

Abstract
Numerous therapeutic applications have been proposed for molecules that bind heparin-binding proteins. Development of such compounds has primarily focused on optimizing the degree and orientation of anionic groups on a scaffold, but utility of these polyanions has been diminished by their typically large size and nonspecific interactions with many proteins. In this study, N-arylacyl O-sulfonated aminoglycosides were synthesized and evaluated for their ability to selectively inhibit structurally similar bacterial and human topoisomerases. It is demonstrated that the structure of the aminoglycoside and of the N-arylacyl moiety imparts selective inhibition of different topoisomerases and alters the mechanism. The results here outline a strategy that will be applicable to identifying small, structurally defined oligosaccharides that bind heparin-binding proteins with a high degree of selectivity.
Keywords: Topoisomerase inhibitor, glycosaminoglycan mimic, sulfated oligosaccharide, aminoglycoside derivatives, ciprofloxacin-induced DNA cleavage
Topoisomerases are enzymes that wind and unwind DNA by breaking and then religating the DNA. Type II topoisomerases, such as DNA gyrase and topoisomerase IV (Topo IV), make a double stranded break in DNA and pass unbroken DNA through the break, creating a net change of two in the linking number.1 Type I topoisomerases, conversely, make single strand breaks that allow the DNA to swivel around the unbroken strand.2,3 Topoisomerases are targets for antibacterial and anticancer agents.4,5
Heparin, a highly sulfated human glycosaminoglycan (GAG) has been shown to inhibit topoisomerase I (Topo I), a type I topoisomerase.6 The polyanionic polysaccharide is thought to mimic the phosphate backbone of DNA and bind to the DNA binding site.6 Heparin and heparan sulfate (HS) have been shown to inhibit type I topoisomerase, while HS has an as-yet unexplained inductive effect on type II topoisomerases.7−9 Inhibition of topoisomerase with fractionated heparin mixtures demonstrated that highly sulfated heparin chains are better at inhibiting Topo I.8 Heparin was also a much more potent inhibitor of Topo I than HS, a less sulfated, though structurally similar GAG.9 Other oligosaccharide and polysaccharide GAG mimics have also been shown to inhibit topoisomerases, including suramin, a hexa-sulfated poly aromatic compound,10,11 a sulfated polysaccharide isolated from microalgae in red tide,12 and an unsulfonated dextran derivative that was modified to various degrees by the addition of aromatic carboxymethyl-benzylamides.13
Previous work in our laboratory evaluated replacement of N-sulfo groups on heparin and other GAGs with nonanionic-N-acyl groups to afford reduced-charge structures capable of competing with unmodified heparin for binding endogenous HS-binding proteins.14,15 Replacement of N-sulfo groups with select N-aryl groups afforded heparin derivatives with maintained or increased affinity for HS-binding proteins.14−16 As an intracellular target, topoisomerases are not readily accessed by sulfated saccharides. However, there is a significant degree of structural homology among topoisomerases,17 which makes topoisomerase inhibition a rigorous test for evaluating new types of GAG mimics for their ability to selectively interact with structurally similar heparin-binding proteins.
In this study, a panel of bacterial (E. coli gyrase and E. coli Topo IV) and human (hTopo I and hTopo II) topoisomerases was used to evaluate N-arylacyl O-sulfonated aminoglycoside derivatives for their ability to selectively inhibit the different topoisomerases. The goal of this work was to determine the level of selectivity that could be achieved for these novel trisaccharide and tetrasaccharide heparin mimics to inhibit function of such structurally similar heparin-binding proteins and to test this strategy for creating novel, small heparin mimics that selectively bind heparin and modulate function of heparin-binding proteins. In addition, the identification of derivatives that inhibit topoisomerases would represent new lead structures for further elaboration as possible anticancer or antibacterial agents.
Synthesis of the O-sulfonated N-arylacyl aminoglycosides was accomplished in two steps (Scheme 1, panel A). The aminoglycosides apramycin, kanamycin, and neomycin were first converted to their N-acetyl, N-benzoyl (N-bz), N-benzyloxycarbonyl (N-cbz), and N-phenylacetyl (N-pha) derivatives by coupling with the corresponding N-(acyl) succinimide (NHS ester) or acyl chloride under alkaline conditions. Synthesis and characterization of per N-cbz derivatives of neomycin,18 kanamycin,19 and apramycin20 have been reported. The per-N-acyl aminoglycosides were sulfonated with pyridine sulfur trioxide complex (Pyr•SO3) or chlorosulfonic acid (ClSO3H). The degree of sulfonation for each derivative was determined as previously described (see also Supporting Information for compound synthesis, purification, and characterization).21 The acetyl derivatives were not assigned an average degree of sulfation because they lack a chromophore for calculating peak areas with HPLC and are instead characterized by a range of sulfated states (e.g., “3–5” indicates the sample contains a mixture of tri-, tetra-, and penta- sulfated states).
Scheme 1. Synthesis of N-Arylacylated O-Sulfonated Aminoglycosides.

(Panel A) Methods used for the per N-acylation and O-sulfonation of aminoglycosides (kanamycin shown as example). (a) NHS ester, NaHCO3, H2O, 23 °C, 12-16 h, 30-85%; (b) acyl chloride, NaHCO3, H2O; (c) i: Pyr·SO3, DMF, anhydrous pyr, 66 °C, 5-7 h; ii: H2O, 10 mM NaOH, 4 °C, 45-65%; (d) i: ClSO3H, pyr, 57 °C, 4-6 h; ii: H2O, NaHCO3, 35-95%. (Panel B) Structures of apramycin and neomycin. (Panel C) The O-sulfonation reactions give individual products having varied degrees of sulfation. For each aminoglycoside, final products obtained are noted by the core aminoglycoside (n, neomycin; k, kanamycin; a, apramycin), the N-arylacyl group (a, acetyl; b, benzoyl; z, benzyloxycarbonyl; p, phenylacetyl), and the calculated average number of sulfate groups per product obtained (e.g., nz5.6) or range of sulfate groups for the acetyl derivatives (e.g., aa3-5).
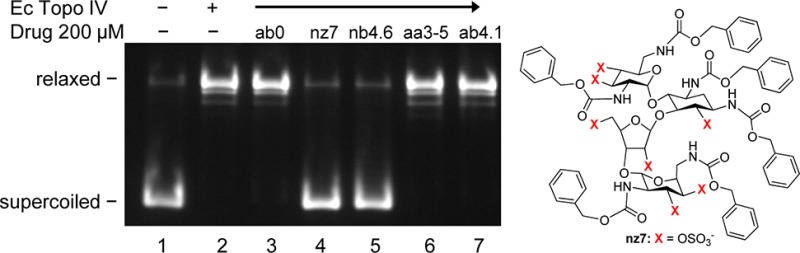
The N-arylacyl O-sulfonated aminoglycosides were initially screened to determine inhibition of E. coli gyrase-mediated supercoiling of relaxed DNA and E. coli Topo IV-mediated relaxation of supercoiled DNA and decatenation of kinetoplast DNA (kDNA) (see Figure 1; methods in Supporting Information). For inhibition of gyrase-mediated supercoiling, gyrase was incubated with relaxed DNA and either no test compound or 200 μM of one of the following compounds: ab0, nz7, nb4.6, aa3–5, or ab4.1 (Figure 1A). In the absence of a test compound, E. coli gyrase converted relaxed DNA into supercoiled DNA (lane 2). Of the compounds screened, only nz7 (lane 4) blocked the supercoiling activity of gyrase. For Topo IV-mediated relaxation of supercoiled DNA, Topo IV was incubated with negatively supercoiled DNA in the absence (lane 2) or presence of 200 μM of one of the test compounds: ab0, nz7, nb4.6, aa3–5, or ab4.1. Topo IV-mediated relaxation of supercoiled DNA was inhibited by both nz7 (lane 4) and nb4.6 (lane 5) (Figure 1B). While nz7 inhibited both supercoiling by gyrase and relaxation by Topo IV, nb4.6 only inhibited topo-IV-mediated relaxation. For Topo IV-mediated decatenation of kDNA, E. coli Topo IV was incubated with kDNA and 200 μM of one of the test compounds (Figure 1C). Only nz7 completely blocked the decatenation reaction (lane 4), while nb4.6 partially inhibited decatenation (lane 5). Thus, Both nz7 and nb4.6 inhibited Topo IV activity in both relaxation and decatenation assays, though the inhibitory effect of nb4.6 was more pronounced in the relaxation assay.
Figure 1.

Inhibition of E. coli type II topoisomerases. In all panels, enzyme and test compound (drug) are absent (−) in lane 1, enzyme present (+) and drug absent in lane 2, and enzyme (arrow) and drug present in lanes 3–7 (in D, 3–13). (Panel A) Supercoiling activity of E. coli gyrase was blocked by nz7 but not affected by the other modified aminoglycosides. (Panel B) Relaxation activity of E. coli Topo IV was inhibited by either nz7 (lane 4) or nb4.6 (lane 5). (Panel C) Decatenation activity of Topo IV was completely blocked by nz7 (lane 4) and partially blocked by nb4.6 (lane 5). (Panel D) Concentration-dependent inhibition of gyrase supercoiling by nz5.6 and nz7.
The observed differential inhibition of bacterial type-II topoisomerases by select N-arylacyl O-sulfonated aminoglycosides may be due to the degree of sulfation, the N-arylacyl moiety, the core aminoglycoside, or a combination these structural differences. As shown in Figure 1A, nz7 inhibited the gyrase-mediated supercoiling reaction. Since more highly sulfated heparin derivatives are more potent inhibitors of topo I,8N-cbz O-sulfonated neomycin with an average degree of sulfation of 5.6 (nz5.6) was screened along with persulfated nz7 to determine concentration-dependent inhibition of gyrase-mediated supercoiling of relaxed DNA (Figure 1D). Although 100 μM of nz5.6 only partially inhibited gyrase activity (lane 6), 200 μM of nz5.6 completely inhibited the supercoiling activity of gyrase (lane 7). Comparatively, 50 μM of nz7 partially inhibited gyrase (lane 11), while 100 μM of nz7 completely inhibited gyrase supercoiling activity (lane 12). Thus, nz7 is about twice as potent as nz5.6. While this result demonstrates the importance of sulfate groups for these particular neomycin derivatives to inhibit gyrase, the overall data in Figure 1 clearly shows the influence of N-arylacyl moieties and the core aminoglycoside for selective inhibition of the bacterial type-II topoisomerases.
To more fully evaluate the role of sulfate groups, core structure, and N-arylacyl group on selective topoisomerase interactions, the complete panel of N-arylacyl and N-acetyl aminoglycosides, both O-sulfonated and bearing no sulfate, were screened for inhibitory activity with four topoisomerases: E. coli gyrase, E. coli Topo IV, hTopo II, and hTopo I (Table 1). None of the unsulfated N-arylacyl aminoglycosides (azo, kb0, kz0, kp0, na0, nz0, and np0) inhibited any topoisomerase (data not shown). The presence of sulfate groups on the N-acyl aminoglycosides was not alone sufficient to afford topoisomerase inhibition; neither of the N-acetyl O-sulfonated aminoglycosides (aa3–5 and na1–6) inhibited any of the topoisomerases. Indeed, sulfated dextran oligosaccharides with a degree of polymerization of 14 are capable of inhibiting hTopo-I.22 Some DNA-binding and heparin-binding proteins, such as fibroblast growth factor-I, will bind persulfated disaccharides like sucrose octasulfate and sulfonated polyaromatics like suramin with weak affinity. However, most DNA-binding and heparin-binding proteins have poor affinity for sulfated polysaccharides less than octasaccharide in length.
Table 1. Percent Inhibition of Catalytic Activity for Different Type I and Type II Topoisomerases.
| druga | gyrase | Topo IV | hTopo II | hTopo I |
|---|---|---|---|---|
| az4.9 | 90% | 95% | 95% | >95% |
| kb6.4 | 85% | 80% | >95% | 60% |
| kz6 | 75% | >95% | ||
| kp5.8 | b | 70% | ||
| na1–6 | ||||
| nz5.6 | >95% | >95% | >95% | |
| np5.9 | 95% | >95% | >95% | >95% |
Compounds screened at 200 μM.
Blank spaces: <5% inhibition.
Only select N-arylacyl O-sulfonated aminoglycoside that have both N-arylacyl groups and sulfate groups on a certain aminoglycoside scaffold inhibited different topoisomerases to different degrees (Table 1 and Figure 1). This result is consistent with previous studies for N-desulfonated, N-acyl derivatives of heparin, where only select N-arylacyl groups could be substituted in place of N-sulfo groups to maintain or increase affinity for select heparin-binding proteins.14−16 In addition to the aryl groups on these tri- and tetra-saccharide-sized compounds being necessary for topoisomerase inhibition, these results show that both the structure of the core aminoglycoside and the structure of the N-arylacyl moiety imparts selectivity for which the topoisomerase(s) are inhibited. For example, among the N-cbz O-sulfonated aminoglycosides, only az4.9 showed greater than 90% inhibition of all four topoisomerases tested (Table 1). The N-cbz kanamycin derivative kz6, which is more highly sulfated than az4.9, showed 75% inhibition of gyrase and greater than 95% inhibition of hTopo I but did not inhibit either Topo IV or hTopo II. The cognate N-cbz neomycin derivative, nz5.6, showed greater than 95% inhibition of gyrase, hTopo II, and hTopo I, but it did not inhibit Topo IV.
To better understand the mechanism of topoisomerase inhibition, the N-arylacyl O-sulfonated aminoglycosides were coadministered with ciprofloxacin to evaluate their ability to interfere with ciprofloxacin-induced DNA cleavage. Ciprofloxacin is a known topoisomerase poison that, when incubated with a bacterial type II topoisomerase and DNA, induces DNA cleavage by arresting the enzyme and its DNA substrate in a ternary complex that prevents religation of the double stranded break in the DNA.23−25
Incubation of E. coli Topo IV with a plasmid DNA affords mainly supercoiled DNA, and small amounts of nicked and linear DNA (Figure 2, lane 2). The addition of ciprofloxacin stimulates the production of linear, cleaved DNA (Figure 2, lane 3). Ciprofloxacin-induced DNA cleavage is completely blocked by nz7 (Figure 2, lane 5), while ab0 and nb4.6 produce a slight decrease (Figure 2, lanes 4 and 6). No effect on ciprofloxacin-induced DNA cleavage was seen with aa3–5 or ab4.1 (Figure 2, lanes 7 and 8).
Figure 2.
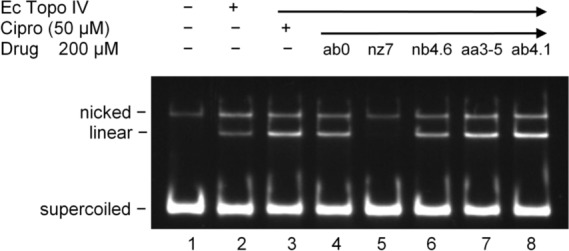
Select N-arylacyl O-sulfonated aminoglycosides block ciprofloxacin-induced cleavage of DNA by TopoIV.
The effect of N-arylacyl O-sulfonated aminoglycosides on ciprofloxacin-induced DNA cleavage by gyrase was similarly investigated (Table 2). With gyrase, kz6 and nz5.6 completely blocked the production of ciprofloxacin-induced linear DNA, while np5.9, kb6.4, and az4.9 partially blocked ciprofloxacin-induced DNA cleavage. These results indicate that, despite the structural homologies between gyrase and Topo IV,17 compounds az4.9, kb6.4, and np5.9 block ciprofloxacin-induced DNA cleavage with Topo IV and not with gyrase and thus may have different binding sites on Topo IV and gyrase. In the absence of ciprofloxacin, the N-arylacyl O-sulfonated aminoglycosides did not induce production of linear DNA, and thus, they do not act as topoisomerase poisons (see Supporting Information). This unique ability of some N-arylacyl O-sulfonated aminoglycosides to block ciprofloxacin-induced DNA cleavage is unexpected and unusual.
Table 2. Inhibition of Ciprofloxacin-Induced DNA Cleavagea.
| compound | Ec gyrase | Sa gyrase | Ec Topo IV |
|---|---|---|---|
| az4.9 | - | - | + |
| kb6.4 | - | - | + |
| kz6 | + | + | + |
| kp5.8 | ND | ND | - |
| nz5.6 | + | + | + |
| np5.9 | - | - | + |
ND: not determined; + indicates greater than 95% inhibition.
To further test for mechanistic differences, kb6.4 and kz6, were compared for their ability to inhibit the supercoiling activity of E. coli gyrase and to block ciprofloxacin-induced DNA cleavage (Figure 3). At 20 μM, neither kb6.4 nor kz6 inhibited gyrase supercoiling activity (Figure 3A, lanes 3 and 5), while at 200 μM, both inhibited the supercoiling activity of gyrase (Figure 3A, lanes 4 and 6). In contrast, kb6.4 showed no apparent effect on ciprofloxacin-induced cleavage of DNA (Figure 3B, lanes 3 and 5), while kz6 blocked the effect of ciprofloxacin on gyrase-mediated DNA cleavage (Figure 3B, lanes 3 and 7). This suggests that inhibition of gyrase by kz6 and kb6.4 may occur at different steps during the gyrase-mediated supercoiling reaction. One possible explanation for this result would be that kb6.4 does not inhibit the binding of the G-segment but that it inhibits either the binding of the T-segment or the binding of ATP. In contrast, kz6 and other oligosaccharides inhibit the supercoiling activity of gyrase by blocking the binding of the G-segment.
Figure 3.

Inhibition of gyrase supercoiling does not correlate with the effect on ciprofloxacin-induced DNA cleavage for different N-arylacyl O-sulfonated aminoglycosides. (A) Supercoiling reaction with kb6.4 and kz6 at either low (L, 20 μM) or high (H, 200 μM) concentration. (B) Ciprofloxacin-induced DNA cleavage reaction with kb6.4 and kz6 at 200 μM.
Studies are currently underway to evaluate selective binding of these novel small molecule heparin mimics with extracellular heparin-binding proteins that play a role in a number of disease states. In addition, while the different topoisomerases were employed in this study as a class of test proteins to evaluate selective interaction of test compounds with structurally similar heparin-binding proteins, select compounds possess unexpectedly unique activity. Although these N-arylacyl O-sulfonated aminoglycosides do not appear to readily enter human or bacterial cells, as evidenced by a lack of cytotoxicity at 200 μM (see Supporting Information), understanding how these novel agents uniquely bind and inhibit the different topoisomerases will further the design, synthesis, and development of smaller, lower charge structures that might penetrate cells and have therapeutic utility as novel topoisomerase inhibitors. The apparent lack of cellular penetration and cytotoxicity for the present compounds is a positive characteristic in the context of pursuing agents to selectively bind and modulate activity of extracellular heparin-binding proteins.
Acknowledgments
This work was supported in part by NIH grant AI087671 to R.J.K., an NIH Predoctoral Traineeship in the Pharmacological Sciences to A.M.F. (T32 GM067795), an ACS Division of Medicinal Chemistry Predoctoral Fellowship sponsored by Bristol-Myers Squibb to A.M.F., and a Predoctoral Fellowship from the American Foundation for Pharmaceutical Education to A.M.F.
Glossary
Abbreviations
- bz
benzoyl
- cbz
benzyloxycarbonyl
- cipro
ciprofloxacin
- ClSO3H
chlorosulfonic acid
- Ec
E. coli
- GAG
glycosaminoglycan
- h
human
- HS
heparan sulfate
- kDNA
kinetoplast DNA
- N-(acyl)succinimide
R-NHS
- pha
phenylacetyl
- Pyr·SO3
pyridine sulfur trioxide complex
- topo
topoisomerase
Supporting Information Available
Synthesis and purification of N-arylacyl O-sulfonated aminoglycosides, HPLC and LC–MS chromatograms, and topoisomerase assay conditions. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Levine C.; Hiasa H.; Marians K. J. DNA gyrase and topoisomerase IV: biochemical activities, physiological roles during chromosome replication, and drug sensitivities. Biochim. Biophys. Acta 1998, 1400, 29–43. [DOI] [PubMed] [Google Scholar]
- Schvartzman J. B.; Stasiak A. A topological view of the replicon. EMBO Rep. 2004, 5, 256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. C. DNA topoisomerases. Annu. Rev. Biochem. 1985, 54, 665–697. [DOI] [PubMed] [Google Scholar]
- Gellert M.; O’Dea M. H.; Itoh T.; Tomizawa J. Novobiocin and coumermycin inhibit DNA supercoiling catalyzed by DNA gyrase. Proc. Natl. Acad. Sci. U.S.A. 1976, 73, 4474–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugino A.; Peebles C. L.; Kreuzer K. N.; Cozzarelli N. R. Mechanism of action of nalidixic acid: purification of Escherichia coli nalA gene product and its relationship to DNA gyrase and a novel nicking-closing enzyme. Proc. Natl. Acad. Sci. U.S.A. 1977, 74, 4767–4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii K.; Katase A.; Andoh T.; Seno N. Inhibition of topoisomerase I by heparin. Biochem. Biophys. Res. Commun. 1982, 104, 541–547. [DOI] [PubMed] [Google Scholar]
- Harisi R.; Dudas J.; Timar F.; Pogany G.; Timar J.; Kovalszky I.; Szendroi M.; Jeney A. Invasive growth and topoisomerase-switch induced by tumorous extracellular matrix in osteosarcoma cell culture. Cell. Biol. Int. 2005, 29, 959–967. [DOI] [PubMed] [Google Scholar]
- Ishii K.; Futaki S.; Uchiyama H.; Nagasawa K.; Andoh T. Mechanism of inhibition of mammalian DNA topoisomerase I by heparin. Biochem. J. 1987, 241, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalszky I.; Dudas J.; Olah-Nagy J.; Pogany G.; Tovary J.; Timar J.; Kopper L.; Jeney A.; Iozzo R. V. Inhibition of DNA topoisomerase I activity by heparan sulfate and modulation by basic fibroblast growth factor. Mol. Cell. Biochem. 1998, 183, 11–23. [DOI] [PubMed] [Google Scholar]
- Bojanowski K.; Lelievre S.; Markovits J.; Couprie J.; Jacquemin-Sablon A.; Larsen A. K. Suramin is an inhibitor of DNA topoisomerase II in vitro and in Chinese hamster fibrosarcoma cells. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 3025–3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onda T.; Toyoda E.; Miyazaki O.; Seno C.; Kagaya S.; Okamoto K.; Nishikawa K. NK314, a novel topoisomerase II inhibitor, induces rapid DNA double-strand breaks and exhibits superior antitumor effects against tumors resistant to other topoisomerase II inhibitors. Cancer Lett. 2008, 259, 99–110. [DOI] [PubMed] [Google Scholar]
- Umemura K.; Yanase K.; Suzuki M.; Okutani K.; Yamori T.; Andoh T. Inhibition of DNA topoisomerases I and II, and growth inhibition of human cancer cell lines by a marine microalgal polysaccharide. Biochem. Pharmacol. 2003, 66, 481–487. [DOI] [PubMed] [Google Scholar]
- Bittoun P.; Avramoglou T.; Vassy J.; Crepin M.; Chaubet F.; Fermandjian S. Low-molecular-weight dextran derivatives (f-CMDB) enter the nucleus and are better cell-growth inhibitors compared with parent CMDB polymers. Carbohydr. Res. 1999, 322, 247–255. [DOI] [PubMed] [Google Scholar]
- Huang L.; Kerns R. J. Diversity-oriented chemical modification of heparin: Identification of charge-reduced N-acyl heparin derivatives having increased selectivity for heparin-binding proteins. Bioorg. Med. Chem. 2006, 14, 2300–2313. [DOI] [PubMed] [Google Scholar]
- Huang L.; Fernandez C.; Kerns R. J. Different protein-binding selectivities for N-acyl heparin derivatives having N-phenylacetyl and heterocycle analogs of N-phenylacetyl substituted in place of N-sulfo groups. Bioorg. Med. Chem. Lett. 2007, 17, 419–423. [DOI] [PubMed] [Google Scholar]
- Fernandez C.; Hattan C. M.; Kerns R. J. Semi-synthetic heparin derivatives: chemical modifications of heparin beyond chain length, sulfate substitution pattern and N-sulfo/N-acetyl groups. Carbohydr. Res. 2006, 341, 1253–1265. [DOI] [PubMed] [Google Scholar]
- Champoux J. J. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [DOI] [PubMed] [Google Scholar]
- Kumar V.; Remers W. A. Aminoglycoside antibiotics. 4. Regiospecific partial synthesis of ribostamycin and 4″-thioribostamycin. J. Org. Chem. 1981, 46, 4298–4300. [Google Scholar]
- Chen G.; Pan P.; Yao Y.; Chen Y.; Meng X.; Li Z. Regioselective modification of amino groups in aminoglycosides based on cyclic carbamate formation. Tetrahedron 2008, 64, 9078–9087. [Google Scholar]
- Igarashi K.; Honma T.. Aprosamine derivatives. Belgian Patent 890.873, Feb 15, 1982.
- Fenner A. M.; Kerns R. J. Synthesis, separation, and characterization of amphiphilic sulfated oligosaccharides enabled by reversed-phase ion pairing LC and LC–MS methods. Carbohydr. Res. 2011, 346, 2792–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii K.; Futaki S.; Uchiyama H.; Nagasawa K.; Andoh T. Mechanism of inhibition of mammalian DNA topoisomerase I by heparin. Biochem. J. 1987, 241, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece R. J.; Maxwell A. DNA gyrase: structure and function. Crit. Rev. Biochem. Mol. Biol. 1991, 26, 335–375. [DOI] [PubMed] [Google Scholar]
- Froelich-Ammon S. J.; Osheroff N. Topoisomerase poisons: harnessing the dark side of enzyme mechanism. J. Biol. Chem. 1995, 270, 21429–21432. [DOI] [PubMed] [Google Scholar]
- Hiasa H. The Glu-84 of the ParC subunit plays critical roles in both topoisomerase IV-quinolone and topoisomerase IV-DNA interactions. Biochemistry 2002, 41, 11779–11785. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.