

Abstract
The estimation of isoform abundances from RNA-Seq data requires a time-intensive step of mapping reads to either an assembled or previously annotated transcriptome, followed by an optimization procedure for deconvolution of multi-mapping reads. These procedures are essential for downstream analysis such as differential expression. In cases where it is desirable to adjust the underlying annotation, for example, on the discovery of novel isoforms or errors in existing annotations, current pipelines must be rerun from scratch. This makes it difficult to update abundance estimates after re-annotation, or to explore the effect of changes in the transcriptome on analyses. We present a novel efficient algorithm for updating abundance estimates from RNA-Seq experiments on re-annotation that does not require re-analysis of the entire dataset. Our approach is based on a fast partitioning algorithm for identifying transcripts whose abundances may depend on the added or deleted isoforms, and on a fast follow-up approach to re-estimating abundances for all transcripts. We demonstrate the effectiveness of our methods by showing how to synchronize RNA-Seq abundance estimates with the daily RefSeq incremental updates. Thus, we provide a practical approach to maintaining relevant databases of RNA-Seq derived abundance estimates even as annotations are being constantly revised.
Availability and implementation: Our methods are implemented in software called ReXpress and are freely available, together with source code, at http://bio.math.berkeley.edu/ReXpress/.
Contact: lpachter@math.berkeley.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Two major bottlenecks in RNA-Seq analysis are the mapping of reads to transcripts, which is a prerequisite for quantification and differential analysis, and abundance estimation following mapping. The latter step is particularly complex when multi-mapping reads need to be resolved, which is necessary for estimating isoform-level abundances, or when genes have been duplicated (Trapnell et al., 2012). Popular programs for multi-read assignment, such as Cufflinks (Trapnell et al., 2010) and RSEM (Li and Dewey, 2011), have large memory and time requirements [see Fig. 1 of (Roberts and Pachter, 2013)]. Alternative approaches, such as eXpress (Roberts and Pachter, 2013), which uses a streaming algorithm for assignment, are faster with a low-memory footprint but must still re-process all the data from scratch when the underlying annotation is adjusted. For large datasets, such as the 3.5 billion reads of (Graveley et al., 2010), a complete run of read mapping with Bowtie, followed by abundance estimation with eXpress, takes 11 days (with 44 cores used for the mapping).
Fig. 1.

Overview of the approach: Reads are initially aligned to a set of known transcript sequences and these alignments are used to probabilistically assign multi-mapping reads and to estimate abundances of the transcripts. The result is a set of relative abundances, for example, in fragments per kilobase per million mapped (FPKM) units. When a new annotation is given, differences are identified. Reads are mapped to any added transcripts, and the ambiguity graph, where vertices correspond to transcripts and edges correspond to pairs of transcripts to which reads have mapped ambiguously, is updated (deleted transcripts in red and added transcripts in blue). The ‘affected’ transcripts whose abundance must be re-computed are obtained from a partitioning in the graph. Finally, the subset of affected transcripts have their abundances re-computed using the relevant reads, and abundances for the transcriptome are re-computed
In cases where an annotation of transcripts in a genome may change after mapping, current analysis pipelines require re-mapping of all reads followed by a complete recomputation of abundances (Schultheiss et al., 2011; Trapnell et al., 2013). This has made it time-consuming and impractical to determine the effects of the addition of possibly novel transcripts on results or the impact of removal of transcripts that appear to be incorrect. Moreover, in cases of model organisms, it has resulted in the ‘freezing’ of analyses with respect to specific annotation sets, even though re-annotation efforts are resulting in continuous changes to ‘reference’ transcriptomes (Ouzouonis and Karp, 2002).
The problem we solve in this article is how to update quantification of transcript abundances in cases where annotations change, without remapping all reads to all transcripts and running abundance estimation procedures from scratch. This problem is non-trivial for two reasons:
Multi-mapping: Frequently reads map to multiple transcripts, and therefore the removal or addition of transcripts may change the posterior probabilities associated to read mappings. In particular, the addition of a single transcript may require re-quantification of many other related transcripts.
Abundance estimates from RNA-Seq are relative and not absolute: Because RNA-Seq abundance estimates are relative, a change in the abundance estimate of a single transcript affects all other transcripts.
Given a change in the underlying transcripts, we show that abundance estimates can be updated by a procedure that only involves mapping reads to a small subset of the transcripts and re-computing assignment probabilities of multi-mapping reads for a similarly small set (Fig. 1). This is made possible by isolating a small relevant set of transcripts using a partitioning algorithm on a graph constructed from read alignments. When abundance estimation is subsequently performed using a fast online algorithm, the updating of estimates is particularly fast when the change to the underlying annotation is small.
An implication of this result is that it is possible to easily update RNA-Seq abundance estimates for annotations that are continuously updated, as is the case with the nightly Reference Sequence (RefSeq) updates. RefSeq is a large database of sequences that includes widely used reference transcripts for many organisms. RefSeq is updated nightly to reflect improvements in annotations, and although the changes are small, we show that they can affect abundance estimates in RNA-Seq analyses. Our results demonstrate that it is possible, with our algorithm, to analyze an RNA-Seq dataset by building up the annotation one transcript at a time. In particular, our tool ReXpress allows scientists to routinely update abundance estimates for RNA-Seq analyses to reflect best possible results at any time. Although ReXpress is designed to work with formats produced by the eXpress RNA-Seq quantification tool, the program is general and suitable for use with many mapping and abundance estimation methods.
2 APPROACH AND RESULTS
2.1 The ambiguity graph
RNA-Seq quantification consists of estimating abundances for a fixed set of transcripts from sequenced reads, i.e. given a set of transcripts 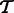 , quantification is an estimate of abundances
, quantification is an estimate of abundances 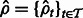 , where
, where 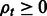 and
and 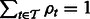 from alignments of a set of fragments
from alignments of a set of fragments 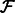 to
to 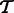 .
.
A widely used method for quantifying abundances is by statistical inference using the Expectation Maximization (EM) algorithm (Li and Dewey, 2011; Roberts and Pachter, 2013; Trapnell et al., 2010). In these methods, the likelihood function for a generative model is maximized over the 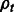 values given
values given 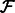 and
and 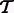 . To make the optimization tractable, a read aligner is used to remove unlikely alignments from consideration, thus providing, for each fragment
. To make the optimization tractable, a read aligner is used to remove unlikely alignments from consideration, thus providing, for each fragment 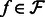 , a subset of likely transcripts the fragment is derived from. We denote the mapping by
, a subset of likely transcripts the fragment is derived from. We denote the mapping by 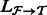 , where
, where 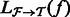 is the set of transcripts that fragment
is the set of transcripts that fragment 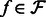 is aligned to.
is aligned to.
The approximation based on these alignments introduces sparsity to the inference, and allows the likelihood function to be factorized. This factorization can then be used to reduce the computation necessary to update abundance estimates after re-annotation.
To algorithmically leverage the sparsity of alignments, we make use of an ‘ambiguity graph’. In this graph, vertices represent transcripts, and two vertices are connected by an edge when there is at least one ambiguous fragment aligning to the two transcripts. The ambiguity graph is defined formally as follows: It is the undirected graph 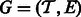 , where
, where 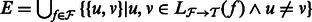 . It is easy to show that each of the components of G define a factorization of the likelihood functions used in most RNA-Seq inference algorithms (Pachter, 2011). Specifically, the set of transcripts in each component can be considered independently when assigning ambiguous fragments and computing abundances.
. It is easy to show that each of the components of G define a factorization of the likelihood functions used in most RNA-Seq inference algorithms (Pachter, 2011). Specifically, the set of transcripts in each component can be considered independently when assigning ambiguous fragments and computing abundances.
An example of an ambiguity graph obtained for a dataset of 60 million reads (see Methods) is shown in Supplementary Figure S1 and summarized in Figure 2. The graph is highly structured, and in what follows we show how this can be used to allow for rapid updates of abundance estimates upon re-annotation without extensive read mapping or numerical optimization to estimate abundances.
Fig. 2.

The distribution of component sizes in the ambiguity graph for the 60 hour time point in (Trapnell et al., 2010) using ∼30 million mapped 75 bp paired-end reads. The largest component (shown in Supplementary Fig. S1) exhibits substantial structure and the existence of many small clusters within it is the reason for the effectiveness of the partitioning algorithm we describe to reduce the complexity of the update algorithm
2.2 Incremental adjustment of abundance estimates
We begin by describing the adjustments that are required to update an RNA-Seq analysis with respect to a re-annotation. An outline of the algorithm is provided in Figure 1. We will assume that there already exists an initial annotation, alignments, abundances and ambiguity graph. In our software implementation, we assume that the files are in eXpress format (Roberts and Pachter, 2013), but eXpress itself does not have to be used to generate the output.
Given an updated FASTA file containing a re-annotation, the newly added and deleted transcripts are detected. In some cases, re-annotation can involve simply renaming existing transcripts, and this case is checked for and ignored if detected (after names/identifiers are correctly updated). A modified transcript can always be described in terms of a transcript deletion followed by an addition. For each difference between annotations, the nature of the edit is recorded.
The set of reads are then aligned to the added transcripts, and the ambiguity graph is updated with new nodes representing these transcripts and edges induced by the new alignments. The transcripts and alignments associated with independent components of the ambiguity graph containing added and deleted transcripts are extracted. The abundances for all transcripts in these components are then re-quantified separately using the updated annotation. Finally, the new annotation, alignments, abundances (for all transcripts) and ambiguity graph are output to be used with the next re-annotation update. Below is a more formal description that explains in detail the steps. Proofs of correctness follow trivially from the factorization of the standard likelihood function used in RNA-Seq, and are omitted.
We require two fields from the output of an RNA-Seq quantification program after it has been used to estimate abundances for a set of transcripts 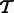 : the estimates
: the estimates 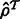 and the ‘ambiguity graph’ (defined in Section 2.1) of
and the ‘ambiguity graph’ (defined in Section 2.1) of 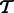 , which we denote by
, which we denote by 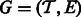 . We assume that
. We assume that 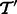 consists of
consists of 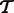 with the addition of a set of transcripts
with the addition of a set of transcripts 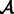 and the deletion of a set of transcripts
and the deletion of a set of transcripts 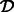 so that
so that 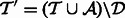 . Finally, we will need the stored alignments from
. Finally, we will need the stored alignments from 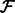 to
to 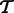 , which we denote by
, which we denote by 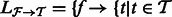 and f aligns to
and f aligns to 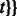 .
.
To simplify the presentation, we explain separately the case of adding transcripts (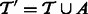 ) and the case of deletion (
) and the case of deletion (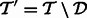 ). Additions and deletions can be handled in two stages or in a single, combined pass (details omitted). For simplicity, we restrict the exposition to the case of addition/deletion of a single transcript in the description below.
). Additions and deletions can be handled in two stages or in a single, combined pass (details omitted). For simplicity, we restrict the exposition to the case of addition/deletion of a single transcript in the description below.
Given a set of transcripts 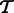 , let
, let 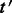 be a transcript with
be a transcript with 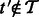 . The updating of estimates when
. The updating of estimates when 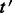 is added to the annotation is performed as follows:
is added to the annotation is performed as follows:
Align the reads in
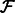 to
to 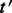 and denote the subset of reads of
and denote the subset of reads of 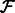 that align to
that align to 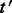 by
by 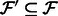 . Denote the alignments of
. Denote the alignments of 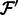 as
as 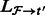 .
.Extract the read alignments for the reads in
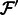 from
from  and denote as
and denote as 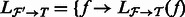 for all
for all 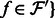 . In addition, denote by
. In addition, denote by 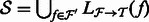 the set of transcripts in
the set of transcripts in 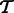 that appear in
that appear in 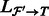 .
.Create the updated ambiguity graph
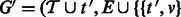 for all
for all 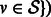 .
.Let
 . Extract the alignments in
. Extract the alignments in 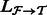 that consist of a read mapping to a transcript in
that consist of a read mapping to a transcript in 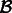 as
as 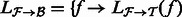 for all
for all 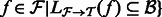 .
.Merge the alignments to create
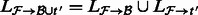 .
.Perform quantification on the set of transcripts
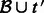 using the alignments
using the alignments 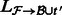 . This produces a set of estimates
. This produces a set of estimates  .
.Compute
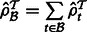 . Set
. Set 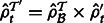 for all
for all 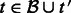 .
.
Deletion is performed via a similar procedure. Let 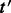 be a transcript with
be a transcript with 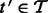 .
.
Let
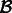 be the component in G that contains
be the component in G that contains 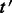 .
.Extract the alignments from
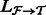 that contain reads mapping to transcripts in
that contain reads mapping to transcripts in 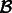 , denoted by
, denoted by 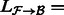
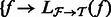 for all
for all 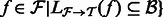 .
.Remove the alignments of reads to
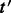 from
from 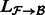 as
as 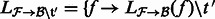 for all
for all 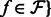 .
.Perform quantification on the set of transcripts
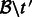 using the alignment file
using the alignment file 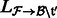 . This produces a set of estimates
. This produces a set of estimates 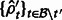 .
.Compute
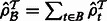 . Set
. Set 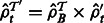 for all
for all 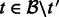 .
.Create the updated ambiguity graph
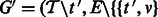 for all
for all 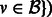 .
.
Note that in the rare case when there is a change in the total number of aligned fragments after the addition or deletion of a target, an additional step is required to renormalize the relative abundances between components. This step is trivial and fast, and the details are omitted.
2.3 Improving performance by approximating the affected set
There is another issue that can hurt performance in practice: the affected component 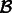 can be large (Fig. 2 and Supplementary Fig. S1). In typical RNA-Seq experiments, as much as one-fifth of all transcripts can lie in a single component of the ambiguity graph (Roberts and Pachter, 2013). This component typically consists of large gene families and multiple isoform genes that share common sequence. To improve performance, it is therefore desirable to restrict the re-quantification to a smaller subset without sacrificing important information in the form of fragment alignments. We do this by partitioning a weighted generalization of the ambiguity graph, obtained by the addition of edge weights representing the number of ambiguous alignments between each pair of transcripts. For a given mapping
can be large (Fig. 2 and Supplementary Fig. S1). In typical RNA-Seq experiments, as much as one-fifth of all transcripts can lie in a single component of the ambiguity graph (Roberts and Pachter, 2013). This component typically consists of large gene families and multiple isoform genes that share common sequence. To improve performance, it is therefore desirable to restrict the re-quantification to a smaller subset without sacrificing important information in the form of fragment alignments. We do this by partitioning a weighted generalization of the ambiguity graph, obtained by the addition of edge weights representing the number of ambiguous alignments between each pair of transcripts. For a given mapping 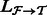 and induced ambiguity graph G, we let the weight between two transcripts
and induced ambiguity graph G, we let the weight between two transcripts 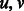 be
be 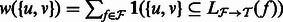 . Given these weights, we wish to partition around
. Given these weights, we wish to partition around 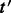 such that the total weight of edges crossing the partition cut is small compared with the weight of edges inside the block. Moreover, it is desirable that the block containing
such that the total weight of edges crossing the partition cut is small compared with the weight of edges inside the block. Moreover, it is desirable that the block containing 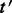 is small.
is small.
Many sophisticated objective functions and algorithms exist for partitioning graphs (Bichot and Siarry, 2011). A detailed exploration of the applications of these methods to our problem is outside the scope of this article. Instead, to demonstrate the feasibility of a partitioning scheme for improving the performance of our method with large components, we chose to use the greedy approach outlined below, which is motivated by the objective of removing edges that correspond to the ‘least informative’ alignments.
First, we define the density of a block S, d(S), as the total weight of edges incident to a node in the block and a node outside of the block divided by the total weight of edges incident to the nodes in the block. Intuitively, this is the ratio of edges crossing the cut to all of the edges incident to nodes in the block. Formally,
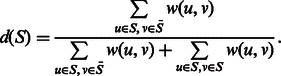 |
Our objective is to find, for a given transcript 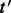 , a block S that contains t such that
, a block S that contains t such that 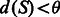 for a given threshold
for a given threshold 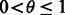 . We do so using the following greedy update.
. We do so using the following greedy update.
Begin with
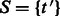 .
.Iteratively add node
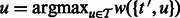 to S until
to S until 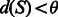 .
.
It is easy to show that for any valid θ, this algorithm will terminate. As we show below in Section 2.4, the method is empirically both fast and accurate.
2.4 Accuracy of partitioning approximation
To validate the performance of our greedy partitioning algorithm, we randomly selected with replacement 250 transcripts from the largest partition (3898 transcripts) in the RefSeq annotation and simulated their addition at some earlier date. We used the set of reads produced for (Trapnell et al., 2010), which consisted of RNA-Seq performed on C212, a mouse moblast cell line. Each selected transcript was removed from the FASTA, alignment file and ambiguity graph, and was then re-added using a single step of our algorithm. We show the results of this update using 20 different values of θ in Supplementary Figure S2. There is a clear tradeoff between the accuracy of the approximation and the size of the resulting block selected by different values of θ. We note that for 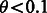 , the correlation is already reasonably close to the accuracy of the eXpress algorithm demonstrated in (Roberts and Pachter, 2013).
, the correlation is already reasonably close to the accuracy of the eXpress algorithm demonstrated in (Roberts and Pachter, 2013).
2.5 Application to RefSeq incremental update
To demonstrate the effectiveness of our approach, we applied it to the same large C2C12 RNA-Seq dataset as used above. These data were first analyzed in 2010, but since then the mouse RefSeq annotation has been updated numerous times. Specifically, as a proof-of-concept, we applied ReXpress (our implementation of the methods above) to 34 days of the RefSeq incremental update (RIU), which is a daily update of the RefSeq annotation database (see Methods, Fig. 3a).
Fig. 3.

(a) Updates to the mouse RefSeq transcriptome over the course of 34 days. Transcripts that kept the same name but changed sequence were treated as an addition and a deletion. (b) ReXpress run time, in minutes, on each RefSeq update, with and without partitioning. Initial run time consists of Bowtie2 alignment time (24 cores) and eXpress abundance estimation time (3 cores), without ReXpress. Partitioning was done when a changed transcript was part of a component larger than 300 transcripts, which occurred seven times over the 34-day period
Using 24 free cores, 644 min were required for the initial Bowtie2 alignment, 505 min for 20 repetitions of abundance estimation with eXpress and 11 min for building the ambiguity graph. Across the entire month of RefSeq updates, a size 3910 component was affected seven times, while components of size 15 or less were affected 37 times.
Each subsequent update required, on average, 55 min to complete our re-annotation pipeline. This is compared with the ∼644 + 505 = 1149 min that would be required for alignment and abundance estimation from scratch with Bowtie2 and eXpress after each re-annotation.
The abundance estimates for the final time point had a Spearman rank correlation of 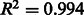 with those calculated from scratch (Supplementary Fig. S3). The small discrepancy is due to the fact that the online EM method in eXpress approximates the maximum likelihood solution, and therefore is not expected to be exact.
with those calculated from scratch (Supplementary Fig. S3). The small discrepancy is due to the fact that the online EM method in eXpress approximates the maximum likelihood solution, and therefore is not expected to be exact.
Because some of the transcripts added and deleted over the time period affected the large components in the ambiguity graph, we also ran the analysis using the greedy partitioning scheme described above (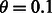 ). While the speed of the updates was greatly improved by the partitioning by reducing the size of the (approximate) affected components (Fig. 3b), the results were nearly identical (Supplementary Fig. S4).
). While the speed of the updates was greatly improved by the partitioning by reducing the size of the (approximate) affected components (Fig. 3b), the results were nearly identical (Supplementary Fig. S4).
3 METHODS
3.1 Datasets
The annotations used for mouse were based on RefSeq. The RefSeq database is updated incrementally every night at 3:30 EST. All updates over the 34-day period between November 9 and December 13, 2012 were used for this analysis. The RNA-Seq data used was based on (Trapnell et al., 2010). We restricted ourselves to analysis of the 60 hour time point, for which 60 million reads were available.
3.2 Read mapping
Reads were mapped with Bowtie2 version 2.1.0 (Langmead and Salzberg, 2012) using the parameters -k 1000, –rdg 6,5, –rfg 6,5, –score-min L,-0.6,-0.4, –no-discordant and –no-mixed. With these options, 47% of the reads were mapped concordantly.
3.3 Abundance estimation
Abundances were estimated with eXpress version 1.3.0 (Roberts and Pachter, 2013), initially using no optional parameters and then the –aux-param-file for re-estimation using previously computed auxiliary parameters after editing of the annotation. A forgetting factor of 0.85 (the default) was used for the full dataset and 0.75 used for the smaller update datasets.
3.4 Software
The methods have been implemented in a software program called ReXpress. ReXpress is a Python script that takes as input an annotation and its update, reads, their alignments to the initial annotation, abundance estimates and the ambiguity graph. It outputs updated versions of all of the input (Fig. 1 for exact files input and output). ReXpress makes heavy use of pySAM (pySAM, 2012; Li et al., 2009) and is based on Bowtie2 and eXpress for quantification (Roberts and Pachter, 2013), although many alignment and quantification tools can be substituted.
4 CONCLUSION
Despite the difficulties in storing, processing and distribution of high-throughput sequence data (Sboner et al., 2011), repositories such as the Gene Expression Omnibus have led to an explosion in publicly available genome-wide expression data. However, numerous technical challenges that arise in re-using data have limited the utility of publicly archived RNA-Seq reads (Rung and Brazma, 2013).
Our results show that it is possible to efficiently update RNA-Seq abundance estimates on re-annotation, thus removing a major obstacle to re-using publicly available data. This should prove to be particularly useful in newly sequenced organisms whose annotations are not stable and undergo periodic revision, and also in human cancer transcriptomics where structural alterations can be tumor specific (Asmann et al., 2012; Yorukoglu et al., 2012). We also believe that ReXpress will be particularly useful for sequencing centers providing analysis services. Instead of producing one-time output, it should now be possible to refresh analyses as annotations improve, without expensive hardware or compute time needed as user bases and datasets grow.
Other applications of our work include a randomized approach to optimization of transcriptome assembly in conjunction with abundance estimation (Li and Jiang, 2012; Li et al., 2011; Mezlini et al., 2013), and the development of an RNA-Seq quantification database for publicly available datasets that is automatically updated as annotations improve.
Moreover, our work on component identification in and partitioning of the ambiguity graph can be used to develop more efficient batch methods for abundance estimation. A recurring issue in the commonly used batch EM solutions (Li and Dewey, 2011; Trapnell et al., 2010) is the necessity of iterating over a large number of reads, which has a memory bottleneck as shown in (Roberts and Pachter, 2013). Trapnell et al. (2010) attempts to avoid the bottleneck by treating all genomic loci as independent blocks and using a heuristic ‘rescue method’ to partially correct for the approximation. A better solution for the memory bottleneck in the batch method is to iterate over approximately independent partitions of the ambiguity graph whose associated reads can be fit into memory. Because most components are often small, only the largest will need to be partitioned as in our method above. The blocks can then be processed in parallel only a single machine or distributed over a cluster.
Finally, in conjunction with the streaming algorithm for quantification in (Roberts and Pachter, 2013), the present method provides an online algorithm in both the reads and the targets in any setting where probabilistic assignment of multi-mapping reads is a bottleneck in analysis of high-throughput sequencing data.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Isabelle Stanton for her advice on graph partitioning.
Funding: AR was partly funded by an NSF graduate fellowship. AR and LP were partially funded by NIH R01 HG006129.
Conflict of Interest: none declared.
REFERENCES
- Asmann YW, et al. Detection of redundant fusion transcripts as biomarkers or disease-specific therapeutic targets in breast cancer. Cancer Res. 2012;72:1921–1928. doi: 10.1158/0008-5472.CAN-11-3142. [DOI] [PubMed] [Google Scholar]
- Bichot CE, Siarry P, editors. Graph Partitioning. Hoboken, NJ, USA: Wiley; 2011. [Google Scholar]
- Graveley BR, et al. The developmental transcriptome of Drosophila melanogaster. Nature. 2010;471:473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey C. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, et al. IsoLasso: a LASSO regression approach to RNA-Seq based transcriptome assembly. J. Comput. Biol. 2011;18:1693–1707. doi: 10.1089/cmb.2011.0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Jiang T. Transcriptome assembly and isoform expression level estimation from biased RNA-Seq reads. Bioinformatics. 2012;28:2914–2921. doi: 10.1093/bioinformatics/bts559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezlini AM, et al. iReckon: simultaneous isoform discovery and abundance estimation from RNA-seq data. Genome Res. 2013;23:519–529. doi: 10.1101/gr.142232.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouzouonis CA, Karp PD. The past, present and future of genome-wide re-annotation. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-2-comment2001. comment2001.1–comment2001.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachter L. Models for Transcript Quantification from RNA-Seq. 2011. arXiv:1104.3889. [Google Scholar]
- Pruitt KD, et al. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40:D130–D135. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- pySAM. 2012 http://code.google.com/p/pysam/ (December 2012, date last accessed) [Google Scholar]
- Roberts A, Pachter L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat. Methods. 2013;10:71–73. doi: 10.1038/nmeth.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rung J, Brazma A. Reuse of public genome-wide gene expression data. Nat. Rev. Genet. 2013;14:89–99. doi: 10.1038/nrg3394. [DOI] [PubMed] [Google Scholar]
- Sboner A, et al. The real cost of sequencing: higher than you think! Genome Biol. 2011;12:125. doi: 10.1186/gb-2011-12-8-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultheiss SJ, et al. Oqtans: a Galaxy-integrated workflow for quantitative transcriptome analysis from NGS Data. BMC Bioinformatics. 2011;12 A7. [Google Scholar]
- Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorukoglu D, et al. Dissect: detection and characterization of novel structural alterations in transcribed sequences. Bioinformatics. 2012;28:i179–i187. doi: 10.1093/bioinformatics/bts214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.