

Abstract
Alzheimer’s disease (AD) is by far the most common neurodegenerative disease. AD is histologically characterized not only by extracellular senile plaques and vascular deposits consisting of β-amyloid (Aβ) but also by accompanying neuroinflammatory processes involving the brain’s microglia. The importance of microglia is still in controversial discussion, which currently favors a protective function in disease progression. Recent findings by different research groups highlighted the importance of strain-specific and mitochondria-specific genomic variations in mouse models of cerebral β-amyloidosis. Here, we want to summarize our previously presented data and add new results that draw attention towards the consideration of strain-specific genomic alterations in the setting of APP transgenes. We present data from APP-transgenic mice in commonly used C57Bl/6J and FVB/N genomic backgrounds and show a direct influence on the kinetics of Aβ deposition and the activity of resident microglia. Plaque size, plaque deposition rate, and the total amount of Aβ are highest in C57Bl/6J mice as compared to the FVB/N genomic background, which can be explained at least partially by a reduced microglia activity toward amyloid deposits in the C57Bl/6J strain.
Keywords: microglia, neuroinflammation, genomic background, mouse models, Alzheimer’s disease, neurodegeneration
Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disorder and histologically characterized by the accumulation of toxic amyloid-β peptides (Aβ) that form extracellular senile plaques and vascular deposits, and intracellular neurofibrillary tangles composed of hyperphosphorylated tau [1]. The resulting loss of neuronal and synaptic function leads to a gradual decline of cognitive abilities and the development of clinical overt dementia [2–4]. Today, the widely accepted hypothesis implies that aggregated moieties of Aβ, especially oligomeric species, are the initial cause for the long pathogenesis process of 20–30 years that leads to the development of ‘senile dementia’ [5, 6]. Different Aβ species are generated by the cleavage of the transmembrane amyloid precursor protein (APP). Thereby, the longer variant – Aβ42 – is more prone to aggregate than the shorter Aβ40 variant and its self-aggregation results in different forms of small and larger Aβ aggregates [7, 8]. Here, the small, soluble oligomeric assemblies were detected to be far more toxic than the insoluble fibrillar forms of Aβ [5, 9–11]. Their clearance from the brain tissue is widely acknowledged as one important strategy to stop AD progression and clinical onset [12–19]. In fact, a first physiological defense mechanism to prevent the formation of amyloid plaques is provided by the resident immune effector cells of the central nervous system – the microglia. Their main function is to ensure the brain’s protection against several pathogens and pathological conditions [12, 20]. In animal models of amyloidosis, microglia group around amyloid plaques [21], have been demonstrated to be beneficial due to their ability to phagocytize toxic Aβ peptides [21–23], and secrete neurotrophic factors [24]. Until today, it is not fully verified whether microglia only have a protective or also a partly neurotoxic effect, as they were shown to release pro-inflammatory cytokines like IL1 and TNFα and thereby may also promote disease progression [25–28]. Currently, it is widely believed that microglia undergo morphological alterations during activation and also change the expression of several cell surface receptors and the secretion of cytokines and chemokines. Initial neuroprotective effects are based on the microglia’s activity which contributes to the restriction of the amyloid plaques’ growth. Later, microglial effects may reverse as a result of chronic activation during AD progression and culminate in neurotoxicity (reviewed in ref. [29]). Although the precise role of microglia is still unclear, there is wide evidence that microglia activation plays a crucial role in disease pathogenesis [19, 21, 30–33].
Transgenic mouse models are an important tool that allows to create distinct pathological conditions and to investigate new hypotheses of disease mechanisms and also novel therapy strategies. As important risk factors, besides the age, rare mutations in the APP, PS1, and PS2 genes lead to the early-onset, inherited form of AD (familiar AD-FAD), which are often used to generate β-amyloidosis in mouse models [34]. In doing so, the genomic background was found to importantly influence the transgene expression pattern and the morphological phenotype, thus, vastly affecting the experimental outcomes. This issue came more and more into the focus of the research when different labs presented controversial or at least vigorously different results using the same mouse models in different or even mixed genomic backgrounds, e.g. as shown in behavioral tests and histochemical analyses concerning mossy fiber distribution in fragile-X mental retardation protein knockout (Fmr1) mice in either C57Bl/6J or FVB background [35]. In neurodegeneration research, varying genomic backgrounds were shown to affect APP processing [36], to lead to diversified brain [36–38] and plasma levels of Aβ [36] and as a consequence in total plaque number [36, 37, 39]. Also, the time course of Aβ deposition was found to be strain-specifically altered [36, 37]. Moreover, clearance processes are restricted, as both insulin-degrading enzyme expression [39] and neprilysin activity [40] were found to be influenced by the genomic background. As a result, life expectancy varies considerably [37, 41]. Interestingly, C57Bl/6J mice were identified to be genetically disadvantageous in models of cerebral amyloidosis as compared to other strains like 129S1/SvImJ [36], DBA/2J [36], A/J [37], SJL [41] and Swiss Webster [40]. Furthermore, induced neuronal degeneration was found to be highly strain dependent with different vulnerability for neuronal death [42]. Notably, controversial findings were made concerning microglia distribution in rats of different genomic origin. By comparing SHR72 and WKY72 rats, no significant strain-specific differences of total microglia and astrocyte number were observed. However, the levels of MHCII-immunoreactive microglia differ rigorously in both models [43]. Additionally, the neurofibrillary tangle load is apparently independent of transgene expression levels [44], but is strongly related to the genomic background [43].
One widely used model of cerebral β-amyloidosis with varying genomic backgrounds is the APP/PS1 transgenic mouse (in the following referred to as APPtg) [45]. It carries a human APP transgene with the Swedish double mutation (Lys670Asn, Met671Leu) and a mutated presenilin 1 (Leu166Pro), as a functional part of the γ-secretase complex involved in Aβ production, expressed postnatally by the Thy1 promoter, which ensures a widespread neuronal expression. Various findings were made using these mice in the context of AD. In 2011, we described that the ABC transporter ABCC1 extensively influences the intracerebral aggregation profile of Aβ [46] using APPtg mice in the FVB/N background (common background of ABC transporter-deficient strains). Just recently, we published how inbred mitochondrial DNA polymorphisms (conplastic mitochondria) influence Aβ accumulation and microglial activity in mouse models with the genomic DNA C57Bl/6J background [38]. Taken together, the data of the control groups of both publications, strain-specific differences in Aβ concentration at distinct time points of 100 and 200 days in the C57Bl/6J or FVB/N backgrounds became evident. As we used the same analytic methods in both projects, we thereby realized the background-specific alterations in Aβ kinetics, which we then further investigated. Here, we want to integrate and review the recently published data [21, 38, 46] in this new context and substantiate the results with a set of new time-series data to show how differences in Aβ concentration, aggregation and microglia response are indeed background specific.
C57Bl/6J and FVB/N genomic background effects on β-amyloid plaque formation
With advancing age, soluble Aβ peptides processed from APP accumulate in the extracellular medium and aggregate into oligomers, protofibrils, and insoluble fibrils prior to the formation of diffuse and dense-cored plaques (reviewed in ref. [47]). In the APPtg mice, transgene-related APP expression is driven by the same promoter in both C57Bl/6J (B6) and FVB/N (FVB) background mouse strains, resulting in nearly identical Aβ production. Thus, it seemed most likely that they do not differ in the initial Aβ concentration and the resulting amyloid plaque formation process. Starting from this premise, we time-dependently examined specific parameters in APPtg mice from both strains using standard methods (ELISA, immunohistochemistry) and found that Aβ has distinct deposition kinetics in both strains that indeed lead to differences in the number of plaques, size distribution of plaques and total Aβ concentration.
To compare the plaque burden of APPtg mice with different genomic backgrounds, we determined the mean number of plaques in the cortex, as one of the first affected regions of the brain by immunohistochemistry (Fig. 1). Tissue preparation and semi-automated, 230 nm high-resolution, whole-slide analysis was performed as previously described [21]. Analysis started at the age of 50 days when first plaques appeared and ended at 200 days of age, the oldest time point studied. Here, we found that APPtgB6 mice have significantly more plaques at the age of 175 and 200 days as compared to APPtg-FVB mice. Additionally, as a first sign of difference, APPtg-B6 mice show already significantly higher plaque burden at 75 days of age. APPtg-B6 mice displayed a continuous increase until the age of 150 days with a subsequent sharp increase in older age. In contrast, APPtg-FVB mice revealed a sharp increase at early time points followed by a plateau phase starting at 125 days of age till the latest time point measured (200 days).
Fig. 1.

Genomic background-specific variations lead to differences in plaque burden. The number of plaques is significantly increased in APPtg mice in the C57Bl/6J background (filled bars) as compared to the FVB/N background (white bars) at the age of 75, 175, and 200 days. APPtg-B6 mice show a continuous increase of the mean number of plaques, whereas APPtg-FVB mice reach a plateau phase at the age of 125 days with no further increase afterwards. Data are presented as means ± SEM (n ≥ 5 per group), *p ≤ 0.05; **p ≤ 0.01
These data are strongly correlated to the intracerebral Aβ42 concentrations measured with ELISA (as described in ref. [46]) (Fig. 2). Here, approximately even twice the amount of soluble (monomeric and oligomeric) and insoluble (fibrillar) Aβ42 was found at 200 days of age in APPtg-B6 mice, according to the exponential increase using this volumetric measure method. Even the significant increases in total plaque number at 75 and 175 days of age correspond to the obtained Aβ concentrations; again confirming the differences between both background strains.
Fig. 2.
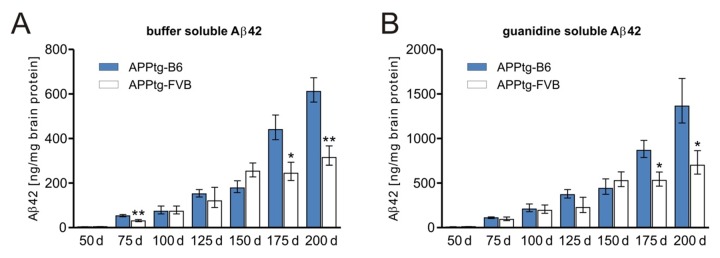
The genomic background influences the amount of soluble and insoluble Aβ species. Aβ42 measurements of APPtg mice in two different background strains indicate distinct deposition kinetics. Until day 150, C57Bl/6J- and FVB/N-background strains exhibit similar quantities of buffer-soluble Aβ (monomers and small oligomers) and guanidine-soluble Aβ (fibrils). In APPtg-FVB mice, a stationary phase is detectable in which the amount of Aβ stays nearly constant. In contrast, APPtg-B6 mice display a large increase in monomeric, oligomeric and fibrillar Aβ. Data are presented as means ± SEM (n ≥ 5 per group), *p ≤ 0.05; **p ≤ 0.01
Furthermore, plaque size distribution was examined (Fig. 3). Plaques were categorized [21, 48] according to the covered area; plaques smaller than 400 µm2 were referred to as ‘small’, whereas these larger than 700 µm2 were denoted ‘large’. ‘Medium’-sized plaques cover an area between both limits. Here, APPtg-B6 mice have significantly less ‘small’ but higher numbers of ‘large’ plaques starting at an age of 75 days till 200 days as compared to APPtg-FVB mice. ‘Medium’ plaques only differ significantly at the last time point measured. Additionally, at old age (200 days), a uniform distribution of ‘small’ and ‘large’ plaques is detectable in APPtg-B6 mice in contrast to the APPtg-FVB strain, in which the ‘small’ plaques comprise the largest plaque population throughout all ages. Moreover, the plaque size distribution reaches a phase at 150 days of age in APPtg-B6 mice where the plaque categories show only little changes. The distribution kinetic in APPtg-FVB mice reveals a sharp decrease in ‘small’ and only a slight increase in ‘large’ plaques between 50 days and 200 days of age.
Fig. 3.

The relative number of different-sized amyloid plaques in the cortex strongly depends on the genomic background. The number of ‘small’ and ‘large’ plaques shows significant variations when comparing APPtg-B6 and APPtg-FVB strains. Mice in the C57Bl/6J background reveal a uniform distribution of ‘small’ and ‘large’ plaques at old age (150 days – 200 days). ‘Small’ plaques are the largest population through all ages in APPtg-FVB mice. Data are presented as means ± SEM (n ≥ 5 per group), *p ≤ 0.05; **p ≤ 0.01
The β-amyloid-directed microglia response depends on the genomic background
Microglia represent the first barrier of the brain’s immune system and are located in the closed vicinity of developing and mature amyloid depositions [21, 49, 50]. Microglia are also capable to phagocytoze Aβ in vitro [22, 23]. However, in mouse models that lack microglia [51], it was shown that no changes occur in Aβ load, plaque morphology or plaque size distribution. These mostly HSV-TK-driven, microglia-deficient models show substantial caveats for the investigation of the microglia function and, thus, diverging interpretations have been obtained during the recent years. Especially, microglia’s role for the maintenance of amyloid plaques has been controversially discussed [24, 52]. Here, we wanted to examine genomic background-related differences in microglia activation in the C57BL/6J and FVB/N strains. To determine the amount of activated microglia, their coverage of amyloid plaques was detected by means of immunohistochemistry (Fig. 4). Tissue preparation and 230 nm high-resolution analysis were performed as previously described [21]. We used the number of plaques covered at least by 50% by microglia for the evaluation, a value that was empirically defined as a marker of microglia activity [21].
Fig. 4.

Plaque burden and microglial response is altered by the genomic background in β-amyloidosis mouse models. Plaque number and plaque size are significantly increased in APPtg-B6 mice (A and C) versus APPtg-FVB mice (B and D) at 100 days (A and B) and 200 days (C and D) of age. Insets show examples of microglia (brown) located near amyloid deposits (red) (scale bars: 100 µm)
Figure 5 highlights the age-related changes in microglia coverage of plaques in APPtg-B6 and APPtg-FVB mice. A rapid microgliosis occurs prior and during the first phase of plaques development, shown by the rapid increase from 50 to 75 days of age, especially prominent in the FVB background strain. In the APPtg-B6 strain the coverage gradually increases till 150 days of age but remains far below the APPtg-FVB strain levels. In the APPtg-FVB strain the coverage increases rapidly (~60–70% of all plaques are highly covered) and stays at this high level till the latest time point studied (200 days of age). These results coincide with the significantly higher number of ‘small’ plaques in the APPtg-FVB strain (Fig. 3a). In APPtg-FVB mice, the continuous growth is restricted due to the early effects of the activated microglia leading to higher numbers of ‘small’ plaques. Concomitant with the latest hypothesis, in which microglia undergo morphological changes and become ‘neurotoxic’, the coverage and degradation of amyloid deposits decreases in later time points and reaches a plateau (Fig. 5) not only in APPtg-FVB but also in APPtg-B6 mice. Plaque growth in APPtg-B6 mice shows no restriction during the investigated points in time (Fig. 1), indicating a reduced function of microglia. Although microglia can be found located around amyloid plaques, it is not possible to conclusively distinguish between secreting and phagocytizing phenotype so far [29, 48, 50]. After migrating to the amyloid plaque and the phenotypic shift into round and activated microglia, the microglia cell body is placed peripheral of the amyloid plaque near the ends of the star-shaped branches, and some of the amyloid fibrils can also be found inside the microglial cytoplasm [48]. However, the normal activation process of these macrophages includes formation of vacuoles, lysosomes, lipid droplets, which cannot be found in amyloid plaque-associated microglia [48, 50], indicating a more secreting phenotype in the first place.
Fig. 5.

Relative amount of plaque-related microglia in APPtg mice with different genomic backgrounds. The relative number of plaques that are more than 50% covered by microglia is significantly increased in APPtg-FVB mice at most time points. As shown in the previous figures, the APPtg-FVB mice have lower amounts of plaques and amyloid levels at distinct time points (75, 175, and 200 days) that may be explained by the increased activity of microglia in this specific strain. Data are presented as means ± SEM (n ≥6 per group), **p ≤ 0.01; ***p ≤ 0.001
Whether the observed differences in our strains depend on microglial activity or whether microglial activity just correlates with the number and size of plaques in the first place cannot be clarified, here. We see vigorous variations in these examined parameters in mouse models of AD with distinct genomic backgrounds. These findings are based on various differences in nuclear and mitochondrial genomes in both strains. The identification of specific genes that are involved in this differentially regulated Aβ kinetics and deposition in the FVB/N mice in contrast to C57Bl/6J mice could help to identify new targets for the treatment or modulation of AD.
Summary
Here, we show that different genomic backgrounds influence amyloid pathology and microglia reaction and could confirm data from earlier experiments using YACs [36]. Thus, it is of importance to compare results only from genetically identical mouse models. However, till now, experimental data of mixed or incompatible background strains are still being published although the results are nearly incomparable and interpretation is almost impossible. The detection of true biological changes requires careful analysis to become useful for the improvement of the patient’s situation and lead to effective new discoveries.
Footnotes
Declaration. The authors declare no conflict of interest.
Author contributions.
Conceived and designed experiments: MK, JP
Performed experiments: CF, KP, JoS, JaS
Analyzed data: CF, KP, JoS, JaS, JP
Contributed reagents/materials/analysis tools: none
Wrote manuscript: CF, KP, JoS, MK, HJH, JP
Contributor Information
Christina Fröhlich, 1University of Magdeburg, Department of Neurology, Neurodegeneration Research Lab (NRL), Leipziger Str. 44, 39120 Magdeburg, Germany.
Kristin Paarmann, 1University of Magdeburg, Department of Neurology, Neurodegeneration Research Lab (NRL), Leipziger Str. 44, 39120 Magdeburg, Germany.
Johannes Steffen, 1University of Magdeburg, Department of Neurology, Neurodegeneration Research Lab (NRL), Leipziger Str. 44, 39120 Magdeburg, Germany.
Jan Stenzel, 1University of Magdeburg, Department of Neurology, Neurodegeneration Research Lab (NRL), Leipziger Str. 44, 39120 Magdeburg, Germany.
Markus Krohn, 1University of Magdeburg, Department of Neurology, Neurodegeneration Research Lab (NRL), Leipziger Str. 44, 39120 Magdeburg, Germany.
Hans-Jochen Heinze, 1University of Magdeburg, Department of Neurology, Neurodegeneration Research Lab (NRL), Leipziger Str. 44, 39120 Magdeburg, Germany; 2German Center for Neurodegenerative Diseases (DZNE) Magdeburg, Leipziger Str. 44, 39120 Magdeburg, Germany; 3Leibniz Institute for Neurobiology (LIN), Department of Behavioral Neurology, Brennecke Str. 6, 39118 Magdeburg, Germany.
Jens Pahnke, 1Department of Neurology, Neurodegeneration Research Laboratory (NRL), University of Magdeburg, Leipziger Str 44, Bldg 15, 39120 Magdeburg, FAX: +49 (391) 67 24528, Phone: +49 (391) 67 24514, Germany, Email: jens.pahnke@gmail.com; 2German Center for Neurodegenerative Diseases (DZNE) Magdeburg, Leipziger Str. 44, 39120 Magdeburg, Germany; 3Leibniz Institute for Neurobiology (LIN), Department of Behavioral Neurology, Brennecke Str. 6, 39118 Magdeburg, Germany.
References
- 1.DJ Selkoe. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001 Apr;81(2):741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 2.Ma T, Klann E. Amyloid β: linking synaptic plasticity failure to memory disruption in Alzheimer's disease. J Neurochem. 2012 Jan;120(Suppl 1):140–148. doi: 10.1111/j.1471-4159.2011.07506.x. Epub 2011 Nov 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nature medicine. 2008 Aug 1;14(8) doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Annals of neurology. 1990 May 1;27(5) doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 5.Larson ME, Lesné SE. Soluble Aβ oligomer production and toxicity. J Neurochem. 2012 Jan;120(Suppl 1):125–139. doi: 10.1111/j.1471-4159.2011.07478.x. Epub 2011 Nov 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nature reviews. Molecular cell biology. 2007 Feb 1;8(2) doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 7.Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten I, Van Der Werf K, Subramaniam V, Braeken D, Callewaert G, Bartic C, D'Hooge R, Martins IC, Rousseau F, Schymkowitz J, De Strooper B. Neurotoxicity of Alzheimer's disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 2010 Oct 6;29(19):3408–3420. doi: 10.1038/emboj.2010.211. Epub 2010 Sep 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Findeis MA. The role of amyloid beta peptide 42 in Alzheimer's disease. Pharmacology & therapeutics. 2007 Nov 1;116(2) doi: 10.1016/j.pharmthera.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 9.Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimer's disease conundrum? Trends in neurosciences. 2001 Apr 1;24(4) doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 10.Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. 2007 Jun;101(5):1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. Epub 2007 Feb 5. [DOI] [PubMed] [Google Scholar]
- 11.Hung LW, Ciccotosto GD, Giannakis E, Tew DJ, Perez K, Masters CL, Cappai R, Wade JD, Barnham KJ. Amyloid-beta peptide (Abeta) neurotoxicity is modulated by the rate of peptide aggregation: Abeta dimers and trimers correlate with neurotoxicity. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008 Nov 12;28(46) doi: 10.1523/JNEUROSCI.3916-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Napoli I, Neumann H. Microglial clearance function in health and disease. Neuroscience. 2009 Feb 6;158(3) doi: 10.1016/j.neuroscience.2008.06.046. [DOI] [PubMed] [Google Scholar]
- 13.Mandrekar S, Jiang Q, Lee CY, Koenigsknecht-Talboo J, Holtzman DM, Landreth GE. Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009 Apr 1;29(13) doi: 10.1523/JNEUROSCI.5572-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pahnke J, Wolkenhauer O, Krohn M, Walker LC. Clinico-pathologic function of cerebral ABC transporters - implications for the pathogenesis of Alzheimer's disease. Current Alzheimer research. 2008 Aug 1;5(4) doi: 10.2174/156720508785132262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science. 2012 Mar 23;335(6075):1503–1506. doi: 10.1126/science.1217697. Epub 2012 Feb 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. 2012 Aug 15;4(147):147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.RJ Bateman. Amyloid-beta production and clearance rates in Alzheimer’s disease. Alzheimer’s and Dementia. 2010;6:S101. [Google Scholar]
- 18.Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science (New York, N.Y.) 2010 Dec 24;330(6012) doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee CY, Landreth GE. The role of microglia in amyloid clearance from the AD brain. Journal of neural transmission (Vienna, Austria : 1996) 2010 Aug 1;117(8) doi: 10.1007/s00702-010-0433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Filippo M, Chiasserini D, Tozzi A, Picconi B, Calabresi P. Mitochondria and the link between neuroinflammation and neurodegeneration. Journal of Alzheimer's disease : JAD. 20 Suppl 2 doi: 10.3233/JAD-2010-100543. [DOI] [PubMed] [Google Scholar]
- 21.Scheffler K, Stenzel J, Krohn M, Lange C, Hofrichter J, Schumacher T, Brüning T, AS Plath, Walker L, Pahnke J. Determination of spatial and temporal distribution of microglia by 230nm-high-resolution, high-throughput automated analysis reveals different amyloid plaque populations in an APP/PS1 mouse model of Alzheimer's disease. Current Alzheimer research. 2011 Nov 1;8(7) doi: 10.2174/156720511797633179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koenigsknecht J, Landreth G. Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004 Nov 3;24(44) doi: 10.1523/JNEUROSCI.2557-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rogers J, Strohmeyer R, Kovelowski CJ, Li R. Microglia and inflammatory mechanisms in the clearance of amyloid beta peptide. Glia. 2002 Nov 1;40(2) doi: 10.1002/glia.10153. [DOI] [PubMed] [Google Scholar]
- 24.Bessis A, Béchade C, Bernard D, Roumier A. Microglial control of neuronal death and synaptic properties. Glia. 2007 Feb 1;55(3) doi: 10.1002/glia.20459. [DOI] [PubMed] [Google Scholar]
- 25.Jimenez S, Baglietto-Vargas D, Caballero C, Moreno-Gonzalez I, Torres M, Sanchez-Varo R, Ruano D, Vizuete M, Gutierrez A, Vitorica J. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer's disease: age-dependent switch in the microglial phenotype from alternative to classic. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008 Nov 5;28(45) doi: 10.1523/JNEUROSCI.3024-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paranjape GS, Terrill SE, Gouwens LK, Ruck BM, Nichols MR. Amyloid-β(1-42) Protofibrils Formed in Modified Artificial Cerebrospinal Fluid Bind and Activate Microglia. J Neuroimmune Pharmacol. 2013 Mar;8(1):312–322. doi: 10.1007/s11481-012-9424-6. Epub 2012 Dec 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer's disease, role of cytokines. TheScientificWorldJournal. 2012 Apr 1;2012 doi: 10.1100/2012/756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meda L, Cassatella MA, Szendrei GI, Otvos L Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995 Apr 13;374(6523) doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 29.Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nature reviews. Neurology. 2010 Apr 1;6(4) doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- 30.Naert G, Rivest S. The role of microglial cell subsets in Alzheimer's disease. Current Alzheimer research. 2011 Mar 1;8(2) doi: 10.2174/156720511795256035. [DOI] [PubMed] [Google Scholar]
- 31.Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013 Jan 31;493(7434) doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vom Berg J, Prokop S, Miller KR, Obst J, Kälin RE, Lopategui-Cabezas I, Wegner A, Mair F, Schipke CG, Peters O, Winter Y, Becher B, Heppner FL. Inhibition of IL-12/IL-23 signaling reduces Alzheimer's disease-like pathology and cognitive decline. Nature medicine. 2012 Dec 1;18(12) doi: 10.1038/nm.2965. [DOI] [PubMed] [Google Scholar]
- 33.Karlmark K, et al. Monocytes in health and disease. Eur J Microbiol Immunol. 2012;2:97–102. doi: 10.1556/EuJMI.2.2012.2.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teipel SJ, Buchert R, Thome J, Hampel H, Pahnke J. Development of Alzheimer-disease neuroimaging-biomarkers using mouse models with amyloid-precursor protein-transgene expression. Progress in Neurobiology. 2011 Dec 1;95(4) doi: 10.1016/j.pneurobio.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 35.Ivanco TL, Greenough WT. Altered mossy fiber distributions in adult Fmr1 (FVB) knockout mice. Hippocampus. 12(1) doi: 10.1002/hipo.10004. [DOI] [PubMed] [Google Scholar]
- 36.Lehman EJ, Kulnane LS, Gao Y, Petriello MC, Pimpis KM, Younkin L, Dolios G, Wang R, Younkin SG, Lamb BT. Genetic background regulates beta-amyloid precursor protein processing and beta-amyloid deposition in the mouse. Human molecular genetics. 2003 Nov 15;12(22) doi: 10.1093/hmg/ddg322. [DOI] [PubMed] [Google Scholar]
- 37.Sebastiani G, Krzywkowski P, Dudal S, Yu M, Paquette J, Malo D, Gervais F, Tremblay P. Mapping genetic modulators of amyloid plaque deposition in TgCRND8 transgenic mice. Human molecular genetics. 2006 Aug 1;15(15) doi: 10.1093/hmg/ddl157. [DOI] [PubMed] [Google Scholar]
- 38.Scheffler K, Krohn M, Dunkelmann T, Stenzel J, Miroux B, Ibrahim S, von Bohlen Und Halbach O, Heinze HJ, Walker LC, Gsponer JA, Pahnke J. Mitochondrial DNA polymorphisms specifically modify cerebral β-amyloid proteostasis. Acta Neuropathol. 2012 Aug;124(2):199–208. doi: 10.1007/s00401-012-0980-x. Epub 2012 Apr 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glazner KA, Odero GL, Anema E, Motnenko A, Schapansky J, Grossman D, Oliver DR, Glazner GW, Albensi BC. Strain specific differences in memory and neuropathology in a mouse model of Alzheimer's disease. Life sciences. 2010 Jun 19;86(25-26) doi: 10.1016/j.lfs.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 40.Carter TL, Pedrini S, Ghiso J, Ehrlich ME, Gandy S. Brain neprilysin activity and susceptibility to transgene-induced Alzheimer amyloidosis. Neuroscience letters. 2006 Jan 16;392(3) doi: 10.1016/j.neulet.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 41.Carlson GA, Borchelt DR, Dake A, Turner S, Danielson V, Coffin JD, Eckman C, Meiners J, Nilsen SP, Younkin SG, Hsiao KK. Genetic modification of the phenotypes produced by amyloid precursor protein overexpression in transgenic mice. Human molecular genetics. 1997 Oct 1;6(11) doi: 10.1093/hmg/6.11.1951. [DOI] [PubMed] [Google Scholar]
- 42.Liu L, Hamre KM, Goldowitz D. Kainic acid-induced neuronal degeneration in hippocampal pyramidal neurons is driven by both intrinsic and extrinsic factors: analysis of FVB/N↔C57BL/6 chimeras. J Neurosci. 2012 Aug 29;32(35):12093–12101. doi: 10.1523/JNEUROSCI.6478-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stozicka Z, Zilka N, Novak P, Kovacech B, Bugos O, Novak M. Genetic background modifies neurodegeneration and neuroinflammation driven by misfolded human tau protein in rat model of tauopathy: implication for immunomodulatory approach to Alzheimer's disease. Journal of Neuroinflammation. 2010 Oct 12;7 doi: 10.1186/1742-2094-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koson P, Zilka N, Kovac A, Kovacech B, Korenova M, Filipcik P, Novak M. Truncated tau expression levels determine life span of a rat model of tauopathy without causing neuronal loss or correlating with terminal neurofibrillary tangle load. The European Journal of Neuroscience. 2008 Jul 1;28(2) doi: 10.1111/j.1460-9568.2008.06329.x. [DOI] [PubMed] [Google Scholar]
- 45.Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jäggi F, Wolburg H, Gengler S, Haass C, Ghetti B, Czech C, Hölscher C, Mathews PM, Jucker M. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Reports. 2006 Sep 1;7(9) doi: 10.1038/sj.embor.7400784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krohn M, Lange C, Hofrichter J, Scheffler K, Stenzel J, Steffen J, Schumacher T, Brüning T, Plath AS, Alfen F, Schmidt A, Winter F, Rateitschak K, Wree A, Gsponer J, Walker LC, Pahnke J. Cerebral amyloid-β proteostasis is regulated by the membrane transport protein ABCC1 in mice. J Clin Invest. 2011 Oct;121(10):3924–3931. doi: 10.1172/JCI57867. Epub 2011 Sep 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pahnke J, Walker LC, Scheffler K, Krohn M. Alzheimer's disease and blood-brain barrier function-Why have anti-beta-amyloid therapies failed to prevent dementia progression? Neuroscience and Biobehavioral Reviews. 2009 Jul 1;33(7) doi: 10.1016/j.neubiorev.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008 Apr 16;28(16) doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. The American journal of pathology. 1999 Jun 1;154(6) doi: 10.1016/S0002-9440(10)65423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perlmutter LS, Barron E, Chui HC. Morphologic association between microglia and senile plaque amyloid in Alzheimer's disease. Neuroscience Letters. 1990 Oct 30;119(1) doi: 10.1016/0304-3940(90)90748-x. [DOI] [PubMed] [Google Scholar]
- 51.Grathwohl SA, Kälin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, Odenthal J, Radde R, Eldh T, Gandy S, Aguzzi A, Staufenbiel M, Mathews PM, Wolburg H, Heppner FL, Jucker M. Formation and maintenance of Alzheimer's disease beta-amyloid plaques in the absence of microglia. Nature neuroscience. 2009 Nov 1;12(11) doi: 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harry GJ, Kraft AD. Neuroinflammation and microglia: considerations and approaches for neurotoxicity assessment. Expert Opinion on Drug Metabolism & Toxicology. 2008 Oct 1;4(10) doi: 10.1517/17425255.4.10.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]