-phase HPLC of this material showed only the presence of O-(2-naphthacyl) ester of valic acid (D-10b) that co-eluted with an authentic standard. Therefore, D-valic acid is present in 3, and the complete configuration of the peptide is revealed.
Peptide 3 is most similar to grassystatins A, B (2a,b), however, important differences in the structures of 3 and 2a,b are apparent. Compounds 2a,b contain two units of S valic acid whereas 3 has an R valic acid residue. Hydrolysis of statine-like residues is sometimes accompanied by epimerization at C-3. We find that under conditions of acid hydrolysis of 3, the statine residue undergoes only partial epimerization at C-3, unlike that of grassypeptin A (2a), or the homologated-Phe γ-amino acid residue in stictamides A-C,xviii or the isostatine residue in didemnin B.xix In contrast, the C-4 stereocenter (pyrrole numbering) of 4a (Scheme 2) does not epimerize under similar conditions. Consequently, the observed ratio of 5a:5b in the hydrolysate of 3 suggests the major diastereomer retains the configuration (3S,4S) of the intact peptide.
The configurational analysis of DMAAs has, in the past, relied on total synthesis (e.g. the need for total synthesis of 1, its diastereomers and systematic spectroscopic comparisonsxx) or methods of lower sensitivityxxi based in chiral-phase HPLC (penicillamine-bonded phase, elution with Cu2+, aq. CH3CN). The 2-naphthacyl ester approach provides an independent, highly sensitive tool for configurational analysis of DMAAs. Our finding that the α-CH carbon 1 in the terminal N,N-Me2-Ile has the D configuration refutes an earlier assumption, “the precedent that all marine natural products containing an N,N-dimethyl terminal amino acid residue possess the L configuration at this center”,ia and offers a more satisfying experimental solution for independent verification of DMAA configuration.
Symplocin A (3) lacked significant cytotoxicity against cultured tumor cells (HCT-116) but exhibited potent activity as an inhibitor of the protease enzyme, cathepsin Exxii (IC50 300 pM), comparable to that of pepstatin.xxiii This is consistent with the findings by Luesch and coworkers who reported inhibition of cathepsin E by grassystatins A and B (IC50 886 pM and 354 pM, respectively).ic
In conclusion, a new peptide, symplocin A (3) from Symploca sp., has been characterized and shown to contain the N,N-Me2-D-Ile terminal amino acid residue and an acetate-homologated γ-amino acid, (3S,4S)-statine. A new method to assign the absolute configurations of N,N-dimethylamino acids and 2-hydroxy acids was deployed, along with Marfey’s analysis, to secure the complete stereostructure of 3.
Experimental Section
General Experimental Procedures
General procedures are described elsewhere.xxiv
Microbial Material
A mixed cyanobacterial assemblage (10-10-039) was collected at San Salvador Island, Bahamas, in July 2010, at a depth of 25 m, and frozen until required. The following characteristics of the major filamentous components (>70%) were observed by comparative microscopy: presence of sheath, cell dimensions: width ~7.5 μm, length ~5 μm, shallow constrictions with a single trichome, lack of calyptra, which are characteristic of Symploca sp.xxv Based on general appearance of the type sample slide, approximately 75–80% of the type sample appears to be Symploca and the remaining sample is made up of diatoms, other cyanobacteria resembling the genus Lyngbya spp. A voucher specimen is archived at UC San Diego, Department of Chemistry and Biochemistry.
Extraction and Isolation
A sample of a cyanobacterial assemblage (116 g wet wt.) was extracted with MeOH (2 × 900 mL over 8 h). The concentrated extract was partitioned between EtOAc (3 × 700 mL) and H2O (300 mL) and the organic layer concentrated under reduced pressure to give a green solid (130.0 mg). The EtOAc extract (121.5 mg) was subjected to Sephadex LH-20 chromatography eluting with 100% MeOH to give 45 fractions. Fractions 12–14 (22.9 mg) were combined, dried under reduced pressure, and subjected to semi-preparative reversed-phase HPLC (C18, 2 mL/min, gradient, 40:60 to 100:0 CH3CN + 0.1% aq. TFA: H2O +0.1% aq. TFA over 40 min) to give 3 (3.1 mg).
Colorless amorphous solid; [α]D22.5 +16.0 (c 2.18, MeOH); FTIR (ATR): ν 3311, 2972, 1745, 1671, 1518, 1447, 1204, 1138 cm−1; 1H and 13C NMR, see Table 1. HRESIMS m/z 1095.6330 [M+H]+ (calcd for C56H86N8O14, 1095.6336).
N,N-Dimethyl-L-isoleucinexxvi
A mixture of L-isoleucine (0.020 g in 0.8 mL water), formaldehyde (0.049 mL, 37% aq.) and 10% Pd-C (20 mg, 0.188 mmol) was stirred vigorously overnight under 1 atm of H2. After venting, the mixture was heated to reflux, filtered hot (x2), and the filtrate concentrated under reduced pressure to afford N,N-dimethyl-L-isoleucinexxvi as a colorless solid; 1H NMR (300 MHz, D2O) δ 3.39 (1H, d, J = 10.2 Hz), 3.30 (1H, s), 2.87 (6H, s), 2.01 (1H, m), 1.41 (1H, m), 1.03 (3H, d, J = 7.3 Hz), 0.89 (3H, t, J = 7.4 Hz).
N-Boc-L-Phenylalanine Ethyl Ester
To a solution of L-phenylalanine ethyl ester (1.0 g in 10 mL CH2Cl2) was added di-tert-butyl dicarbonate (1.31 g, 5.57 mmol) followed by dry, distilled diisopropylethylamine (10 mL, 7.42 mmol). The mixture was stirred at rt (0.5 h), extracted (3 × 25 mL H2O), dried over MgSO4, and concentrated under reduced pressure to yield 1.0 g N-Boc-L-phenylalanine ethyl ester; 1H NMR (300 MHz, CDCl3) δ 7.12–7.33 (m, 5H), 4.97 (1H, m), 4.56 (1H, m), 4.15 (3H, q, J = 7.1 Hz), 3.07 (1H, br s), 1.52 (3H, s), 1.41 (9H s), 1.22 (3H, t, J = 7.2 Hz).
N-Methyl-L-Phenylalanine Hydrochloride
A mixture of N-Boc-phenylalanine ethyl ester (0.4 g, 1.4 mmol), freshly prepared Ag2O (1.28 g, 5.5 mmol) and CH3I (1.54 mL, 11.0 mmol) in DMF (15 mL) was stirred overnight at rt. The mixture was diluted with H2O (300 mL) and extracted (200 mL CHCl3, 3x) and the combined organic extracts dried over MgSO4, and concentrated. The product was purified by flash chromatography (silica, 1:10 acetone:hexane, flow rate: 25 mL/min) to obtain N-Boc-N-methyl-L-phenylalanine ethyl esterxxvii (0.236 g, 56%). A solution of N-Boc-N-methyl-L-phenylalanine ethyl ester (1 mg) in MeOH (0.25 mL) was treated with HCl (3 M HCl aq, 0.25 mL). The reaction mixture was stirred for 2 h at room temperature, and extracted with EtOAc (3 × 1 mL). The combined organic extracts were concentrated under a stream of N2 to give pure N-methyl-L-phenylalanine hydrochloride 1H NMR data were in agreement with literature values.xxvii
(4S,5S)-4-Hydroxypyrrolidin-2-one (4a)
To a solution of N-Boc-L-leucine (0.5 g, 2.16 mmol) in CH2Cl2 (5 mL) was cooled to 0 °C and treated with Meldrum’s acid (0.809 g, 2.16 mmol), DMAP (0.369 g, 3.2 mmol), and freshly triturated DCC (0.5 g, 2.51 mmol). The solution was warmed to rt and left to stir for 3 h and poured into cold EtOAc (50 mL). The solution was then filtered and the filtrate was washed with cold aqueous NaHSO4 (8 mL, 5% w/v), followed by brine (8 mL). After concentration under reduced pressure, the resulting solid was redissolved in EtOAc (20 mL) and heated at reflux (0.5 h). After removal of volatiles, the residue was purified by flash chromatography (silica, gradient: 1:9 to 1:1 EtOAc: hexane). Fractions identified by TLC were then combined and concentrated to yield the known pyrrolidin-2,4-dioneix (0.42g, 42%) which was used directly in the next step. 1H NMR (300 MHz, CDCl3) δ 4.40 (1H, m), 3.28 (1H, d, J = 18.9 Hz), 3.16 (1H, d, J = 18.9 Hz), 1.87–2.01 (1H, m), 1.56 (9H, s), 1.1–1.4 (1H, m), 0.95 (6H, d, J = 6 Hz) in agreement with literature values.ix
A solution of the above described pyrrolidin-2,4-dione (0.186 g, 0.71 mmol in CH2Cl2-AcOH (3.5 mL, 9:1) was cooled to 0 °C and treated with NaBH4 (0.055 g, 1.4 mmol) in two portions. The mixture was stirred at 0 °C for 30 min, concentrated, and redissolved in EtOAc (7 mL). The organic solution was washed with 5% aq NaHCO3 (3 × 3 mL). The crude product was then purified by flash chromatography (silica, gradient 3:1 EtOAc/hexane to EtOAc) to yield known 4-hydroxypyrrolidinone 4aix (55.5 mg, 30% yield). 1H NMR (400 MHz, DMSO) δ 5.29 (1H, d, 9.66), 4.30 (1H, m), 4.00 (1H, m), 2.45 (2H, dq, J = 16.8, 6.9), 1.72 (2H, m), 1.44 (9H, s), 0.90 (6H, m), in agreement with literature values.ix
(4R,5S)-4-Hydroxypyrrolidin-2-one (4b)
To a suspension of pyrrolidin-2,4-dione (0.128 g, 0.78 mmol) in CH2Cl2 (1 mL) was added TFA (1 mL) and stirred for 20 min. The solution was concentrated under a stream of N2 to provide crude product (0.121 g) that was dissolved in CH2Cl2-AcOH (9:1, 4 mL). NaBH4 (60 mg, 1.56 mmol) was added at 0 °C in two portions and the mixture was allowed to warm to rt over 45 min with stirring. The organic solution was washed with 5% NaHCO3 aq. (3× 5mL), and the organic phase dried over MgSO4, concentrated and purified by flash chromatography (silica, 9:1 CH2Cl2-MeOH) to yield a mixture of 4a:4b (62:38, 38.0 mg, 30%).
(3S,4S)-Statine TFA Salt (5a)
A mixture of 4a (55.5 mg in 1 mL MeOH) and NaOMe (0.5 mL, 0.44 M) was stirred for 1 h, concentrated under reduced pressure and taken up in EtOAc (10 mL). The EtOAc solution was then washed with H2O (5 mL) and brine (5mL). Acidification of the aqueous layer (1M HCl, 0.5 mL) yielded pure (3S,4S)-N-Boc statineix (21.4 mg, 38%) with data consistent with literature values. 1H NMR (300 MHz, CDCl3) δ 4.02 (1H, m), 3.61 (1H, m), 2.55 (2H, d, J = 3.0 Hz), 1.43 (9H, s), 0.91 (6H, m). A sample of the latter compound (1.0 mg, 0.36 μmole) was stirred in CH2Cl2:TFA (1:1, 0.5 mL) at rt for 2 h to yield, after removal of volatiles, (3S,4S)-statine TFA salt (5a, quant). 1H NMR (400 MHz, D2O) δ 4.10 (1H, m), 3.31 (1H, m), 2.66 (2H, m), 1.68 (1H, m), 1.50 (2H, m), 0.91 (6H, m).ix
(3R,4S)-Statine TFA Salt (5b)
S-N-Boc Pyrrolidin-2,4-one was subject to deprotection (TFA), followed by NaBH4 reductionx and hydrolysis (6M HCl, 110 °C, 2.5 h) to give a mixture of diastereomers 5a:5b (62:38). LRMS m/z 176.4 [M+H]+ (calcd 176.2).
(S)-Valic Acid
To an ice-cold solution of L-valine (100 mg, 0.854 mmol) in 2M H2SO4 (0.97 mL) was added NaNO2 (116 mg) in H2O (0.155 mL) over 4 h, and the mixture allowed to warm to rt with stirring over 6 h. The reaction mixture was saturated with NaCl, and extracted with EtOAc (10 × 20 mL) and the combined organics dried over MgSO4. Removal of the volatiles under reduced pressure afforded S-valic acid (9a) as a viscous oil (0.053 g, 53%). 1H NMR (300 MHz, CDCl3) δ 4.16 (1H, d, J = 3.4 Hz), 2.16 (1H, m), 1.07 (3H, d, J = 6.9), 0.93 (3H, d, J = 6.9 Hz).xxviii
(S)-Valic Acid 2-Naphthacyl Ester 10a
To a solution of S-valic acid (9a, 20 mg, 0.17 mmol in 0.50 mL EtOAc) was added α-bromo-2-acetonaphthone (0.042 g, 0.17 mmol) in EtOAc (2.5 mL) followed by Et3N (23 μL). The solution was stirred at rt for 3 h followed by extraction with H2O (3 × 2 mL). The combined organic layers were washed, sequentially, with 10% citric acid (1 mL), 7% NaHCO3 (1 mL), and brine (1 mL) and the volatiles removed under reduced pressure. The residue was dissolved in EtOAc and separated by preparative TLC (1:1 hexane:EtOAc). and the middle-eluting product band was extracted from silica gel (2 mL EtOAc), to yield L-valic acid 2-naphthacyl ester (10a, 5.0 mg), colorless solid; [α]D26 +5.0 (c 1.01, EtOAc); UV (MeOH), λmax (log ε) 248 (4.34), 283 (3.65), 338 (2.95) nm; FTIR (ATR): ν 3349, 2921, 2851, 1741, 1695 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.42 (1H, s), 7.94 (5H, m), 7.63 (2H, m), 5.69 (1H, d, J = 21.6 Hz), 5.52 (1H, d, J = 21.6 Hz, 5.18 (1H, d, J = 5.8 Hz), 2.53 (1H, m), 1.20 (6H, m). 13C (100 MHz, CDCl3) δ 191 (C), 168 (C), 136 (C), 133 (C), 131 (C), 129.9 (CH), 129.8 (CH), 129.3 (CH), 129.2 (CH), 128 (CH), 127 (CH), 123 (CH), 84.1 (CH), 67.0 (CH2), 30.0 (CH), 19.0 (CH3), 17.4 (CH3). HRESIMS m/z 309.1098 [M+Na]+ (calcd for C17H18O4Na, 309.1097).
(±)-D,L-10a,b was prepared from (±)-valic acid in a similar manner. See below for chiral-phase HPLC retention times.
N,N-Dimethyl-L-isoleucine 2-Naphthacyl Ester 8a
To a solution of N,N-dimethyl-L-isoleucine (6a, 1.18 mg, 11.4 μmol) in EtOH (200 μL) was added NaHCO3 (3 mg, 23 μmol) followed by α-bromo-2-acetonaphthone (7, 4 mg, 11.4 μmol). The mixture was stirred at room temperature overnight and dried down and separated by preparative TLC (9:1 hexane: EtOAc). The band corresponding to product was extracted with CH2Cl2 to yield N,N-dimethyl-L-isoleucine naphthacyl ester (8a, 0.8 mg, 40%). [α]D23 +12.2 (c 0.35, CH3CN); UV (CH3CN) λmax 248 (log ε 4.45), 283 (3.73), 338 (3.00) nm; FTIR (ATR): ν 2920, 2851, 2346, 1680, 1466 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.45 (1H, s), 7.93 (4H, m), 7.60 (2H, m), 5.53 (2H, s), 3.06 (2H, d, J = 10.1), 2.41 (6H, s), 1.93 (1H, m), 1.72 (1H, m), 1.21 (2H, m), 0.99 (3H, d, J = 6.6), 0.93 (3H, t, J = 7.5 Hz). 13C (100 MHz, CDCl3) δ 192 (C), 171 (C), 136 (C), 132 (C), 131 (C), 130 (CH), 129 (CH), 128 (CH), 127 (CH), 123 (CH), 72.5 (CH), 65.6 (CH2), 41.6 (CH), 33.6 (CH), 25.3 (CH2), 15.8 (CH3), 10.7 (CH3); HRESIMS m/z 328.1909 [M+H]+ (calcd for C20H26NO3, 328.1907).
Chiral-Phase HPLC Analysis of N,N-Dimethyl-L-isoleucine 2-Naphthacyl Esters 8a-d
Stereoisomerically pure and mixed N,N-Me2 isoleucines [allo-L-Ile, 6c, 1:1 mixture of L-Ile, 6a, and allo-D-Ile (6d, Sigma), and a 1:1:1:1 mixture of 6a-d (Sigma)] were converted to the corresponding naphthacyl esters 8a-d, as described above for pure S-6a, and analyzed by chiral-phase HPLC (Chiralcel OD-H, 250 × 4.60, 0.5 mL.min−1, 2:98 i-PrOH+0.1% TFA/hexane) and gave the following retention times, tR = 28.60, 29.91, 27.42, 25.23 min, respectively.
Hydrolysis of Symplocin A
Symplocin A (3, 200 μg) was subjected to acid hydrolysis (6N HCl, 120 °C, 20 h), the mixture was cooled, and a portion was extracted with EtOAc. The organic layer was dried and used in the preparation of 10a (see below) and the remainder of the aqueous hydrolysate was dried under a stream of nitrogen and high vacuum before conversion to Marfey’s derivatives or 2-naphthacyl esters as described below.
Preparation of 2-Naphthacyl Esters from the Peptide Hydrolysate of 3
An aliquot of the peptide hydrolysate of 3 (~100 μg in 200 μL EtOAc) of the ethyl acetate-soluble fraction was treated with α-bromo-2-acetonaphthone (200 μg) followed by triethylamine (~10 μL). The reaction was stirred overnight at rt followed by isolation by preparative TLC (1:1 EtOAc:hexane). The extracted band was identified as product by LRMS (m/z 287.5 [M+H]+; calc 287.1) as valic acid 2-naphthacyl ester 10. The product was concentrated under a stream of N2 and redissolved (1 mg/mL, 1:1 hexane:i-PrOH) and analyzed by chiral-phase HPLC (see below).
An aliquot of the total hydrolysate (2.0 mg) was dissolved in EtOH (250 μL) and adjusted to pH = 6 (aq. LiOH). To the reaction mixture was added α-bromo-2-acetonaphthone (4.2 mg). The reaction mixture was left to stir at rt overnight. The reaction mixture was then dried under a stream of N2 and redissolved in CH2Cl2 (0.5 mL) and separated by preparative TLC (1:9 EtOAc:hexane). The product band containing 8 (LRMS, m/z 328.52 [M+H]+; calc 328.18) was concentrated under a stream of N2 and re-constituted (1 mg/mL, 1:1 hexane:i-PrOH) and analyzed by chiral-phase HPLC (Chiracel OD-H, 250 × 4.6 mm, 0.5 mL.min−1, 2:98 i-PrOH+0.1%TFA:hexane) along with standard naphthacyl esters 8a-d prepared as described above. The natural product-derived N,N-dimethylisoleucine 2-naphthacyl ester was identified as (2R,3R)-8b (tR=28.75) and confirmed by coinjection with standard 8b.
Chiral-Phase HPLC Analysis of 2-Naphthacyl Valic Acid Esters
An aliquot of the EtOAc-soluble hydrolysate of peptide 3 (~100 μg) was derivatized with α-bromo-2-acetonaphthone (see above) and compared with the standard 2-naphthacyl esters (10a,b) (prepared from (±)-valic acid 9a,b, see above) by chiral-phase HPLC (Chiracel OD-H, 250 × 4.60, 0.5 mL.min−1, 25:75 i-PrOH/hexane), giving the following respective retention times. tR = 18.17, 30.14 and 18.16 min. The identity of the natural product-derived S-10b and the synthetic standard was confirmed by coinjection.
Marfey’s Analysis of 3
Aliquots (~100 μg) of hydrolysed symplocin A (3) were dissolved in 1M aq. NaHCO3 (400 μL) and treated, separately, with L-FDAA or D-FDAA (50 μL, 2 mg/mL in acetone). The mixture was stirred at 80 °C for 30 min followed by the addition of HCl (2 M, 200 μL) and dilution with CH3CN (100 μL). The freshly prepared Marfey’s derivatives of the peptide hydrolysate and standard amino acids were analyzed under two sets of conditions. Condition A (RP HPLC, Agilent Eclipse XDB-C18, 1.0 mL/min, gradient elution with 1:4 to 1:1 CH3CN/H2O + 0.1% HCOOH over 45 min, monitoring at 340 nm) was used to assign Ser, Tyr, and N-MePhe. Condition B (RP HPLC, Agilent Eclipse XDB-C18, 1.0 mL/min, gradient elution with 1:9 to 100% CH3CN/H2O + 0.1% formic acid over 45 min, monitoring at 340 nm) was used to assign Pro and Val. The retention times (min) of authentic L-FDAA derivatives were L-Ser (22.39), D-Ser (23.6), L-Pro (31.25), D-Pro (33.0), L-Tyr (26.08), D-Tyr (30.79), L-Val (33.42), D-Val (37.84), L-N-MePhe (33.35), and D-N-MePhe (44.46). The Marfey’s derivatives of hydrolyzed 3 gave retention times of 22.00, 31.92, 27.37, 33.21, and 43.71 min, corresponding to L-Ser, L-Pro, L-Try, L-Val, and D-N-MePhe, respectively, which were confirmed by co-injections.
Supplementary Material
Acknowledgments
We thank the UC Davis Molecular Structure Facility for amino acid composition analysis, and Y. Su of the UC San Diego Small Molecular Mass Spectrometry facility for MS fragmentation data. The 500 MHz NMR spectrometers were purchased with a grant from the NSF (CRIF, CHE0741968). This work was generously supported by the NIH (AI039987).
Footnotes
Dedicated to Dr. Gordon M. Cragg, formerly Chief, Natural Products Branch, National Cancer Institute, Frederick, Maryland, for his pioneering work on the development of natural product anticancer agents
Supporting Information Available: 1H, 13C NMR and 2D NMR spectra of 3, and 1H,13C NMR of naphthacyl esters 8a, and 10a. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- i.a) Linington RG, Clark BR, Trimble EE, Almanza A, Ureña LD, Kyle DE, Gerwick WH. J Nat Prod. 2009;72:14–17. doi: 10.1021/np8003529. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Luesch H, Moore RE, Paul VJ, Mooberry SL, Corbett T. J Nat Prod. 2001;64:907–910. doi: 10.1021/np010049y. [DOI] [PubMed] [Google Scholar]; c) Kwan JC, Eksioglu EA, Liu C, Paul VJ, Luesch H. J Med Chem. 2009;52:5732–5747. doi: 10.1021/jm9009394. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Simmons TL, McPhail KL, Ortega-Barría E, Mooberry SL, Gerwick WH. Tetrahedron Lett. 2006;47:3387–3390. [Google Scholar]
- ii.Pettit GR, Kamano Y, Dufresne C, Cerny RL, Herald CL, Schmidt JM. J Org Chem. 1989;54:6005–6006. [Google Scholar]
- iii.(a) Pettit GR, Kamano Y, Herald CL, Tuinman AA, Boettner FE, Kizu H, Schmidt JM, Baczynskyj L, Tomer KB, Bontems RJ. J Am Chem Soc. 1987;109:6883–63885. [Google Scholar]; (b) Pettit GR, Singh SB, Hogan F, Lloyd-Williams P, Herald DL, Burkett DD, Clewlow PJ. J Am Chem Soc. 1989;111:5463–5465. [Google Scholar]
- iv.Pitot HC, McElroy EA, Reid JM. Clin Cancer Res. 1999;5:523–531. [PubMed] [Google Scholar]
- v.(a) MacMillan JB, Ernst-Russell MA, de Ropp JS, Molinski TF. J Org Chem. 2002;67:8210–8215. doi: 10.1021/jo0261909. [DOI] [PubMed] [Google Scholar]; (b) Dalisay DS, Rogers EW, Edison A, Molinski TF. J Nat Prod. 2009;72:732–738. doi: 10.1021/np8007649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vi.Hutchinson CR. Proc Natl Acad Sci USA. 2003;100:3010–3012. doi: 10.1073/pnas.0730689100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vii.Marfey P. Carlsberg Res Commun. 1984;49:591–596. [Google Scholar]
- viii.Stahl GL, Walter R, Smith CW. J Org Chem. 1978;43:2285–2286. [Google Scholar]
- ix.Schmidt U, Riedl B, Haas G, Griesser H, Vetter A, Weinbrenner S. Synthesis. 1993:216–220. [Google Scholar]
- x.Galeotti N, Poncet J, Chiche L, Jouin P. J Org Chem. 1993;58:5370–5376. [Google Scholar]
- xi.Hosseini M, Kringelum H, Murray A, Tonder JE. Org Lett. 2006;8:2103–2106. doi: 10.1021/ol060500i. [DOI] [PubMed] [Google Scholar]
- xii.The lower diastereoselectivity of NaBH4 reduction of the unprotected pyrrolidindione has been noted before (ref. x).
- xiii.Other investigators (e.g. references ib) have noted a tendency for peptides containing statine-like acetate-homologated γ-aminoacid residues to undergo partial or complete epimerization at C3, most likely by reversible acid-catalyzed elimination–Michael addition of H2O.
- xiv.Fujii K, Ikai Y, Mayumi T, Oka H, Suzuki M, Harada KI. Anal Chem. 1997;69:3346–3352. [Google Scholar]
- xv.Bray AM, Maej NJ, Valerio RM, Campbell RA, Geysen HM. J Org Chem. 1991;56:6659–6666. [Google Scholar]
- xvi.Bowman RE, Stroud HH. J Chem Soc. 1950:1322–1345. [Google Scholar]
- xvii.Muller J, Feifel SC, Schmiederer T, Zocher R, Sussmuth RD. ChemBioChem. 2009;10:323–328. doi: 10.1002/cbic.200800539. [DOI] [PubMed] [Google Scholar]
- xviii.Liang Z, Sorribas A, Sulzmaier FJ, Jimenez JI, Wang X, Sauvage T, Yoshida WY, Wang G, Ramos JW, Williams PG. J Org Chem. 2011;76:3635–3643. doi: 10.1021/jo200241h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xix.Rinehart KL, Sakai R, Kishore V, Sullins DW, Li KM. J Org Chem. 1992;57:3007–3013. [Google Scholar]
- xx.Pettit GR, Singh SB, Hogan F, Lloyd-Williams P, Herald DL, Burkett DD, Clewlow PJ. J Am Chem Soc. 1989;111:5463–5465. [Google Scholar]
- xxi.Assignment of configuration of the N,N-Me2-L-Val residues in grassystatin A (Ref. ib) and belamide A (Ref. 1d) were made by chiral-phase LC, but no experimental details were given for the latter. Configurational assignment of the N,N-Me2-L-Ile residue in symplostatin (Ref. ib) was also made by chiral-phase LC, but not explicitly assigned in gallinamide A; the latter was assumed to be L- based on “precedence” (Ref. ia).
- xxii.Zuidi N, Hermann C, Herrmann T, Kalbacher H. Biochem Biophys Res Commun. 2008;377:327–330. doi: 10.1016/j.bbrc.2008.10.034. [DOI] [PubMed] [Google Scholar]
- xxiii.Cathepsin E inhibition assays were carried out by Reaction Biology Corporation, Malvern, Pennsylvania.
- xxiv.Dalisay DS, Rogers EW, Edison A, Molinski TF. J Nat Prod. 2009;72:732–738. doi: 10.1021/np8007649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxv.Boone DR, Castenholz RW. Bergey’s Manual of Systematic Bacteriology. 2. Vol. 1. Springer; New York: 2001. p. 541. [Google Scholar]
- xxvi.Ingram VM. J Biol Chem. 1953;202:193–201. [PubMed] [Google Scholar]
- xxvii.Kawabata T, Wirth T, Yahiro K, Suzuki H, Fuji K. J Am Chem Soc. 1994;116:10809–10810. [Google Scholar]
- xxviii.Honda Y, Ori A, Tsuchihashi G. Bull Chem Soc Jpn. 1987;60:1027–1036. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.