

Abstract
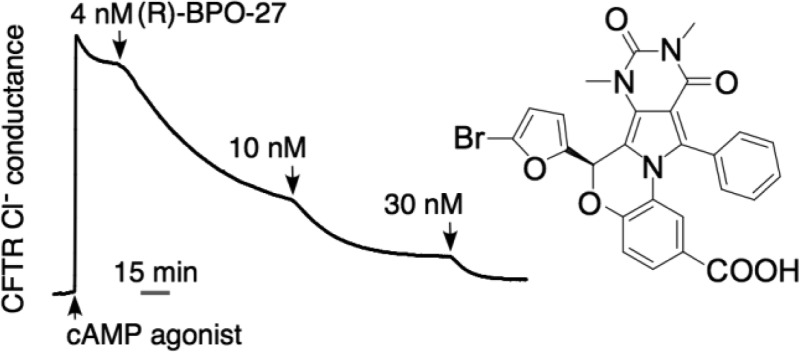
We previously reported benzopyrimido-pyrrolo-oxazinedione (BPO) inhibitors of the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel and showed their efficacy in a model of polycystic kidney disease. Here, we separated the enantiomers of lead compound BPO-27 (1), which contains a single chiral center, and determined their absolute configuration, activity, and metabolic stability. Following separation by chiral supercritical fluid chromatography, the R enantiomer, as determined by X-ray crystallography, inhibited CFTR chloride conductance with IC50 ≈ 4 nM, while S enantiomer was inactive. In vitro metabolic stability in hepatic microsomes showed both enantiomers as stable, with <5% metabolism in 4 h. Following bolus interperitoneal administration in mice, serum (R)-1 decayed with t1/2 ≈ 1.6 h and gave sustained therapeutic concentrations in kidney.
Keywords: Cystic fibrosis, polycystic kidney disease, secretory diarrhea, crystallography
The cystic fibrosis transmembrane conductance regulator (CFTR) protein is a cAMP-regulated chloride channel expressed in secretory and absorptive epithelia.1 Loss of function mutations in CFTR cause the genetic disease cystic fibrosis (CF). Excess activation of CFTR in intestinal enterocytes occurs in secretory diarrheas that are caused by bacterial enterotoxins, such as cholera.2,3 CFTR is also involved in fluid secretion into cysts in autosomal dominant polycystic kidney disease (ADPKD).4,5 CFTR is thus an important drug target, with CFTR activators (potentiators and correctors) for cystic fibrosis therapy and CFTR inhibitors for therapy of secretory diarrheas and ADPKD,6 and for CF research.
We previously identified several classes of small-molecule CFTR inhibitors, including thiazolidinones (e.g., CFTRinh-172),5,7−9 glycine hydrazides (e.g., GlyH-101),10−14 and pyrimido-pyrrolo-quinoxalinediones (e.g., PPQ-102),15 and showed their efficacy in models of polycystic kidney disease and cholera toxin-induced secretory diarrhea. We recently reported an analogue of PPQ-102, the benzopyrimido-pyrrolo-oxazinedione BPO-27 (1) (Scheme 1),16 which inhibited CFTR with IC50 ≈ 8 nM and had greatly improved in vitro metabolic stability and aqueous solubility compared to PPQ-102.15 Compound 1, which was synthesized and tested as a racemic mixture (R/S 50:50), prevented and reversed renal cyst formation in an embryonic kidney culture model of ADPKD.16
Scheme 1. Transformations of BPO-27.

Reagents and conditions: (a) RegisCell 4.6 × 100 mm chiral column, 75% CO2, 25% EtOH, 0.1% 2-propylamine, 4 mL/min, 100 bar, 25 °C, yield 73%; (b) (i) HCl, H2O, EtOAc 3× extraction 89% yield; (c) (i) CH2Cl2, EDC, DMAP, EtOH, 24 h, (ii) aq. HCl H2O, 2× extraction, (iii) SiO2 flash column (2:3 EtOAc/hexane), yield 79%.
We separated 1 into its enantiomers with ≥98.6% enantiomeric excess (e.e.), determined their absolute configuration by X-ray crystallography, and measured their CFTR inhibition activity, metabolic stability, and in vivo pharmacology in mice. A single enantiomer of 1 strongly inhibited CFTR chloride conductance with IC50 ≈ 4 nM, while the other enantiomer was inactive.
Separation of ∼1.0 g racemic (±)-1 was carried out utilizing chiral supercritical fluid chromatography (SFC) on a RegisCell 3.0 × 25.0 cm column using a combination of CO2 and ethanol containing 1% 2-propylamine. Two distinct peaks were detected at 230 nm following elution (Figure 1A). Fraction 1 contained 413 mg with 99.5% e.e. (Figure 1B), and fraction 2 contained 396 mg with 98.6% e.e. (Figure 1C). As a consequence of the separation process, the isolated material was not the acid 1, but the 2-proplyamine carboxylic salt 2. Optical rotation measurements revealed fraction 1 to be (+)-2 and fraction 2 to be (−)-2. When dissolved in aqueous buffer under physiological conditions, both 2 and 1 convert to the same carboxylate salt form.
Figure 1.
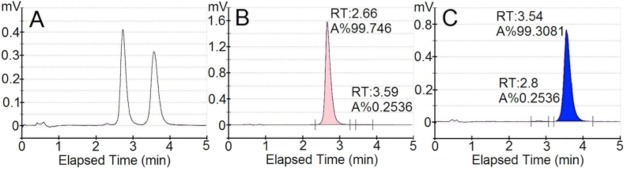
Chromatograms of purified BPO-27 enantiomers following chiral HPLC separation. (A) Analytical chromatogram following preparative separation of ∼1 g (±)-1. (B) Chromatogram of fraction 1. (C) Chromatograph of fraction 2, showing retention time (RT) and % area (A%).
CFTR inhibition potency was measured by short-circuit current analysis in FRT epithelial cells expressing human CFTR in the presence of a transepithelial chloride gradient and in which the basolateral membrane was permeabilized with amphotericin B. Under these conditions, short-circuit current is proportional to CFTR chloride conductance. Figure 2A shows no significant inhibition by (−)-2 at 100 nM, whereas (+)-2 at 100 nM completely inhibited current. Figure 2B shows the (+)-2 concentration-dependence, giving an IC50 ≈ 4 nM, as compared to ∼8 nM for (±)-1 as reported previously.16
Figure 2.
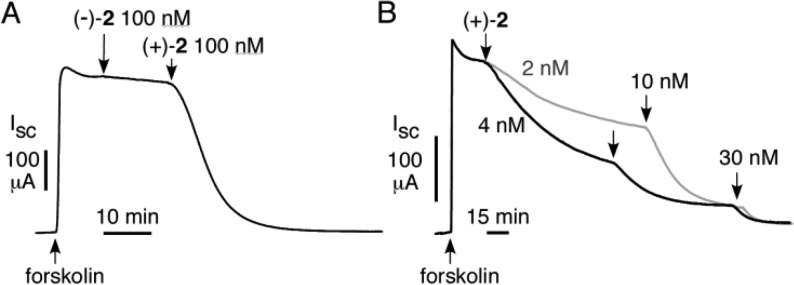
CFTR inhibition by enantiopure (+)-2 and (−)-2. Short-circuit current was measured in FRT cells expressing human wild-type CFTR in the presence of a transepithelial chloride gradient and following permeabilization of the basolateral membrane. CFTR chloride conductance was activated by 10 μM forskolin. (A) (−)-2 and then (+)-2 (each 100 nM) were added where indicated. (B) (+)-2 added at indicated concentrations, deduced IC50 ≈ 4 nM.
The absolute configuration of the inactive enantiomer was determined by X-ray crystallography. Attempts to crystallize (−)-2 failed to yield X-ray quality crystals, as did the corresponding carboxylic acid 1, which was isolated by aqueous acidification and organic extraction (Scheme 1, step b). We found that the ethyl ester 3 dissolved in multiple solvents and readily formed large crystals. Chiral ester 3 was thus prepared from inactive (−)-2 (Scheme 1).
X-ray quality crystals of 3 were obtained by vapor diffusion crystallization in toluene and hexane. X-ray analysis revealed the absolute structure to be (S)-3 (Figure 3); therefore, the active enantiomer has the R configuration. Bioassay of the remaining (S)-3 crystals confirmed no CFTR inhibition activity at 100 nM, whereas racemic 3 strongly inhibited CFTR.16
Figure 3.
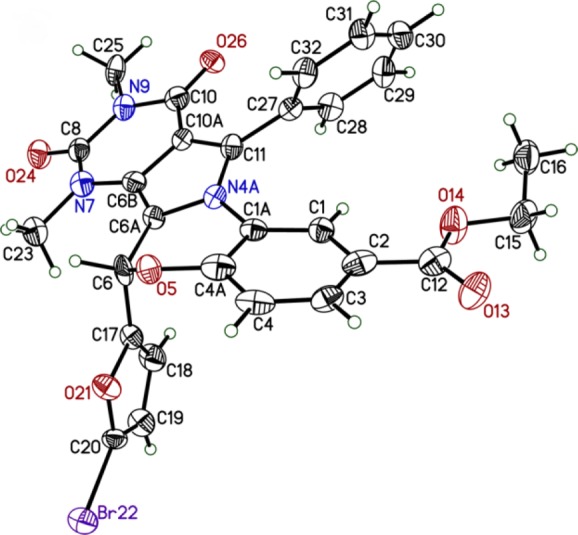
ORTEP representation of the crystal structure of (S)-3.
Although enantiopure (−)-2 was exposed to both acid and base conditions during its purification and conversion to (S)-3, racemization did not occur to any significant extent, as it would not have been possible to generate chiral crystals from racemized 3. The absence of activity of the prepared (S)-3 also argues against racemization, as racemic 3 would strongly inhibit CFTR at 100 nM. The chiral integrity of (−)-2 was further confirmed by a hydrogen–deuterium exchange experiment. (−)-2 was incubated under relevant physiological conditions in the presence of deuterium oxide. No reduction was found in the integrated signal of the singlet peak corresponding to the proton on the chiral carbon (see Supporting Information).
The in vitro metabolic stability of (R)-1 and (S)-1 was determined using rat hepatic microsomes. Following compound incubation with hepatic microsomes in the presence of NADPH, the nonmetabolized compound was quantified by LC/MS. Figure 4A shows little metabolism of either enantiomer.
Figure 4.

Metabolic stability of compound 1. (A) In vitro metabolic stability of 1 measured in rat hepatic microsomes supplemented with NADPH. (left) LC/MS chromatograms of nonmetabolized enatiomers of 1 at indicated times. (right) Percentage nonmetabolized 1 remaining (S.E., n = 4). (B) In vivo pharmacokinetics of (R)-1 in mice following bolus intraperitoneal injection at 10 mg/kg body weight. (left) Reference measurement. LC/MS chromatograms in which known amounts of (R)-1 were added to blood and then extracted. Inset shows assay linearity. (right) LC/MS chromatograms of blood at indicated times after bolus intraperitoneal injection. (R)-1 ion chromatograms (m/z = 548 [M + H]+) are shown along with summary of serum concentration data (mean ± S.E., n = 3).
The pharmacokinetics of (R)-1 was measured in mice by bolus intraperitoneal administration in an aqueous formulation consisting of 5% DMSO, 2.5% Tween-80, and 2.5% PEG400 in water. The concentration of (R)-1 was measured by LC/MS in the blood and kidney. Figure 4B (left) shows ion current in calibration studies in which specified amounts of (R)-1 were added to mouse blood. The assay was linear with a detection sensitivity of better than 500 nM (R)-1. Figure 4B (right) shows serum (R)-1 concentration following bolus intraperitoneal administration. The t1/2 for the disappearance of the original compound was ∼1.6 h. (R)-1 concentration remained at predicted therapeutic levels (≫IC50 of 4 nM) for many hours following single dose administration. Using a CFTR inhibition bioassay to distinguish between active (R)-1 and inactive (S)-1, we confirmed no measurable interconversion of the enantiomers in serum at 4 h. LC/MS analysis of kidney homogenate at 4 h showed a concentration of 0.6 ± 0.3 μM (3 mice).
In summary, chiral chromatography separated (±)-1 into nearly pure enantiomers, one of which was ∼2-fold more potent at inhibiting the CFTR chloride current than the racemic mixture, while the other enantiomer was inactive. The absolute configuration of the active enantiomer is R as determined by X-ray crystallography. The target of (R)-1 and its analogues is likely CFTR itself, as these compounds inhibit CFTR chloride current in response to different types of agonists, including activators that target CFTR directly.15,16 Definitive determination of the (R)-1 binding site will require mutagenesis, molecular modeling, and/or biochemical studies.
Glossary
Abbreviations
- ADPKD
autosomal dominant polycystic kidney disease
- BPO
benzopyrimido-pyrrolo-oxazinedione
- CFTR
cystic fibrosis transmembrane conductance regulator
- DMAP
dimethylaminopyrdine
- EDC
1-ethyl-3-(3-dimethylamino-propyl)carbodiimide HCl
- EtOAc
ethyl acetate
- EtOH
ethanol
- PKD
polycystic kidney disease
- PPQ
pyrimido-pyrrolo-quinoxaline-dione
- SiO2
silica gel
- SFC
supercritical fluid chromatography
- TLC
thin layer chromatography
Supporting Information Available
Detailed description of chromatography parameters, chemical synthesis, bioassay procedures, crystallographic information, and NMR. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
All authors contributed to writing of this manuscript and approved the final version.
Supported by NIH grants DK72517, DK86125, HL73856, EB00415, DK35124, and EY13574 and a Research Development Program grant from the Cystic Fibrosis Foundation. The dual-source X-ray diffractometer was funded by the National Science Foundation (grant 0840444)
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Riordan J. R. CFTR function and prospects for therapy. Annu. Rev. Biochem. 2008, 77, 701–726. [DOI] [PubMed] [Google Scholar]
- Thiagarajah J. R.; Verkman A. S. CFTR pharmacology and its role in intestinal fluid secretion. Curr. Opin. Pharmacol. 2003, 3, 594–599. [DOI] [PubMed] [Google Scholar]
- Venkatasubramanian J.; Ao M.; Rao M. C. Ion transport in the small intestine. Curr. Opin. Gastroenterol. 2010, 26, 123–128. [DOI] [PubMed] [Google Scholar]
- Torres V. E.; Harris P. C.; Pirson Y. Autosomal dominant polycystic kidney disease. Lancet 2007, 369, 1287–1301. [DOI] [PubMed] [Google Scholar]
- Yang B.; Sonawane N. D.; Zhao D.; Somlo S.; Verkman A. S. Small-molecule CFTR inhibitors slow cyst growth in polycystic kidney disease. J. Am. Soc. Nephrol. 2008, 19, 1300–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkman A. S.; Galietta L. J. Chloride channels as drug targets. Nat. Rev. Drug Discovery 2009, 8, 153–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T.; Thiagarajah J. R.; Yang H.; Sonawane N. D.; Folli C.; Galietta L. J.; Verkman A. S. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J. Clin. Invest. 2002, 110, 1651–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajah J. R.; Song Y.; Haggie P. M.; Verkman A. S. A small molecule CFTR inhibitor produces cystic fibrosis-like submucosal gland fluid secretions in normal airways. FASEB J. 2004, 18, 875–877. [DOI] [PubMed] [Google Scholar]
- Sonawane N. D.; Muanprasat C.; Nagatani R. Jr.; Song Y.; Verkman A. S. In vivo pharmacology and antidiarrheal efficacy of a thiazolidinone CFTR inhibitor in rodents. J. Pharm. Sci. 2005, 94, 134–143. [DOI] [PubMed] [Google Scholar]
- Muanprasat C.; Sonawane N. D.; Salinas D.; Taddei A.; Galietta L. J.; Verkman A. S. Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure–activity analysis, and in vivo efficacy. J. Gen. Physiol. 2004, 124, 125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonawane N. D.; Hu J.; Muanprasat C.; Verkman A. S. Luminally active, nonabsorbable CFTR inhibitors as potential therapy to reduce intestinal fluid loss in cholera. FASEB J. 2006, 20, 130–132. [DOI] [PubMed] [Google Scholar]
- Sonawane N. D.; Zhao D.; Zegarra-Moran O.; Galietta L. J.; Verkman A. S. Lectin conjugates as potent, nonabsorbable CFTR inhibitors for reducing intestinal fluid secretion in cholera. Gastroenterology 2007, 132, 1234–1244. [DOI] [PubMed] [Google Scholar]
- Sonawane N. D.; Verkman A. S. Thiazolidinone CFTR inhibitors with improved water solubility identified by structure–activity analysis. Bioorg. Med. Chem. 2008, 16, 8187–8195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonawane N. D.; Zhao D.; Zegarra-Moran O.; Galietta L. J.; Verkman A. S. Nanomolar CFTR inhibition by pore-occluding divalent polyethylene glycol-malonic acid hydrazides. Chem. Biol. 2008, 15, 718–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tradtrantip L.; Sonawane N. D.; Namkung W.; Verkman A. S. Nanomolar potency pyrimido-pyrrolo-quinoxalinedione CFTR inhibitor reduces cyst size in a polycystic kidney disease model. J. Med. Chem. 2009, 52, 6447–6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder D. S.; Tradtrantip L.; Yao C.; Kurth M. J.; Verkman A. S. Potent, metabolically stable benzopyrimido-pyrrolo-oxazine-dione (BPO) CFTR inhibitors for polycystic kidney disease. J. Med. Chem. 2011, 54, 5468–5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.