

Abstract
Motivation: Protein domains are subunits that can fold and evolve independently. Identification of domain boundary locations is often the first step in protein folding and function annotations. Most of the current methods deduce domain boundaries by sequence-based analysis, which has low accuracy. There is no efficient method for predicting discontinuous domains that consist of segments from separated sequence regions. As template-based methods are most efficient for protein 3D structure modeling, combining multiple threading alignment information should increase the accuracy and reliability of computational domain predictions.
Result: We developed a new protein domain predictor, ThreaDom, which deduces domain boundary locations based on multiple threading alignments. The core of the method development is the derivation of a domain conservation score that combines information from template domain structures and terminal and internal alignment gaps. Tested on 630 non-redundant sequences, without using homologous templates, ThreaDom generates correct single- and multi-domain classifications in 81% of cases, where 78% have the domain linker assigned within ±20 residues. In a second test on 486 proteins with discontinuous domains, ThreaDom achieves an average precision 84% and recall 65% in domain boundary prediction. Finally, ThreaDom was examined on 56 targets from CASP8 and had a domain overlap rate 73, 87 and 85% with the target for Free Modeling, Hard multiple-domain and discontinuous domain proteins, respectively, which are significantly higher than most domain predictors in the CASP8. Similar results were achieved on the targets from the most recently CASP9 and CASP10 experiments.
Availability: http://zhanglab.ccmb.med.umich.edu/ThreaDom/.
Contact: zhng@umich.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Protein molecules are composed of domains that fold, function and evolve autonomously. The definition of protein domains is, however, not absolute. Recent studies have shown that protein domains within the same family or superfamily can vary significantly in both structure and function (Dessailly et al., 2010; Reeves et al., 2006). Nevertheless, correct assignment of boundaries of the protein domains is essential for the efficient elucidations of protein function and evolutionary mechanisms.
The most accurate characterization of protein domains is through the analysis of the 3D structures. However, the experimental determination of protein structures is often time and manpower expensive and some proteins are even impossible to solve currently. The computational domain prediction from the amino acid sequence is, therefore, highly demanded. A variety of methods have been recently developed in this regard that can be roughly categorized into three groups: statistical and machine-learning based, homology-based and 3D model-based methods.
The statistical and machine-learning-based methods are probably the most frequently used approaches to protein domain predictions, with examples including DGS (Wheelan et al., 2000), DomCut (Suyama and Ohara, 2003), Armadillo (Dumontier et al., 2005), PPRODO (Sim et al., 2005), DOMPro (Cheng et al., 2006), DomNet (Yoo et al., 2008), DROP (Ebina et al., 2011), DOBO (Eickholt et al., 2011), PRODOM (Servant et al., 2002), ADDA (Heger et al., 2005) and EVEREST (Portugaly et al., 2006). In the DGS, DomCut and Armadillo programs, the statistical regularities seen in the Protein Data Bank (PDB) structures, including domain size distribution and residue propensities, are used to deduce the domain linker and boundary predictions. In PRODOM, ADDA and EVEREST, the domain boundaries are derived by large-scale sequence comparisons followed by clustering analyses. In the rest examples (PPRODO, DOMPro, DomNet, DROP and DOBO), the residue-based statistical features, together with the position-specific scoring matrix from PSI-BLAST search, are trained by machine-learning techniques, including neural network, support vector machine and random forest classifiers. These methods deduce boundary information from sequence only, which can in principle be applied to any proteins. But the overall accuracy is low compared with the homology-based approaches.
In the homology-based methods, e.g. Pfam (Finn et al., 2010), CHOP (Liu and Rost, 2004) and FIEFDOM (Bondugula et al., 2009), target sequences are searched through known protein structure or family libraries by hidden Markov model (HMM) or PSI-BLAST programs. The domain boundary information is then obtained by mapping the domain information from the homologous templates or families following the sequence alignments. The homology-based methods can have a high accuracy of predictions when close templates are identified, but the accuracy decreases sharply when the sequence identity of target and template is low (e.g. <30%).
In the 3D model-based methods, e.g. SnapDRAGON (George and Heringa, 2002), RosettaDom (Kim et al., 2005) and OPUS-DOM (Wu et al., 2009), the authors first construct tertiary structure models of the target by either ab initio folding or knowledge-based coarse-grained modeling simulations. Domain parser tools are then used to assign the domain information on the predicted 3D models. The accuracy of domain assignments relies on the quality of the tertiary models, which usually decreases with the size of the target proteins because of the limited ability of ab initio folding simulations (Zhang, 2008).
Having in mind the improved power of the template-based protein structure predictions and the increasing size of PDB, we propose a new algorithm ThreaDom based on multiple threading algorithms, which aims to significantly improve the reliability of domain predictions in the category of distantly homologous protein targets. Although the threading-based algorithms have been successfully used in the CASP experiments for modeling multiple-domain protein structures where the domain boundaries are usually decided by human-intervened views and interpretations of multiple-threading alignment profiles (Zhang, 2007, 2009), this is the first time to integrate the multiple threading algorithms into an automated pipeline for domain boundary determinations. The key to the algorithm is the development of a sensitive domain boundary profile that can calibrate composite structural and sequence alignment information from the multiple threading templates for precise domain assignment. The method will be systematically benchmarked on large-scale proteins, to examine the weaknesses and strengths in comparison with other widely used domain prediction approaches.
2 METHODS
2.1 Data sets
We collect a non-redundant protein set from PISCES (Wang and Dunbrack, 2003), with the sequence identity cut-off at 25%, resolution <3.0 Å and R-factor <1.0. The domain definitions of these proteins are taken from CATH 3.4 (Orengo et al., 1997). If the template protein does not exist in the CATH, the domain structure is defined by DomainParser (Xu et al., 2000).
All proteins with a chain length <80 residues or domain length <40 residues are removed, which results in a protein set containing 715 multi-domain and 2524 single-domain chains. The 715 multi-domain proteins are divided into training and testing sets, including 400 and 315 chains, respectively. The 2524 single-domain chains are randomly divided into two sets, paired with multi-domain proteins in the training and testing sets.
Based on the significance of threading alignments, the protein chains are categorized into ‘Easy’, ‘Medium’ and ‘Hard’ proteins (see later in the text). Thus, our training set includes 331 × 2 ‘Easy’, 46 × 2 ‘Medium’ and 23 × 2 ‘Hard’ protein sequences, and the testing set contains 261 × 2 ‘Easy’, 36 × 2 ‘Medium’ and 18 × 2 ‘Hard’ non-redundant sequences, where ‘×2’ refers to two sets of single and multiple-domain proteins.
2.2 Multiple template identification by LOMETS
In ThreaDom, LOMETS (Wu and Zhang, 2007) will be used to thread the target sequences through the PDB for structural template identifications. LOMETS contains eight threading programs of complementary approaches, including HHSEARCH (Söding, 2005), MUSTER (Wu and Zhang, 2008), PROSPECT2 (Xu and Xu, 2000), PPA-I (Wu et al., 2007), SAM-T02 (Karplus et al., 1998), SPARKS2 and SP3 (Zhou and Zhou, 2005). In HHSEARCH, we implemented two versions of global and local HMMs, HHSEARCH-1 and HHSEARCH-2. These eight threading programs are displayed in Figure 1 as the LOMETS-based threading server layer.
Fig. 1.

Architecture of ThreaDom. It consists of a library layer, a meta-server threading layer and a domain decision layer from the bottom. Eight threading programs in the meta-server layer access three structure libraries in the library layer to provide alignments for the domain decision layer. Threading Library Map-Adapter calls libraries in the library layer and provides a unified order map. In the decision layer, data flow from A to F
Three independent template libraries are used. First, MUSTER, PPI-1, SAM-T02 and PROSPECT2 use an internal I-TASSER template library with sequence identity <70% from http://zhanglab.ccmb.med.umich.edu/library/; SPARKS2 and SP3 use another internal library of sequence identity <40%; HHSEARCH uses the library downloaded from ftp://ftp.tuebingen.mpg.de/pub/protevo/HHsearch/databases, which has also a 70% sequence identity cut-off. The domain boundaries for all templates are pre-calculated based on CATH3.4 or DomainParser. As residues in the template structures were re-ordered in threading libraries, a map-adapter is established to map all the template libraries into the original entries in the PDB library so that CATH domain definitions can be exploited.
For each LOMETS program, a Z-score cut-off (Z0) is assigned, based on the threading results data of 1190 independent training proteins, so that the well-defined alignments with an average TM-score >0.6 can be achieved when Z-score > Z0. Here, Z-score is a measure of the significance of the target-template alignment, defined as the score difference to the mean in the unit of standard deviation. A protein target is categorized as ‘Easy’ if each of the LOMETS threading programs have at least one template with Z-score > Z0; it is a ‘Hard’ target if there is no template hit with Z-score > Z0 by any programs. Otherwise, it is assigned as a ‘Medium’ target.
2.3 Outline of ThreaDom procedure
Domain predictions in ThreaDom are based on two assumptions: (i) homologous proteins have similar domain structures; (ii) residues in the core regions of domain structures are evolutionally more conserved than that in the boundary (or linker) regions between domains. Following the assumptions, the ThreaDom procedure contains three steps as displayed in Figure 1.
Target sequences are threaded through the PDB by eight LOMETS programs, and a multiple sequence alignment is constructed based on the target sequence (with external inserts/gaps shaved).
A domain conservation score (DCS) is calculated for each residue position based on the LOMETS multiple sequence alignments, which counts for the balance of conservation and gap penalty scores.
Domain boundaries are assigned based on the DCS profile using a target-specific scoring cut-off.
2.4 Domain conservation score
In ThreaDom, the domains of the target sequence are specified by the location and size of linker regions between two domains (e.g. A and B):
| (1) |
where i is the residue number, start(L) is the residue position of the last residue in domain A along the sequence, end (L) is the position of the first residue in domain B and Start(L)<end (L). In ThreaDom, we consider two contributions of template domain boundary structure and target-template alignment gaps, which are used to decide the location and size of the domain linkers of the target sequences.
2.4.1 Template domain linker score
ThreaDom considers T template alignments obtained by LOMETS, where Tgood is the number of templates with a Z-score ≥ Z0, and Tbad is that with a Z-score < Z0. Following CATH (or DomainParser) domain definition, the jth template has a domain split specified by the linker structure 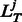 :
:
| (2) |
where 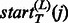 and
and 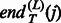 are the starting and ending positions of the linkers on jth template. Considering the alignment error that may result in linker shift, we introduce a distance allowance d to increase the size of template linkers.
are the starting and ending positions of the linkers on jth template. Considering the alignment error that may result in linker shift, we introduce a distance allowance d to increase the size of template linkers.
For residue i, the template domain linker score from the T template alignments is calculated by
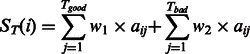 |
(3) |
where w1 and w2 are the weight parameters on ‘good’ or ‘bad’ templates. aij counts for whether the ith residue on the target is aligned with the linker regions of jth template, i.e.
| (4) |
Here, m counts for the confidence of domain assignment of template structures. In our benchmark test, there is an agreement between DomainParser and CATH for ∼80% proteins. We set m = 0.8 if the template domain is assigned by DomainParser, and m = 1.0 if by CATH, as the latter is assisted by human intervention and of a higher accuracy in domain assignment.
2.4.2 Gap penalty score
ThreaDom specifies two types of gap penalties from threading alignments: terminal gap Gterm and internal gap Gint. The terminal gaps on jth template are defined by
| (5) |
where jN and jC are the N- and C-terminal positions of the first and last aligned residues on jth template. The internal gap is defined by
| (6) |
where 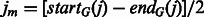 denotes the middle point of the internal gaps, and startG(j) and endG(j) are the starting and ending locations of the gaps. To rule out alignment noise, we only consider the gaps with a size longer than l, i.e.
denotes the middle point of the internal gaps, and startG(j) and endG(j) are the starting and ending locations of the gaps. To rule out alignment noise, we only consider the gaps with a size longer than l, i.e. 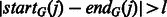 .
.
The total gap penalty score for the residue i is calculated by:
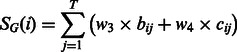 |
(7) |
where w3 and w4 are the weight parameters; bij and cij are the binary values representing whether the ith residue locates in the gap regions of the jth template alignment:
| (8) |
2.4.3 Domain conservation score
The template domain linker score and gap penalty score indicate the degree of variations of multiple threading alignments at the position i. Accordingly, the domain conservation score, S(i), is calculated by
| (9) |
where 1 < i < N, N is the length of the target sequence.
To reduce noise in the DCS assignment that may result in artificial domains with very short length, we smooth the domain conservation score using a 19-residue window:
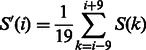 |
(10) |
Meanwhile, S(k) is set to 0 if S(k) <0. Thus, the smoothed domain conservation score S’(i) has a value in (0,1).
2.4.4 Deciding domain linkers by DCS profiles
A putative domain linker L(k) in ThreaDom is an aggregation of the continuous residues that have the conservation score below a certain cut-off Sc, i.e.
| (11) |
where 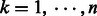 represents the number of linkers. The middle point of the linker,
represents the number of linkers. The middle point of the linker, 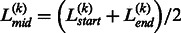 , is noted as the predicted boundary to the linker L(k) in the ThreaDom program.
, is noted as the predicted boundary to the linker L(k) in the ThreaDom program.
As the majority of protein domains in the PDB have a length longer than 40 residues, we consider two length-based domain filters. First, if the distance from 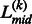 to the terminal of sequence is <40, the L(k) is removed from the putative linker list (i.e. set L(k) = 0). Second, if the distance between two neighboring linkers is too close, i.e.
to the terminal of sequence is <40, the L(k) is removed from the putative linker list (i.e. set L(k) = 0). Second, if the distance between two neighboring linkers is too close, i.e. 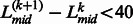 , the linkers will be merged into one linker. The boundary position of the merged linkers is calculated by
, the linkers will be merged into one linker. The boundary position of the merged linkers is calculated by
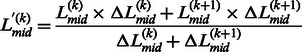 |
(12) |
where 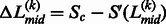 is associated with the confidence of the linker assignment on
is associated with the confidence of the linker assignment on 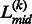 . Based on Equation 12, if the linkers have similar confidence, i.e.
. Based on Equation 12, if the linkers have similar confidence, i.e. 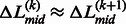 , the boundary position of the merged linker is at the middle of the two linkers,
, the boundary position of the merged linker is at the middle of the two linkers, 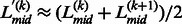 . Otherwise, the boundary of the merged linkers will be biased to the position of the linker with a higher confidence score.
. Otherwise, the boundary of the merged linkers will be biased to the position of the linker with a higher confidence score.
The final continuous domain assignment in the ThreaDom is represented in the form of 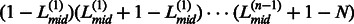 , and the residue range in each pair of parenthesis represents an individual domain. One example of the ThreaDom protein domain prediction is given in Figure 2, where four individual domains are separated by three linkers L(1), L(2) and L(3), with the cutoff Sc = 0.60.
, and the residue range in each pair of parenthesis represents an individual domain. One example of the ThreaDom protein domain prediction is given in Figure 2, where four individual domains are separated by three linkers L(1), L(2) and L(3), with the cutoff Sc = 0.60.
Fig. 2.

An illustration of the domain decision in ThreaDom based on the domain conservation score profile. Four individual domains are separated by three linkers defined by the valleys of the DCS distribution. The vertical dotted lines indicate the start and end locations of each putative linker. The vertical solid lines denote the predicted boundary at the middle of the linkers (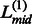 ,
, 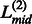 and
and 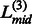 )
)
Equations 1–12 have eight free parameters (w1, w2, w3, w4, d, l, T and Sc), which will be trained on our training proteins of various classes (see later in the text).
2.5 Strategy for detecting discontinuous domains
The term discontinuous domain refers to a domain that contains two or more segments from separated regions of target sequence. ThreaDom detects discontinuous domains based on the DCS profile and the pre-defined domain boundaries of the threading templates, which contains three steps:
Step I: Detecting discontinuous domain sequence. A target is considered to have discontinuous domains if it has >30% templates that have discontinuous domain structure in the LOMETS template collection.
Step II: Clustering the discontinuous domain templates. The discontinuous domain templates are clustered based on their domain boundary locations and domain assignments. The discontinuous templates are clustered into one category if they have the same number of domains with same domain segments number and similar boundaries, where ‘similar boundary’ means that the difference in boundary positions is within ±5 residues after structure alignment of the two templates.
Step III: Boundary refinement and discontinuous domain substitution. After clustering, the DCS-based domain prediction and the domain structure in the first template cluster are combined, i.e. if the domain boundary difference between the DCS prediction and the first template cluster is within ±20 residues, the separated domains in the DCS prediction will be merged into a single domain following the assignment in the first template cluster. Meanwhile, if the number of domains in the DCS prediction is >3 but less than that in the first cluster, we substitute the DCS prediction with the domain information of the first cluster when the domain boundaries in >50% of templates are consistent (i.e. differences are ±20 residues).
2.6 Evaluation criteria
We evaluate the ability of ThreaDom on both the domain number and the domain boundary predictions. The domain number prediction is assessed by counting the accuracy of single- or multi-domain protein classifications. We use specificity, sensitivity and Matthew’s correlation coefficient (MCC) to assess the domain number prediction:
| (13) |
| (14) |
| (15) |
where TP, FP, TN and FN are true positive, false positive, true negative and false negative rates of the classifications, respectively.
To assess the quality of domain boundary predictions, we calculate precision and recall rates, the normalized domain overlap (NDO)-score (Tai et al., 2005) and the domain boundary distance (DBD) score (Tress et al., 2007). The precision has a similar definition to specificity as defined in Equation 13, but boundary prediction is designated as ‘TP’ if it is within ±20 residues of the true boundary; otherwise it is an ‘FP’ prediction. Recall is similar to the sensitivity in Equation 14, which represents the fraction of the target boundaries that are correctly retrieved in the domain predictions. The NDO-score is defined as the normalized overlap rate of all predicted domain and linker regions with the true assignments of the target structure. The DBD-score measures the distance of the predicted boundaries from the true target domain boundaries.
To avoid the contamination of homologous templates that are easy to predict in ThreaDom, we exclude all the templates that have a sequence identity >30% to the target protein or that are detectable by PSI-BLAST with an E-value <0.05. As a control, we implemented five publicly available domain predictors, including FIEFDom (Bondugula et al., 2009), Pfam (Finn et al., 2010), DomPro (Cheng et al., 2006), DROP (Ebina et al., 2011) and PPRODO (Sim et al., 2005), which represent different type of homology- and machine-learning-based methods. These methods are run on the same test set of proteins.
2.7 Parameter training
There are eight free parameters in the ThreaDom scoring Equations (2–11), including four weight parameters (w1, w2, w3 and w4), the linker shift d, the length cut-off of internal gaps l, the number of threading templates T and the DCS cut-off Sc. We trained the parameters by maximizing the precision, recall and NDO scores on the 800 training proteins (400 single-domain + 400 multi-domain proteins, see Section 2.1). The parameters are tuned separately for Easy and Medium/Hard proteins. To increase the efficiency, we projected the parameter values on an 8D system and enumerate various values on the lattices. Parameter values corresponding to the optimal results were selected with results summarized in Table 1. For the 331 × 2 ‘Easy’ targets in our training set, the optimal NDO, precision and recall scores are 0.919, 0.836 and 0.77, respectively, and those for the 69 × 2 ‘Medium/Hard’ targets are 0.821, 0.476 and 0.32, respectively.
Table 1.
The optimized parameters in ThreaDom
| Parameters | Easy | Medium/ Hard |
|---|---|---|
| Weight of good templates (w1) | 2.0 | 2.0 |
| Weight of bad templates (w2) | 0.6 | 0.5 |
| Weight of terminal gaps (w3) | 0.8 | 1.4 |
| Weight of internal gaps (w4) | 0.1 | 0.5 |
| Shift of linkers or gaps (d) | 12 | 10 |
| Minimum length of internal gaps (l) | 15 | 15 |
| Number of used templates (T) | 50 | 50 |
| DCS cut-off (Sc) | 0.6 | 0.76 |
3 RESULTS AND DISCUSSION
3.1 Domain classification prediction
A sequence is considered to be a multi-domain protein if it includes one or more domain linkers. In the test on the 315 × 2 non-homologous proteins that are also non-homologous to the training protein set, ThreaDom correctly classifies proteins as being either single- or multi-domain proteins in 81% of the cases. For the ‘Easy’ protein set, the accuracy is 84.7%, and for ‘Medium/Hard’ test set, the accuracy is 68.5%.
Table 2 shows a summary of ThreaDom performance in control with the other five methods on the single-domain or multi-domain classification. For all the three categories of ‘All’, ‘Easy’ and ‘Medium/Hard’, ThreaDom produces the highest MCC among the five predictors. The MCC values are 54, 60 and 41%, respectively, higher than that by the second best predictor FIEFDom, which is a homologous method-based on PSI-BLAST search. Pfam, a standard HMM-based domain assignment program, has a slightly lower average MCC than FIEFDom.
Table 2.
Single- or multi-domain classifications on CATH domains
| Type | Predictor | MCC | Single-domain | Multi-domain | ||
|---|---|---|---|---|---|---|
| Spec. | Sens. | Spec. | Sens. | |||
| All | ThreaDom | 0.682 | 0.800 | 0.902 | 0.887 | 0.775 |
| FIEFDom | 0.443 | 0.724 | 0.683 | 0.700 | 0.740 | |
| Pfam | 0.378 | 0.645 | 0.813 | 0.747 | 0.552 | |
| DROP | −0.019 | 0.491 | 0.517 | 0.490 | 0.463 | |
| DomPro | 0.287 | 0.571 | 0.917 | 0.790 | 0.311 | |
| PPRODO | 0.076 | 0.800 | 0.025 | 0.505 | 0.994 | |
| Easy | ThreaDom | 0.734 | 0.837 | 0.908 | 0.900 | 0.824 |
| FIEFDom | 0.458 | 0.768 | 0.648 | 0.695 | 0.805 | |
| Pfam | 0.420 | 0.676 | 0.793 | 0.750 | 0.621 | |
| DROP | −0.019 | 0.490 | 0.479 | 0.491 | 0.502 | |
| DomPro | 0.304 | 0.579 | 0.912 | 0.793 | 0.337 | |
| PPRODO | 0.076 | 1.000 | 0.011 | 0.503 | 1.000 | |
| Medium/Hard | ThreaDom | 0.432 | 0.653 | 0.870 | 0.806 | 0.537 |
| FIEFDom | 0.307 | 0.597 | 0.852 | 0.742 | 0.426 | |
| Pfam | 0.178 | 0.538 | 0.907 | 0.706 | 0.222 | |
| DROP | −0.020 | 0.494 | 0.704 | 0.484 | 0.278 | |
| DomPro | 0.199 | 0.537 | 0.944 | 0.769 | 0.185 | |
| PPRODO | 0.113 | 0.714 | 0.093 | 0.515 | 0.963 | |
Note: Bold values denote the best performance in each category.
MCC, Matthew’s correlation coefficient; Spec., specificity; Sens., sensitivity.
Interestingly, the two machine-learning-based methods, DomPro and PPRODO, have top specificity or sensitive values in some categories, but they have a low MCC because of unbalanced classifications. For example, in the ‘Easy’ set, PPRODO has a sensitivity of 100% for multi-domain classification but only 1.1% for single-domain, whereas the corresponding specificity values were 50.3 and 100%, respectively. These data imply that PPRODO classifies almost all sequences as multi-domain protein, which, therefore, leads to a low MCC value of 0.076. Similarly, DomPro tends to classify most chains as being single-domain chains that also results in a modest MCC value, although it has a better balance than PPRODO. In other words, DomPro is an underpredicting method for multiple-domain, whereas PPRODO is overpredicting. DROP, another machine-learning method, has a negative MCC in all three categories of targets because of the low assignment accuracy.
3.2 Domain boundary prediction
For the proteins in the ‘All’, ‘Easy’ and ‘Medium/Hard’ sets, the domain boundary predictions by ThreaDom has the NDO-scores of 0.893, 0.905 and 0.832, DBD-scores of 0.861, 0.887 and 0.737, precisions of 0.784, 0.814 and 0.562 and recalls of 0.670, 0.708 and 0.425, respectively. Figure 3 presents the ThreaDom prediction results together with that by other five control methods, where the y-axis is the value of NDO, DBD, boundary precision and recall scores, and x-axis denotes the categories of protein sets in ‘All’, ‘Easy’ or ‘Medium/Hard’. For proteins in all the three categories, ThreaDom achieves the highest value in NDO-score, DBD-score and boundary precision. ThreaDom also has the highest score in boundary recall for ‘All’ and ‘Easy’ categories, and the second highest score in boundary recall for the ‘Medium/Hard’ proteins.
Fig. 3.

Summary of domain boundary predictions by ThreaDom and the control predictors. (A) Normalized Domain Overlap score; (B) Domain Boundary Distance Score; (C) Precision of predicted boundaries; (D) Recall of pre-defined boundaries
As shown in Figure 3D, PPRODO has a slightly higher recall value than ThreaDom for the ‘Medium/Hard’ targets. This is partly because of the overprediction of PPRODO that predicts most of the single-domain proteins as multi-domain and has, therefore, on average more boundary linkers assigned. This leads to a worse precision value (26.7%) in comparison with that by ThreaDom (56.2%). Because of the unbalanced recall and precision, PPRODO has overall a poor performance when assessed by the NDO- and DBD-scores (Fig. 3A and B).
Different methods have different sensitivities on the category of protein targets. As shown in Figure 3, the predictions by the statistical and machine-learning-based methods (DomPro, DROP and PPRODO) have similar performances in both ‘Easy’ and ‘Medium/Hard’ categories, as these predictions are from sequence only. The HMM-based method, Pfam, also does not show difference between Easy and Hard targets because the domains in Pfam were retrieved from UniProt and ADDA sequence clustering (Heger and Holm, 2003), which are not directly associated with template structures in the PDB library. However, the two template-based methods, ThreaDom and FIEFDom, have an obvious difference between ‘Easy’ and ‘Medium/Hard’ proteins because of the different availability of the template hits in the two category of proteins. Nevertheless, ThreaDom identified much more accurate boundary predictions than FIEFDom in both ‘Easy’ and ‘Medium/Hard’ categories. Particularly in the ‘Medium/Hard’ protein set, the precision and recall are three and five times higher than that in FIEFDom. These improvements are mainly because of (i) the better identification of templates by LOMETS than that by PSI-BLAST or HMM searches and (ii) the sensitive calibration of gaps and alignments by the domain conservation score as designed by ThreaDom. These advantages are essential for ThreaDom to detect efficient domain structures for the weakly- or non-homologous proteins.
In general, for the ‘Easy’ proteins, there are a large number of ‘good’ templates with a high Z-score as detected by various threading programs in ThreaDom. The consensus domain assignments of the template structures dominate the boundary predictions. For the ‘Medium/Hard’ targets, however, there are few consensus ‘good’ templates, and the identification of consensus terminal and internal alignment gaps becomes sensitive. This explains the reason that the weighting parameters of gap penalty score (w3 and w4 in Equation 7) become larger for the ‘Medium/Hard’ targets than that for the ‘Easy’ targets (Table 1). In Supplementary Table S1 of Supplementary Materials, we separated the contributions of template alignment and gap penalty scores in ThreaDom. Although the gap penalty score tends to make more important contribution for Hard targets, a combination of template and gap penalty scores outperforms individual scores in all categories of targets. Thus, using a balanced consensus of template domain assignment and the internal and terminal gaps from multiple template alignments, the DCS system helps erase the incident errors from single template alignment by individual threading programs that are often less reliable.
In Figure 4, we show an illustrative example from the GTP cyclohydrolase I (PDB ID: 1wurA), which is a hard target for which none of the LOMETS threading program has a strong template alignment with Z-score > Z0. The chain is classified as a two-domain protein with boundary structure of (1–55) (56–185) in the CATH database (Fig. 4E). In the control programs, both FIEFDom and DROP incorrectly predicted the protein as a single-domain chain, whereas PPRODO and DomPro correctly assigned it as a multi-domain protein, but the assigned domain boundary is 35 and 45 residues away from the CATH assignment, respectively.
Fig. 4.

An illustrative example of ThreaDom prediction on ‘Hard’ target from the GTP cyclohydrolase I (PDB ID: 1wurA). (A) DCS score distribution. (B–D) Counts of templates with terminal gap, internal gap and template domain assignment along the sequence in the total 50 selected templates. (E) X-ray structure of the target protein with CATH and ThreaDom domain boundaries labeled
As shown in Figure 4D, the alignments among the 50 selected threading templates are divergent and not conclusive: four templates at residue 46, two at residues 65, 69 and 102 and others with domains almost evenly distributed along the sequence. Similarly in Figure 4B and C, the internal and terminal alignment gaps are nearly even-distributed. However, when we combine the contributions from the domain and gap assignments as described in Equations 9 and 10, the overall DCS profile has an obvious valley around residue 60 (Fig. 4A), which is due to the weak but consistent tendency of gap and domain assignments in the multiple threading alignments. Although there are two other valleys in the N- and C-terminals, the locations are <40 residues away from the sequence ends and are ruled out by the default length filter. Finally, the ThreaDom boundary prediction is (1–60) (61–185), which shift by only five residues from the CATH assignment. This example highlights the power of ThreaDom in extracting correct domain information from distantly homologous threading alignments by combining multiple domain and alignment gap/insertion information.
3.3 Domain prediction assessed by alternative domain definitions
One concern of the aforementioned data analyses is on the possible bias of distinctive domain definitions of the training and test proteins, as some methods (e.g. FIEFDom) were trained by domains defined in the SCOP database (Murzin et al., 1995), but the analyses are mainly on CATH definitions, which is what ThreaDom was trained on. In Supplementary Table S2, we present a quantitative analysis of the domain predictions on the 315 test protein pairs with the domains defined by SCOP1.75. Similarly, if a protein cannot be seen in the SCOP library, a definition from DomainParser is used instead. Although some small variations are seen in specific score values, there is no qualitative difference between Supplementary Table S2 and the data shown in Table 1 and Figure 3. These results demonstrate that the distinctive domain definitions of different databases have no impact on the training and testing procedures of domain predictions.
3.4 Discontinuous domain prediction
Domain assignment for the proteins that have domains consisting of segments from separated locations, called discontinuous domains, is a long-standing unsolved problem. Despite the significant importance of discontinuous domains in protein structural determination and function annotations, there is so far no efficient method available for discontinuous domain prediction.
To test the ability of ThreaDom in discontinuous domain prediction, we collect 486 non-homologous multi-domain proteins from CATH 3.4 that include at least one domain with discontinuous segments. These protein domains/segments have >40 residues with a pairwise sequence identity <40%.
Overall, the automated ThreaDom procedure correctly identified 88.9% of the proteins as multi-domain proteins. For the domain boundary prediction, the precision and recall are 83.9 and 64.5%, respectively, which are comparable with that for the continuous domain protein samples (78.4 and 67.0% in precision and recall), although we did not separately train ThreaDom on the discontinuous domain proteins. The success rates of the predictions demonstrate that the segment assembly procedure has efficiently combined the identified domain linkers from separated positions into the discontinuous domains.
To illustrate the procedure, we present in Figure 5 an example of a discontinuous domain protein from the aminopeptidase I in Clostridium acetobutylicum (PDB ID: 2gljE). The domain structure in CATH assignment is (1–105;244–455) (106–243), where the first domain D1 (1–105;244–455) contains two segments S11 (1–105) and S12 (244–455). The second domain D2 is a continuous domain containing one segment S21 from 105 to 243. In Figure 5, the domain boundary residues PRO105 and LYS244 are labeled in blue, ARG106 and GLY243 in magenta. P1 and P2 indicate the positions that split the sequence into the two domains.
Fig. 5.

Illustration of ThreaDom on discontinuous domain prediction for the aminopeptidase I protein (PDB ID: 2gljE). The segments assigned by ThreaDom, s1 (1–103), s2 (104–243) and s3 (244–455), are marked in red, green and yellow, respectively. The separated segments (s1 and s2) are merged into a single domain following the clusters of the discontinuous domain templates. P1 and P2 denote the domain boundary position according to CATH 3.4, and P2 and P3 are that predicted by ThreaDom
As most of the top templates by LOMETS have discontinuous domain structure, ThreaDom classified the target as a discontinuous domain protein. Following the multiple template alignments, the sequence is split into three segments of s1 (1–103), s2 (104–243) and s3 (244–455), which are marked in red, green and yellow, respectively, in Figure 5, where P3 and P2 indicate the splitting positions of the segments. These segments are highly consistent with the first cluster of discontinuous-domain templates that have a domain structure of (27–104;247–454) (105–246). Therefore, the segments of s1 and s3 were merged in a single domain with s2 assigned as the second domain. As a result, there is only a two-residue shift in the domain boundary by the ThreaDom prediction compared with the CATH assignment in this example.
3.5 Benchmark on CASP targets
As publicly available domain predictors are limited, to have a more extensive benchmark with the state-of-the-art methods, we test ThreaDom on the protein targets in CASP8 (Ezkurdia et al., 2009), which is the last community-wide blind experiment on protein domain prediction (DP). The DP section in CASP8 contains seven multi-domain Free Modeling (FM) targets, 29 Multi-Domain Hard (MD-Hard) targets and 20 DisContinuous Domain (DCD) targets. To mimic the CASP procedure, all template proteins, which were solved after the CASP8 experiment, were excluded from the LOMETS threading library when implementing ThreaDom.
In Figure 6, we present the average NDO-score of ThreaDom predictions in the three categories, in control with the seven severs in the CASP8 DP section, which submitted predictions for all the targets (five other servers, which submitted only partial targets, were not shown in the figure). Overall, ThreaDom has an average NDO-score of 0.738, 0.868 and 0.854 for the FM, MD-Hard and DCD targets, respectively. For the entire set of 56 targets, the average NDO-score is 0.847, which is higher than all the predictors in the CASP8 experiment.
Fig. 6.

Average normalized domain overlap (NDO)-score of ThreaDom predictions on the CASP8 domain prediction targets, in comparison with the CASP8 servers that submitted prediction for all 56 DP targets
In Figure 7, we present three typical examples of ThreaDom predictions for the CASP8 targets. First, T0496 and T0397 in Figure 7A and B have the domain boundary defined as (4–123) (124–178) and (1–82) (83–150), respectively, based on the experimental structures. Both targets are FM targets that have no obvious template hit by LOMETS. ThreaDom combined the consensus of template alignments and gap penalty scores, which generated a domain prediction as (1–113) (114–178) and (1–75) (76–150) for the two targets; these correspond to NDO-scores of 0.889 and 0.907, respectively. All the predicted boundaries are within ±10 residues from the native domain definition.
Fig. 7.

Illustrative examples of domain prediction by ThreaDom on CASP8 targets: (A) FM target T0496; (B) FM target T0397; (C) DCD target T0490. The domain boundary from the native structures is labeled by the cutting pars P1–P6 with the adjacent residues marked in blue and magenta. The domain segments predicted by ThreaDom are marked in different colors. In T0490, the neighboring segments are correctly merged into two individual domains
T0490 in Figure 7C is also an FM target but with DCD structure, (5–87|143–227|319–368) (88–142|228–318). The first step of the DCS scan split the target sequence into five segments, i.e. (1–84) (85–142) (143–223) (224–319) (320–369), shown in red, green, orange, cyan and yellow, respectively. After the template domain structure clustering and boundary refinement, the first, third and fifth segments are merged into the first domain and the rest into the second domain. The final prediction (1–85|143–225|319–368) (86–142|226–318) has a DNO-score of 0.97, which is close to the native structure not only in the domain number and boundary but also in the DCD components (Fig. 7C).
Domain prediction tests were not included in the most recent CASP experiments (CASP9 and 10). The FM/Hard targets in the experiments, however, represent a set of well-defined real-time proteins free of homologous contaminations. In Supplementary Table S3, we list the performance of ThreaDom, in control with FIEFDom, Pfam, DROP, Dompro and PPRODO, on 46 FM/MD-Hard targets with 22 from CASP9 and 24 from CASP10. Similarly, all protein templates solved after the CASP experiments were excluded. To examine the impact of different steps of procedures, we implemented two version of ThreaDom, i.e. ThreaDom1 used the Domain Conservation score without linker refinement and DCD detection procedures, whereas ThreaDom2 is a complete ThreaDom implementation, including both procedures.
As shown in Supplementary Table S3, ThreaDom2 obviously outperforms ThreaDom1 in all criteria of precision, recall, DNO- and DBD-scores, which demonstrates the importance of the refinement procedures. Overall, the two ThreaDom programs are ranked as the top two methods in most of the assessments for the CASP9 and CASP10 targets, except for that the DBD-score, and the boundary recall of the ThreaDom programs are slightly lower than that of a few other methods for the CASP9 targets.
3.6 Drawbacks of ThreaDom
ThreaDom is a threading-based method, and the quality of the threading template alignments has a major impact on the performance of the domain predictions. Generally, the success rate for Easy targets with a strong hit is higher than that of Hard/Medium targets. However, there are also cases that strong templates hits can result in incorrect domain assignments. The major sources of errors in ThreaDom come from (i) inconsistent domain order of homologous proteins; (ii) non-specific DCS cut-offs; and (iii) unmatched sequence size between target and templates.
Supplementary Figure S1 shows two examples of the incorrect ThreaDom predictions because of inconsistent domain orders, one from the Talin-1 (PDBID: 3dyjA) and one from the DNA polymerase III subunit β (PDBID: 3d1gA). Target 3dyjA is a two-domain protein with boundary at 164 according to CATH (Supplementary Fig. S1A). LOMETS identified the top template from 2 × 0cA, which has the same architecture to the target but with domains containing swapped segments (Szilagyi et al., 2012) (Supplementary Fig. S1B); this results in an incorrect split (1–98) (99–162) (163–241) (242–311) by ThreaDom (Supplementary Fig. S1C). ThreaDom did not merge the separate segments, as the fraction of hits on 2 × 0cA is below the cut-off (30%) in this example. Target 3d1gA consists of three domains: (1–123) (124–247) (248–366) in CATH (Supplementary Fig. S1D). It has the dominant template alignments from 2awaB with a high TM-score to the target (Supplementary Fig. S1E). However, 2awaB contains discontinuous domain structures (1–138|205–244) (139–204|245–375) in CATH, which results in an incorrect domain assignment (1–123|195–241) (124–194|242–366) following threading mapping (Supplementary Fig. S1F), despite the fact that the overall topology of the two proteins is close.
The domain linker regions in ThreaDom are decided by the interplay of DCS profile and threshold cut-offs. To increase the specificity, the DCS cutoff parameter has been trained in two sets of Easy and Medium/Hard proteins. Nevertheless, a single cut-off score might be still too general, which can result in over- or underprediction of protein domains. Figure 8A is an example of overprediction on 3-methyladenine DNA glycosylase I (PDBID: 2ofkA), which is a single-domain protein target. LOMETS considers it as a Hard target, as no significant template was identified. Among the top 35 template hits, 19 are multiple-domain proteins and 10 have the terminal gap near the residue 120. The DCS profile has, therefore, an artificial valley lower than the DCS threshold 0.76 for Hard/Medium target, which results in an overprediction of (1–120) (121–182) for this target. If the DCS threshold for Easy target (0.6) was taken, this artificial valley could have been ignored.
Fig. 8.

Inappropriate DCS thresholds and template sizes can result in incorrect domain predictions. (A–C) DCS score for 2ofkA, 1sauA and 1e8yA, respectively; (D) histogram of template alignment coverages for 1e8yA
Figure 8B is an example of underprediction because of the inappropriate DCS cut-offs. This protein is from the γ subunit of the dissimilatory sulfite reductase (DsrC) (PDBID: 1sauA), which is a two-domain Hard protein with domain assignment as (1–44) (45–114) in CATH. Although the DCS profile has a well-shaped valley at the correct domain boundary region, ThreaDom mis-predicted it as a single-domain protein because the N-terminal peak of DCS-score is lower than the threshold cut-off 0.76, and the N-terminal domain boundary is, therefore, overseen by ThreaDom. Again, if the DCS threshold for Easy target (0.6) was taken, this valley could have been picked up. The major reason for the low DCS peak is that the domain segment is short, and nearly all the residues undergo a gap penalty because they are too close to the two terminal and internal gaps. As shown in Equations (2, 5 and 6), a distance allowance d (=10/12) was introduced to tolerate the alignment/gap uncertainty; but it also introduces overpenalty for small domains. Generally, ThreaDom is unable to predict small domains with size <20 residues. To enhance the sensitivity for small domains, a size-dependent threshold cut-off might be needed for ThreaDom.
As ThreaDom derives domain information from templates, insufficient coverage of template alignments is another source of errors in ThreaDom prediction. This is particularly a problem for big proteins, such as the armadillo and heat repeats and β-propellers etc, when the solved template proteins cover only part of the repeats. Figure 8C shows the DCS profile of an example of such big proteins from the phosphatidylinositol 3-kinase catalytic subunit (PDBID: 1e8yA), which contains 841 residues with five domains assigned in CATH: (1–166) (168–302) (303–488) (489–646) (647–841). There are overall 31 strong template hits in LOMETS but most of them have a length <550 residues as seen in Figure 8D. As a result, ThreaDom generates a prediction of three domains (1–165) (166–303) (304–841) with only the first two domains correctly assigned. We did notice that there are weak valleys in the third and fourth domain boundary linkers that are from the alignment shifts in the C-terminal region, but they are too weak to pick-up by the current DCS cut-offs (0.6 for Easy proteins). This example highlights on one hand the importance of fine-tuning DCS threshold parameters. On the other hand, the domain prediction for big proteins may be further improved by an iterative threading procedure, i.e. repeating threading on the large single-domain sequences. In this example, if we run LOMETS on the remaining big domain (304–841) recursively, correct domain assignment can be obtained for the third, fourth and fifth domains.
4 CONCLUSION
We developed a multiple-threading-based method, ThreaDom, for protein domain boundary prediction. For a given target, it first threads the sequence through the PDB library to identify homologous and analogous templates. The profile distribution of the DCS, which combines the composite information of template domain structure and terminal/internal alignment gaps, is then derived for identifying the domain boundary locations. If DCDs are detected in the threading alignments, segments from separated sequences will be merged into single domains under the guide of the top template domain clusters and the target-template alignments.
There are several distinct advantages of ThreaDom over the current domain methods in literature. First, for the proteins of homologous templates, the domain assignment from threading alignments achieves a significantly higher accuracy than that from ab initio statistical or machine-learning approaches (Fig. 3). For proteins without close homologies, the LOMETS threading programs often identify multiple alignments or super-secondary structure segments from weakly homologous templates, where the DCS profile can help pull out consensus information between domain structure and alignment gaps. This enables ThreaDom to generate useful domain information for the targets that traditional homology-based approaches have difficulty with. It has also the advantage over the structural modeling-based approaches, as no lengthy modeling simulations are needed, and the approach has basically no limit on the size of protein targets.
ThreaDom was tested on three independent sets of proteins. For the first set of 315 single- and multi-domain protein pairs, ThreaDom achieves MCCs of 0.734 and 0.432 in single-/multi-domain classification compared with the CATH definition for ‘Easy’ and ‘Medium/Hard’ targets, respectively, which are significantly higher than the control methods from homology and machine-learning-based approaches. Similar results are obtained when using an alternative domain definition from SCOP, which demonstrates the reliability of the data analysis. Second, in the test of 486 DCD proteins, ThreaDom has a similar domain assignment accuracy as that in continuous domains with a precision and recall 83.9 and 64.5%, respectively, in the domain boundary prediction. Finally, when tested on the 56 CASP8 targets, ThreaDom has NDO-scores 0.761, 0.868 and 0.854 for the FM, MD-Hard and DCD targets, respectively. The average NDO-score for all targets is 0.847 that is the highest among all CASP8 servers from different categories of homology, machine-learning and ab initio folding-based approaches. Similar achievements are obtained for targets in the CASP9 and CASP10 experiments.
Overall, these data demonstrate a new promising approach that fills up the gaps between the sequence-based and the homology-based methods, which can achieve reliable domain assignments in all categories of template-based and template-free modeling protein targets. Nevertheless, fine-tuning on DCS profile cut-offs and iterative threading are needed for further improvement on small domain recognition and long sequence covering, respectively.
Although ThreaDom uses template-based modeling approach, it is much faster than the normal protein folding simulations, as the threading procedure involves only the sequence alignment search through a subset of the PDB library, which takes ∼20 min for one target protein. This speed makes it fairly feasible to genome-wide applications, as a single threading scan for a middle-size genome of 5000 genes takes <1 day on a 100-core cluster and that using multiple threading programs, such as LOMETS, should take <1 week. An online server, as well as the source code package of ThreaDom, is freely available for academic users at http://zhanglab.ccmb.med.umich.edu/ThreaDom/.
Supplementary Material
ACKNOWLEDGEMENT
The authors are grateful to G. Brandon and Dr J. Yang for discussion, Dr J. Virtanen for reading the manuscript and Dr J. Eichholt, Dr J. Cheng, Dr A. Heger and Dr Y. Kuroda for help in using their software.
Funding: National Science Foundation Career Award (DBI 1027394); National Institute of General Medical Sciences (GM083107 and GM084222); National Natural Science Foundation of China (30700162,61073095).
Conflict of Interest: none declared.
REFERENCES
- Bondugula R, et al. FIEFDom: a transparent domain boundary recognition system using a fuzzy mean operator. Nucleic Acids Res. 2009;37:452–462. doi: 10.1093/nar/gkn944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, et al. DOMpro: protein domain prediction using profiles, secondary structure, relative solvent accessibility, and recursive neural networks. Data Min. Knowl. Discov. 2006;13:1–10. [Google Scholar]
- Dessailly BH, et al. Detailed analysis of function divergence in a large and diverse domain superfamily: toward a refined protocol of function classification. Structure. 2010;18:1522–1535. doi: 10.1016/j.str.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumontier M, et al. Armadillo: domain boundary prediction by amino acid composition. J. Mol. Biol. 2005;350:1061–1073. doi: 10.1016/j.jmb.2005.05.037. [DOI] [PubMed] [Google Scholar]
- Ebina T, et al. DROP: an SVM domain linker predictor trained with optimal features selected by random forest. Bioinformatics. 2011;27:487–494. doi: 10.1093/bioinformatics/btq700. [DOI] [PubMed] [Google Scholar]
- Eickholt J, et al. DoBo: protein domain boundary prediction by integrating evolutionary signals and machine learning. BMC Bioinformatics. 2011;12:43. doi: 10.1186/1471-2105-12-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezkurdia I, et al. Assessment of domain boundary predictions and the prediction of intramolecular contacts in CASP8. Proteins. 2009;77(Suppl. 9):196–209. doi: 10.1002/prot.22554. [DOI] [PubMed] [Google Scholar]
- Finn RD, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George RA, Heringa J. SnapDRAGON: a method to delineate protein structural domains from sequence data. J. Mol. Biol. 2002;316:839–851. doi: 10.1006/jmbi.2001.5387. [DOI] [PubMed] [Google Scholar]
- Heger A, Holm L. Exhaustive enumeration of protein domain families. J. Mol. Biol. 2003;328:749–767. doi: 10.1016/s0022-2836(03)00269-9. [DOI] [PubMed] [Google Scholar]
- Heger A, et al. ADDA: a domain database with global coverage of the protein universe. Nucleic Acids Res. 2005;33:D188–D191. doi: 10.1093/nar/gki096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus K, et al. Hidden Markov models for detecting remote protein homologies. Bioinformatics. 1998;14:846–856. doi: 10.1093/bioinformatics/14.10.846. [DOI] [PubMed] [Google Scholar]
- Kim DE, et al. Automated prediction of domain boundaries in CASP6 targets using Ginzu and RosettaDOM. Proteins. 2005;61(Suppl. 7):193–200. doi: 10.1002/prot.20737. [DOI] [PubMed] [Google Scholar]
- Liu J, Rost B. CHOP proteins into structural domain-like fragments. Proteins. 2004;55:678–688. doi: 10.1002/prot.20095. [DOI] [PubMed] [Google Scholar]
- Murzin AG, et al. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Orengo CA, et al. CATH—a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- Portugaly E, et al. EVEREST: automatic identification and classification of protein domains in all protein sequences. BMC Bioinformatics. 2006;7:277. doi: 10.1186/1471-2105-7-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves GA, et al. Structural diversity of domain superfamilies in the CATH database. J. Mol. Biol. 2006;360:725–741. doi: 10.1016/j.jmb.2006.05.035. [DOI] [PubMed] [Google Scholar]
- Söding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- Servant F, et al. ProDom: automated clustering of homologous domains. Brief. Bioinform. 2002;3:246–251. doi: 10.1093/bib/3.3.246. [DOI] [PubMed] [Google Scholar]
- Sim J, et al. PPRODO: prediction of protein domain boundaries using neural networks. Proteins. 2005;59:627–632. doi: 10.1002/prot.20442. [DOI] [PubMed] [Google Scholar]
- Suyama M, Ohara O. DomCut: prediction of inter-domain linker regions in amino acid sequences. Bioinformatics. 2003;19:673–674. doi: 10.1093/bioinformatics/btg031. [DOI] [PubMed] [Google Scholar]
- Szilagyi A, et al. Intra-chain 3D segment swapping spawns the evolution of new multidomain protein architectures. J. Mol. Biol. 2012;415:221–235. doi: 10.1016/j.jmb.2011.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai CH, et al. Evaluation of domain prediction in CASP6. Proteins. 2005;61(Suppl. 7):183–192. doi: 10.1002/prot.20736. [DOI] [PubMed] [Google Scholar]
- Tress M, et al. Assessment of predictions submitted for the CASP7 domain prediction category. Proteins. 2007;69(Suppl. 8):137–151. doi: 10.1002/prot.21675. [DOI] [PubMed] [Google Scholar]
- Wang G, Dunbrack RL. PISCES: a protein sequence culling server. Biopolymers. 2003;19:1589–1591. doi: 10.1093/bioinformatics/btg224. [DOI] [PubMed] [Google Scholar]
- Wheelan SJ, et al. Domain size distributions can predict domain boundaries. Bioinformatics. 2000;16:613–618. doi: 10.1093/bioinformatics/16.7.613. [DOI] [PubMed] [Google Scholar]
- Wu S, et al. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 2007;5:17. doi: 10.1186/1741-7007-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Zhang Y. MUSTER: improving protein sequence profile-profile alignments by using multiple sources of structure information. Proteins. 2008;72:547–556. doi: 10.1002/prot.21945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ST, Zhang Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids. Res. 2007;35:3375–3382. doi: 10.1093/nar/gkm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, et al. OPUS-Dom: applying the folding-based method VECFOLD to determine protein domain boundaries. J. Mol. Biol. 2009;385:1314–1329. doi: 10.1016/j.jmb.2008.10.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Xu D. Protein threading using PROSPECT: design and evaluation. Proteins. 2000;40:343–354. [PubMed] [Google Scholar]
- Xu Y, et al. Protein domain decomposition using a graph-theoretic approach. Bioinformatics. 2000;16:1091–1104. doi: 10.1093/bioinformatics/16.12.1091. [DOI] [PubMed] [Google Scholar]
- Yoo PD, et al. DomNet: protein domain boundary prediction using enhanced general regression network and new profiles. IEEE Trans. Nanobiosci. 2008;7:172–181. doi: 10.1109/TNB.2008.2000747. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 2007;69:108–117. doi: 10.1002/prot.21702. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Progress and challenges in protein structure prediction. Curr. Opin. Struct. Biol. 2008;18:342–348. doi: 10.1016/j.sbi.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 2009;77:100–113. doi: 10.1002/prot.22588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Zhou Y. Fold recognition by combining sequence profiles derived from evolution and from depth-dependent structural alignment of fragments. Proteins. 2005;58:321–328. doi: 10.1002/prot.20308. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.