

Abstract
Temporal lobe epilepsy (TLE) is defined as the occurrence of spontaneous seizures that involve the limbic system, with the hippocampal formation and associated structures being central to the most prevalent refractory form of adult focal epilepsy. TLE is often associated with psychotic features resembling the hallucinations and delusions that occur with schizophrenia. Given evidence that the ventral hippocampus (vHipp) plays an important role in the maintenance of temporal lobe seizures, we investigated whether an animal model of TLE using intrahippocampal injection of pilocarpine induces alterations in mesolimbic dopamine (DA) neuron activity. We found that in 60% of rats in which pilocarpine induced seizure activity, there was a significant increase in the number of dopamine neurons firing per electrode track. Furthermore, this occurred in concert with an increase in amphetamine-stimulated locomotor activity. Both observations are similar to those observed in a rodent developmental model of psychosis. Therefore, as in animal models of schizophrenia, TLE-associated psychosis is likely due to abnormal hippocampal overdrive of dopamine neuron activity.
Keywords: epilepsy, psychosis, dopamine, hippocampus, electrophysiology, ventral tegmental area
Introduction
The association between epilepsy and schizophrenia –like psychosis has attracted the attention of psychiatrists since the nineteenth century, but many aspects of this relationship remain controversial (Mendez et al., 1993; Qin et al., 2005; Trimble, 1991). Thus, there is a wide variation in the incidence of clinically significant psychiatric symptoms in individuals with epilepsy that depends strongly on the patient sample (Stevens, 1988). Moreover, several studies indicate that schizophrenia-like psychosis is 6–12 times more likely to occur in epileptic patients than in the general population (Slater et al., 1963).
Patients affected by TLE often present symptoms such as aggressive irritability, humorless sobriety, circumstantial preoccupation with detail, nascent religious or philosophical interest, and altered sexual preference (Bear, 1979; Bear and Fedio, 1977; Waxman and Geschwind, 1974). In addition, guilty rumination often leading to depression has been reported. Indeed, depression alone accounts for the most common comorbidity in patient with epilepsy (Harden, 2002; Kanner and Balabanov, 2002). Unfortunately, a consensus on the classification of psychotic syndromes associated with epilepsy is lacking, and neither DSM-IV nor ICD-120 has addressed this issue specifically.
Since clinical seizures are the outstanding feature of epilepsy, psychotic syndromes have traditionally been classified according to their temporal relationship to these events. Interestingly, the hippocampus plays a central role in the pathophysiology of both epilepsy (and particularly TLE) as well as schizophrenia. Thus, in addition to being among the lowest seizure threshold areas in the brain, substantial evidence has linked hippocampal pathology to schizophrenia, such as alterations in hippocampal morphology using magnetic resonance imaging (Lawrie and Abukmeil, 1998), reduced N-acetyl aspartate (Bertolino et al., 1998; Bertolino et al., 1996; Deicken et al., 1999; Maier et al., 1995), and the increased metabolism in the anterior hippocampus (Heckers, 2001; Tamminga et al., 1992) that correlates with psychotic symptoms (Silbersweig et al., 1995).
Animal models of schizophrenia have also implicated hippocampal pathophysiology as a driving force in the abnormal regulation of the dopamine system. Thus, in a developmental animal model of schizophrenia, a loss of parvalbumin interneuron staining in the hippocampus subiculum leads to an overdrive of the subiculum-nucleus accumbens pathway (Lodge et al., 2009). This, in turn, leads to an increase in dopamine neuron population activity, which is proposed to be the basis for dopamine system hyper-responsivity (Lodge and Grace, 2007). Thus, hyperactivity within the hippocampus leads to hyper-responsivity of the dopamine system, with dopamine hyper-responsivity proposed to underlie the psychotic symptoms of schizophrenia (Laruelle and Abi-Dargham, 1999). Indeed, as in this rodent model (Lodge and Grace, 2007), rats with pilocarpine-induced TLE also exhibit a substantially greater behavioral response to amphetamine (Ando et al., 2004).
Induction of TLE with pilocarpine is equally efficacious independent of whether the drug is administered systemically or intra-hippocampally (Furtado Mde et al., 2002), with the intra-hippocampal route associated with a markedly reduced mortality (Furtado Mde et al., 2002). With this method, approximately 70% of the animals are reported to experience status epilepticus (SE) within 30 min after the intra-hippocampal infusion and present spontaneous recurrent seizures (SRSs) after a latent period of 2–30 days. Therefore, this model was used in the current study to examine the relationship between TLE and dopamine system function.
Material and methods
Animals
Adult male Sprague-Dawley rats (175–200 grams) were obtained from Hilltop (Scotdale, PA) and housed in pairs on a 12:12 hour light:dark cycle; food and water were available ad libitum. All experiment were performed in accordance with the guidelines outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
Surgery and pharmacological manipulation
Rats were anesthetized with Nembutal and mounted on a stereotaxic frame using blunt atraumatic ear bars. A stainless steel guide cannula was implanted in the right vHipp (A/P −6.0, M/L +5.3, D/V −4.5 mm from bregma) and fixed in place with dental cement and two or three anchor screws. Once the cement was completely solid, the wound was sutured, the rat removed from the stereotaxic apparatus and monitored closely until conscious. Rats received antibiotic treatment (gentamicin 3mg/Kg, s.c.) and postoperative analgesia (Children’s Tylenol syrup in softened rat chow; 5% v/w) ad libitum for 24 h.
After one week of recovery half of the total number of the animals that received cannula implants were infused intracranially into the vHipp with the proepileptic drug pilocarpine (Sigma, San Louis, MO). Pilocarpine was dissolved in Dulbecco’s phosphate-buffered saline (PBS) and infused (2.4 mg/μl, 1μl; duration around 30s) through a 30 gauge injection cannula protruding 2.0 mm beyond the tip of the guide cannula. The remaining rats were infused with saline solution (0.9 %, 1μl).
Following the infusion of pilocarpine in the vHipp, the progressive evolution of seizures was observed and quantified using the Racine motor scale. All motor activities were recorded using a digital camera. Animals exhibiting SE (defined as an epileptic seizure that lasts more than 30 minutes) were injected with diazepam (5 mg/kg; intraperitoneally) after 120 minutes of SE in order to stop seizure activity. Spontaneous recurrent seizures (SRSs) appear in the animals infused with pilocarpine after a latent period of about 7–30 days. All rats were video monitored continuously after PILO infusion (all during the SE period). After the infusion the rats were video monitored to check for spontaneous recurrent seizures starting one week after the infusion. This monitoring was made daily for 3 hours per day (from 14:00 to 17:00 hours) since previous studies (Raedt et al., 2009) showed that seizures appear more frequently in this time frame with a regular circadian rhythm. Only rats that displayed SRSs at least 2–3 times per day for a total of a week after the infusion of pilocarpine were used in this study. Before the electrophysiological and behavioral test animals were monitored by visual observation 2.3 hours before the experiment to ensure that the recordings were not made in the immediate post-ictal period. Only animals that showed SE and SRSs were evaluated further.
Extracellular recordings
Only drug-naïve animals were used for electrophysiological recordings to circumvent the effects of prior amphetamine exposure on DA neuron activity (Xia et al., 2008; Vanderschuren et al., 1999). Rats were used in the post-ictal period, with recordings made following the appearance of SRSs, which averaged two weeks post-injection and no observable seizures were present for at least 2.3 hours prior to recording, as described above. Animals were anesthetized with 8% chloral hydrate (400 ml/kg; intraperitoneally) and mounted in a Kopf stereotaxic frame. Chloral hydrate was used since, with this drug, DA neuron activity states are consistent with those observed in freely moving rats (Hyland et al., 2002). Anesthesia was maintained by supplemental administration of chloral hydrate as required to maintain suppression of the plantar reflex. Core body temperature was maintained at 37 degrees C using a thermostatically controlled heating pad.
A burr hole was drilled overlying the VTA (A/P −5.1:–5.5, M/L +0.6:+1, D/V −6.5: −8.5 mm from bregma), the dura was resected and glass extracellular microelectrodes (impedance 6–14 MΩ) were lowered into the region using a hydraulic microdrive (Kopf model 640). The activity of the population of DA neurons was obtained by counting the number of spontaneously active neurons encountered while making six to nine vertical passes or tracks separated by 200 μm in a predetermined grid pattern throughout the VTA. Spontaneously active DA neurons were identified using previously well-established electophysiological criteria (Grace and Bunney, 1983) and once isolated their activity was recorded for three minutes.
Three parameters of activity were measured: I) population activity (defined as the number of spontaneously active DA neurons recorded in each electrode track), II) basal firing rate, and III) the percentage of action potentials occurring in bursts (defined as the occurrence of two spikes with an interspike interval of < 80 ms, and the termination of the burst defined as the occurrence of an interspike interval > 160 ms (Grace and Bunney, 1984). The data were acquired, stored and analyzed using custom-designed computer software (Neuroscope, Brian Lowry, Pittsburgh, PA).
Amphetamine-induced locomotion
Behavioral data was collected following the emergence of SRSs, which averaged two weeks post-PILO injection to correspond to the electrophysiology data, with video monitoring visually confirming the absence of an ictal state for a minimum of 2.3 hours prior to the experiment. Epileptic rats and non-epileptic rats (NEC) were placed in an open-field activity monitor (Coulbourn instruments, Allentown, PA) to automatically measure spontaneous locomotor activity in the x-y plane for 60 minutes by beam breaks and recorded with Truscan software (Coulbourn instruments). Rats were then injected with D-amphetamine sulphate (1.5 mg/kg i.p.) and locomotor activity recorded for an additional 120 min. Only rats that displayed SRSs at least three times for day, for a total time of a week after the infusion of pilocarpine in the vHipp, were used for this behavioral test.
Histology
At the cessation of the experiment, the recording site was marked via electrophoretic ejection of Pontamine sky blue dye from the tip of the recording electrode (30 mA constant current, 30–45 minutes). After dye injection, rats were killed by an overdose of chloral hydrate, decapitated and their brains removed, fixed for at least 48 h (8% (w/v) paraformaldehyde in PBS) and cryoprotected (25% (w/v) sucrose in PBS until saturated). Brain were sectioned (60 μm coronal sections), mounted onto gelatin-chrom alum coated slides and stained with cresyl violet for histochemical verification of electrodes sites and thionin for verification of cannula sites. All histology was performed with reference to a stereotaxic atlas (Paxinos and Watson, 1986)
Analysis
Electrophysiological analysis of DA neuron activity was performed using custom-designed computer software (Neuroscope), and locomotor behavior was recorded using TruScan software (Coulbourne Instruments). All data are presented as the mean ± SEM unless otherwise stated. All statistics were calculated using the GraphPad Prism software for Student’s t-test for electrophysiological data and Sigmaplot 11 for Repeated Measures ANOVA for the locomotor data.
Results
Of a total of 50 animals infused in the vHipp with pilocarpine, 1 did not show seizure activity, whereas 49 experienced epileptic seizures. The distribution along the Racine motor scale is summarized in fig 1. Animals in stage 1 (4 in total) were not used, since it was difficult to discriminate between a stage 1 seizure and the normal movements of the head and mouth in these animals. Of the remaining 45 animals, 15 were used for the electrophysiological recordings and 16 were used for amphetamine-induced locomotor activity. Of the 14 remaining animals, 8 had misplaced lesion sites and 6 failed to show SRSs for at least 2–3 times per day, and were excluded from further analyses.
Fig 1.
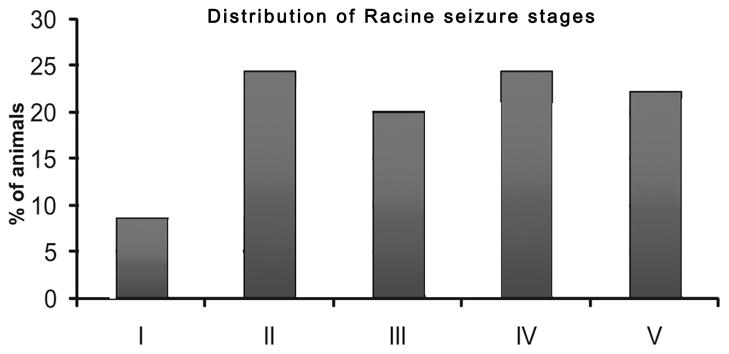
Distribution of the animals (n=49) infused with pilocarpine according to the racine motor scale of epilepsy, with I being mild seizure activity (i.e., mouth and facial movement) and V representing the strongest form of seizure (rearing and falling with forelimb clonus). Only rats scoring II through V were used in the study.
Vhipp infusion of pilocarpine increases ventral tegmental area dopamine neuron activity
Animals that received saline infusion in the vHipp (n = 14 rats, 114 neurons) exhibited an average of 0.85 ± 0.24 spontaneously active DA neurons per electrode track, with an average firing rate of 5.18 ± 2.04 Hz and with 43.0 ± 15.2% of action potentials fired in bursts, which is consistent with previous findings in untreated rats (Floresco et al., 2003; Lodge and Grace, 2006a; Lodge and Grace, 2006b). Approximately 60% (9/15) of the rats infused with pilocarpine into the vHipp and exhibited seizure activity between II and V on the Racine scale (n = 9 rats, 90 neurons) showed significantly greater DA neuron population activity and were labeled as responders (1.62 ± 0.29 cells/track; t=7.018 p<0.001), and showed small but significant reductions in average burst firing (29.8 ± 11.6%; t=2.453, df=22, p<0.05) but not in firing rate (3.81 ±0.5 Hz; t=2.048, df=22, p>0.05) relative to controls (Fig. 2). However, when examined, it was clear that nearly 40% (6/15) of the rats tested exhibited DA neuron population activity nearly identical to that of controls (0.82 ± 0.08 cells/track; 4.4 ± 1.75 Hz; 38.3 ± 13.4 % of bursting cells). These rats were defined as non-responders if their cells/track was <1.1; the resultant group mean for these animals fell within a 5% confidence interval of the control rats (control 5% confidence interval = 0.71–0.99 cells/track) but not for the responders. These groups of rats showed clear separation in DA neuron activity (Fig. 2). There was no correlation between the severity of the epileptic scores with the response status. Such an observation is consistent with what one would predict from the clinical literature regarding TLE, in that only 6–12% of patients with TLE exhibit psychosis. Since in our experimental protocol we made a well-controlled lesion, we expected greater consistency in the impact on the dopamine system.
Fig 2.

Effect of intrahippocampal (i.h.) pilocarpine infusion (2.4 mg/ml) on VTA DA neuron population activity. DA neuron population activity was examined when SRSs appear, which was typically at least 2 weeks following i.h. infusion of pilocarpine. Rats examined electrophysiologically (n=15 PILO and N=14 controls) were divided into two groups: pilocarpine responders (n=9) and pilocarpine non-responders (n=6). The non-responders showed values of cells/track that were within a 5% confidence interval of controls. The PILO responders showed a significant increase in DA neuron population activity (t-test, ***p<0.001) with small but significant decreases in the average percent of burst firing (*p<0.05; B) but not in firing rate (A).
TLE rats exhibit increased behavioral response to amphetamine injection
Previous studies in the gestational methylazoxy methanol (MAM) developmental model of schizophrenia and with amphetamine sensitization (Lodge and Grace, 2007) demonstrated that hyper-activity in the hippocampus is responsible for the increased baseline DA neuron population activity that correlated with behavioral hyper-responsivity to amphetamine injection (Lodge and Grace, 2008). Given evidence of the role of the hippocampus in the regulation of DA neuron population activity, the behavioral responsivity to amphetamine was examined in this model of epilepsy.
Consistent with these previous observations, the pilocarpine-infused rats (n=16) exhibited significantly enhanced locomotor response to d-amphetamine administration when compared with the rats infused with saline solution (n=16; rmANOVA group x time interaction F=7.742, df=35, p<0.001; fig 3). This increase in distance traveled is 75% higher than in controls, and this increase reached maximum approximately 25–30 minutes after the amphetamine injection. After almost 2 hours following the amphetamine injection, the pilocarpine-injected rats recovered to the baseline level of movement observed before the amphetamine injection. Taken together, these data suggest that in pilocarpine-treated rats a pathologically increased DA neuron population activity and the related increased responsivity to amphetamine administration are likely attributable to a hippocampal hyper-activity.
Fig 3.
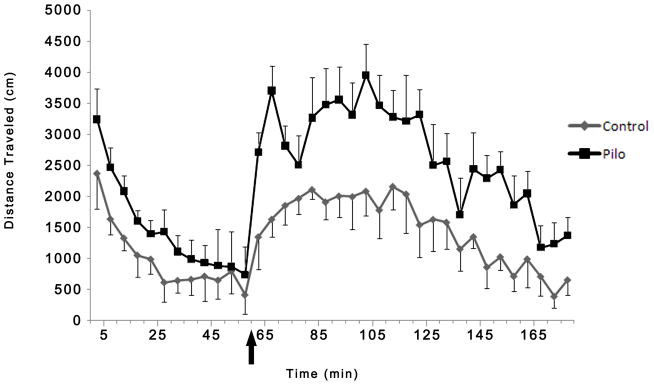
Consistent with a previous study in which DA neuron activity was assessed in an animal model of schizophrenia pilocarpine-treated rats (n=16) displayed a hyperdopaminergic condition reflected by a significant enhancement in locomotor response (repeated measures ANOVA, p<0.001) following administration of 1.5 mg/kg of D-amphetamine (arrow represents the injection of amphetamine) compared to controls (n=16). Taken as a whole these data suggest that in pilocarpine treated rats a pathologically increased DA neuron population activity is associated with increased responsivity to amphetamine.
Discussion
The data presented here demonstrate, for the first time, that pilocarpine-treated rats display an abnormally enhanced DA neuron drive in the form of an increase in DA neuron population activity. Specifically, we demonstrated that most of the pilocarpine-treated rats display a significantly higher number of spontaneously firing DA neurons when compared with control rats. Our studies show that DA neuron population activity, defined as the proportion of spontaneously firing DA neurons, is regulated in normal rats by a ventral subicular-nucleus accumbens-ventral pallidal-VTA pathway (Floresco et al., 2003). Therefore, a TLE-associated pathological hyperactivity in the hippocampus should in turn lead to increased VTA DA neuron activity. Since DA neurons must be spontaneously active in order to exhibit phasic burst firing responses to inputs (Lodge and Grace, 2006a), abnormally high levels of population activity would enable a phasic burst stimulus to elicit burst firing in a greater number of DA neurons, thereby putting the DA system in a hyper-responsive state. Indeed, hippocampal hyperactivity has been linked to psychosis in a rat developmental model of schizophrenia (Lodge and Grace, 2006a; Lodge and Grace, 2008) which, similar to that observed in patients (Laruelle and Abi-Dargham, 1999), is characterized by hyper-responsivity of the dopamine system to amphetamine. Therefore, the link between hippocampal hyperactivity and DA dysregulation leading to psychosis may be a common pathophysiological variable across several disease states. Although there was an increase in population activity in the PILO responsive rats, there was no change in the average firing rate but only a small decrease in percent burst firing, consistent with our previous studies (Lodge & Grace, 2006a, 2008). As suggested in this previous study, this is likely due to recruitment of slowly firing, non-bursting neurons that, when averaged across the population of activated neurons did not result in a substantial difference in rate or pattern.
The source of the hippocampal hyperactivity and resultant DA neuron drive in TLE is not known. Wozny et al suggested that the increased excitability is associated with a pronounced neurodegeneration in layer III of the medial enthorinal cortex (mEC) in patients and in models of temporal lobe epilepsy (Wozny et al., 2005). In fact the EC seems to be critically involved in temporal lobe epilepsy. This limbic region shows an enhanced susceptibility to seizures and epileptiform discharge (Collins et al., 1983; Dasheiff and McNamara, 1982; Spencer and Spencer, 1994). Previous studies showed that SE causes a preferential loss of glutamatergic neurons while sparing GABAergic neurons in layer III of the mEC (Du et al., 1995; Eid et al., 1999; Kobayashi et al., 2003). Interestingly the projection that originates from cells in layer III, the so-called temporoammonic pathway, terminates exclusively in CA1 and the subiculum (Amaral and Witter, 1989; Witter et al., 1989).
In contrast, Knopp et al suggested that the vulnerability of the subicular GABAergic interneurons causes an input-specific disturbance of the subicular inhibitory system. They showed that in the subiculum of pilocarpine-treated animals the density of glutamic acid decarboxylase (GAD) mRNA-positive cells was reduced in all layers. Furthermore they showed a substantial loss of parvalbumin-immunoreactive neurons in the pyramidal cell and in the molecular layer (Knopp et al., 2005). Interestingly we found a similar pattern of parvalbumin-containing interneuron loss in an animal model of schizophrenia (Lodge et al., 2009), which is a robust finding reported in schizophrenia brains post-mortem (Lewis et al., 2008). This deficit in the intrinsic GABAergic signaling may be the origin of the hippocampal hyperactivity that seems to underlie the dopamine dysfunction in psychosis in both TLE and schizophrenia.
These electrophysiological findings are supported by results obtained with the locomotor test activity performed after injection of the psychostimulant D-amphetamine, a well-validated animal model of psychosis. In fact we demonstrate a striking potentiation of stimulant-induced locomotor hyperactivity in pilocarpine-treated rats in accordance with previous studies made in both pilocarpine models and other models of experimental epilepsy (Ando et al., 2004; Jones et al., 2010; Ma and Leung, 2004; Muller et al., 2009; Szyndler et al., 2005). The fact that the use of a cholinergic drug reproduced aspects of TLE seen with other models suggests that it is the epileptogenic properties of the drug, rather than its cholinergic properties, that were significant in this model. Given that the primary mechanism of action of psychostimulants is to increase extracellular DA, this behavioral sensitization is attributable, at least in part, to enhanced activity of the mesolimbic DA pathway (Lodge and Grace, 2008; Pierce and Kalivas, 1997; White and Wang, 1984). Interestingly, in contrast to the electrophysiological results, all pilocarpine-treated rats tested for amphetamine-induced locomotor activity exhibited a heightened response. While the source of this difference is not known, one possibility could be due to the anti-epileptic effects of the anesthetic, causing a proportion of rats to exhibit attenuated hippocampal hyperactivity.
Taken as a whole, the present study demonstrates that the increased responsivity to psychostimulants observed in pilocarpine-treated rats is likely attributable to an increase in tonic DA transmission secondary to augmented activity within the ventral hippocampus. This augmentation of vHipp drive was also found in an animal developmental model of schizophrenia in which endogenous vHipp overdrive also leads to aberrant DA signaling (Lodge and Grace, 2007). One difficulty associated with psychosis in TLE is its resistance to treatment (Chakir et al., 2006; Glien et al., 2002). By understanding the pathological alterations in the dopamine system induced by TLE, we will be in a better position to design effective therapeutic interventions that can address the dysfunction at the source of dopamine overdrive, rather than blocking the postsynaptic consequences of the hyperdopaminergic condition.
Acknowledgments
The authors thank Nicole MacMurdo for technical assistance, Kathryn Gill for her help with the statistical analyses, Brian Lowry for the custom-design software Neuroscope and Dr. Ornella Valenti and Dr. Daniel Lodge for helpful discussions.
This work was supported by Ri.MED Foundation research grant (PC) and USPHS MH57440 (AAG)
Footnotes
Disclosure
Professor Anthony Grace received funds from:
Puretech Ventures, Johnson & Johnson, Merck, Taisho, Lundbeck, Abbott, AstraZeneca, Lilly, Sandoz, GlaxoSmithKline
References
- Amaral DG, Witter MP. The three-dimensional organization of the hippocampal formation: a review of anatomical data. Neuroscience. 1989;31(3):571–591. doi: 10.1016/0306-4522(89)90424-7. [DOI] [PubMed] [Google Scholar]
- Ando N, Morimoto K, Watanabe T, Ninomiya T, et al. Enhancement of central dopaminergic activity in the kainate model of temporal lobe epilepsy: implication for the mechanism of epileptic psychosis. Neuropsychopharmacology. 2004;29(7):1251–1258. doi: 10.1038/sj.npp.1300427. [DOI] [PubMed] [Google Scholar]
- Bear DM. Temporal lobe epilepsy--a syndrome of sensory-limbic hyperconnection. Cortex. 1979;15(3):357–384. doi: 10.1016/s0010-9452(79)80064-7. [DOI] [PubMed] [Google Scholar]
- Bear DM, Fedio P. Quantitative analysis of interictal behavior in temporal lobe epilepsy. Archives of Neurology. 1977;34(8):454–467. doi: 10.1001/archneur.1977.00500200014003. [DOI] [PubMed] [Google Scholar]
- Bertolino A, Callicott JH, Elman I, Mattay VS, et al. Regionally specific neuronal pathology in untreated patients with schizophrenia: a proton magnetic resonance spectroscopic imaging study. Biological Psychiatry. 1998;43(9):641–648. doi: 10.1016/s0006-3223(97)00555-6. [DOI] [PubMed] [Google Scholar]
- Bertolino A, Nawroz S, Mattay VS, Barnett AS, et al. Regionally specific pattern of neurochemical pathology in schizophrenia as assessed by multislice proton magnetic resonance spectroscopic imaging. American Journal of Psychiatry. 1996;153(12):1554–1563. doi: 10.1176/ajp.153.12.1554. [DOI] [PubMed] [Google Scholar]
- Chakir A, Fabene PF, Ouazzani R, Bentivoglio M. Drug resistance and hippocampal damage after delayed treatment of pilocarpine-induced epilepsy in the rat. Brain Research Buletinl. 2006;71(1–3):127–138. doi: 10.1016/j.brainresbull.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Collins RC, Tearse RG, Lothman EW. Functional anatomy of limbic seizures: focal discharges from medial entorhinal cortex in rat. Brain Research. 1983;280(1):2540. doi: 10.1016/0006-8993(83)91170-8. [DOI] [PubMed] [Google Scholar]
- Dasheiff RM, McNamara JO. Electrolytic entorhinal lesions cause seizures. Brain Research. 1982;231(2):444–450. doi: 10.1016/0006-8993(82)90381-x. [DOI] [PubMed] [Google Scholar]
- Deicken RF, Pegues M, Amend D. Reduced hippocampal N-acetylaspartate without volume loss in schizophrenia. Schizophrenia Research. 1999;37(3):217–223. doi: 10.1016/s0920-9964(98)00173-x. [DOI] [PubMed] [Google Scholar]
- Du F, Eid T, Lothman EW, Kohler C, et al. Preferential neuronal loss in layer III of the medial entorhinal cortex in rat models of temporal lobe epilepsy. Journal of Neuroscience. 1995;15(10):6301–6313. doi: 10.1523/JNEUROSCI.15-10-06301.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Schwarcz R, Ottersen OP. Ultrastructure and immunocytochemical distribution of GABA in layer III of the rat medial entorhinal cortex following aminooxyacetic acid-induced seizures. Experimental Brain Research. 1999;125(4):463–475. doi: 10.1007/s002210050704. [DOI] [PubMed] [Google Scholar]
- Floresco SB, West AR, Ash B, Moore H, et al. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nature Neuroscencei. 2003;6(9):968–973. doi: 10.1038/nn1103. [DOI] [PubMed] [Google Scholar]
- de Furtado MA, Braga GK, Oliveira JA, Del Vecchio F, et al. Behavioral, morphologic, and electroencephalographic evaluation of seizures induced by intrahippocampal microinjection of pilocarpine. Epilepsia. 2002;43(Suppl 5):37–39. doi: 10.1046/j.1528-1157.43.s.5.41.x. [DOI] [PubMed] [Google Scholar]
- Glien M, Brandt C, Potschka H, Loscher W. Effects of the novel antiepileptic drug levetiracetam on spontaneous recurrent seizures in the rat pilocarpine model of temporal lobe epilepsy. Epilepsia. 2002;43(4):350–357. doi: 10.1046/j.1528-1157.2002.18101.x. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons--1. Identification and characterization. Neuroscience. 1983;10(2):301–315. doi: 10.1016/0306-4522(83)90135-5. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: burst firing. Journal of Neuroscience. 1984;4(11):2877–2890. doi: 10.1523/JNEUROSCI.04-11-02877.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harden CL. Depression and anxiety in epilepsy patients. Epilepsy and Behavior. 2002;3(3):296. doi: 10.1016/s1525-5050(02)00018-5. [DOI] [PubMed] [Google Scholar]
- Heckers S. Neuroimaging studies of the hippocampus in schizophrenia. Hippocampus. 2001;11(5):520–528. doi: 10.1002/hipo.1068. [DOI] [PubMed] [Google Scholar]
- Hyland BI, Reynolds JN, Hay J, Perk CG, et al. Firing modes of midbrain dopamine cells in the freely moving rat. Neuroscience. 2002;114(2):475–492. doi: 10.1016/s0306-4522(02)00267-1. [DOI] [PubMed] [Google Scholar]
- Jones NC, Martin S, Megatia I, Hakami T, et al. A genetic epilepsy rat model displays endophenotypes of psychosis. Neurobiology of Disease. 2010;39(1):116–25. doi: 10.1016/j.nbd.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Kanner AM, Balabanov A. Depression and epilepsy: how closely related are they? Neurology. 2002;58(8 Suppl 5):S27–39. doi: 10.1212/wnl.58.8_suppl_5.s27. [DOI] [PubMed] [Google Scholar]
- Knopp A, Kivi A, Wozny C, Heinemann U, et al. Cellular and network properties of the subiculum in the pilocarpine model of temporal lobe epilepsy. Journal of Comparative Neurology. 2005;483(4):476–488. doi: 10.1002/cne.20460. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Wen X, Buckmaster PS. Reduced inhibition and increased output of layer II neurons in the medial entorhinal cortex in a model of temporal lobe epilepsy. Journal of Neuroscience. 2003;23(24):8471–8479. doi: 10.1523/JNEUROSCI.23-24-08471.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laruelle M, Abi-Dargham A. Dopamine as the wind of the psychotic fire: new evidence from brain imaging studies. Journal of Psychopharmacology. 1999;13(4):358–371. doi: 10.1177/026988119901300405. [DOI] [PubMed] [Google Scholar]
- Lawrie SM, Abukmeil SS. Brain abnormality in schizophrenia. A systematic and quantitative review of volumetric magnetic resonance imaging studies. British Journal of Psychiatry. 1998;172:110–120. doi: 10.1192/bjp.172.2.110. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Morris HM. Cell and receptor type-specific alterations in markers of GABA neurotransmission in the prefrontal cortex of subjects with schizophrenia. Neurotoxicity Research. 2008;14(2–3):237–248. doi: 10.1007/BF03033813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Behrens MM, Grace AA. A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. Journal of Neuroscience. 2009;29(8):2344–2354. doi: 10.1523/JNEUROSCI.5419-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. The hippocampus modulates dopamine neuron responsivity by regulating the intensity of phasic neuron activation. Neuropsychopharmacology. 2006a;31(7):1356–1361. doi: 10.1038/sj.npp.1300963. [DOI] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. The laterodorsal tegmentum is essential for burst firing of ventral tegmental area dopamine neurons. Proceedings of the National Academy of Sciences U S A. 2006b;103(13):5167–5172. doi: 10.1073/pnas.0510715103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. Journal of Neuroscience. 2007;27(42):11424–11430. doi: 10.1523/JNEUROSCI.2847-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Amphetamine activation of hippocampal drive of mesolimbic dopamine neurons: a mechanism of behavioral sensitization. Journal of Neuroscience. 2008;28(31):7876–7882. doi: 10.1523/JNEUROSCI.1582-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Leung LS. Schizophrenia-like behavioral changes after partial hippocampal kindling. Brain Research. 2004;997(1):111–118. doi: 10.1016/j.brainres.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Maier M, Ron MA, Barker GJ, Tofts PS. Proton magnetic resonance spectroscopy: an in vivo method of estimating hippocampal neuronal depletion in schizophrenia. Psychological Medicine. 1995;25(6):1201–1209. doi: 10.1017/s0033291700033171. [DOI] [PubMed] [Google Scholar]
- Mendez MF, Grau R, Doss RC, Taylor JL. Schizophrenia in epilepsy: seizure and psychosis variables. Neurology. 1993;43(6):1073–1077. doi: 10.1212/wnl.43.6.1073. [DOI] [PubMed] [Google Scholar]
- Muller CJ, Groticke I, Bankstahl M, Loscher W. Behavioral and cognitive alterations, spontaneous seizures, and neuropathology developing after a pilocarpine-induced status epilepticus in C57BL/6 mice. Experimental Neurology. 2009;219(1):284–297. doi: 10.1016/j.expneurol.2009.05.035. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. the rat brain in stereotaxic coordinates. Sydney Academic Australia; 1986. [Google Scholar]
- Pierce RC, Kalivas PW. A circuitry model of the expression of behavioral sensitization to amphetamine-like psychostimulants. Brain Research: Brain Research Reviews. 1997;25(2):192–216. doi: 10.1016/s0165-0173(97)00021-0. [DOI] [PubMed] [Google Scholar]
- Qin P, Xu H, Laursen TM, Vestergaard M, et al. Risk for schizophrenia and schizophrenia-like psychosis among patients with epilepsy: population based cohort study. BMJ. 2005;331(7507):23. doi: 10.1136/bmj.38488.462037.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbersweig DA, Stern E, Frith C, Cahill C, et al. A functional neuroanatomy of hallucinations in schizophrenia. Nature. 1995;378(6553):176–179. doi: 10.1038/378176a0. [DOI] [PubMed] [Google Scholar]
- Raedt R, Van Dycke A, Van Melkebeke D, De Smedt T, et al. Seizures in the intrahippocampal kainic acid epilepsy model: characterization using long-term video-EEG monitoring in the rat. Acta Neurologica Scandinavia. 2009;119(5):293–303. doi: 10.1111/j.1600-0404.2008.01108.x. [DOI] [PubMed] [Google Scholar]
- Slater E, Beard AW, Glithero E. The schizophrenialike psychoses of epilepsy. British Journal of Psychaitry. 1963;109:95–150. doi: 10.1192/bjp.109.458.95. [DOI] [PubMed] [Google Scholar]
- Spencer SS, Spencer DD. Entorhinal-hippocampal interactions in medial temporal lobe epilepsy. Epilepsia. 1994;35(4):721–727. doi: 10.1111/j.1528-1157.1994.tb02502.x. [DOI] [PubMed] [Google Scholar]
- Stevens JR. Epilepsy, psychosis and schizophrenia. Schizophrenia Research. 1988;1(1):79–89. doi: 10.1016/0920-9964(88)90044-8. [DOI] [PubMed] [Google Scholar]
- Szyndler J, Wierzba-Bobrowicz T, Skorzewska A, Maciejak P, et al. Behavioral, biochemical and histological studies in a model of pilocarpine-induced spontaneous recurrent seizures. Pharmacology, Biochemistry and Behavior. 2005;81(1):15–23. doi: 10.1016/j.pbb.2005.01.020. [DOI] [PubMed] [Google Scholar]
- Tamminga CA, Thaker GK, Buchanan R, Kirkpatrick B, et al. Limbic system abnormalities identified in schizophrenia using positron emission tomography with fluorodeoxyglucose and neocortical alterations with deficit syndrome. Archives of General Psychiatryy. 1992;49(7):522–530. doi: 10.1001/archpsyc.1992.01820070016003. [DOI] [PubMed] [Google Scholar]
- Trimble MR. Epilepsy and behaviour. Epilepsy Research. 1991;10(1):71–79. doi: 10.1016/0920-1211(91)90097-y. [DOI] [PubMed] [Google Scholar]
- Vanderschuren LJ, Schmidt ED, De Vries TJ, Van Moorsel CA, et al. A single exposure to amphetamine is sufficient to induce long-term behavioral, neuroendocrine, and neurochemical sensitization in rats. Journal of Neuroscience. 1999;19(21):9579–86. doi: 10.1523/JNEUROSCI.19-21-09579.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Geschwind N. Hypergraphia in temporal lobe epilepsy. Neurology. 1974;24(7):629–636. doi: 10.1212/wnl.24.7.629. [DOI] [PubMed] [Google Scholar]
- White FJ, Wang RY. Electrophysiological evidence for A10 dopamine autoreceptor subsensitivity following chronic D-amphetamine treatment. Brain Research. 1984;309(2):283–292. doi: 10.1016/0006-8993(84)90594-8. [DOI] [PubMed] [Google Scholar]
- Witter MP, Van Hoesen GW, Amaral DG. Topographical organization of the entorhinal projection to the dentate gyrus of the monkey. Journal of Neuroscience. 1989;9(1):216–228. doi: 10.1523/JNEUROSCI.09-01-00216.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozny C, Gabriel S, Jandova K, Schulze K, et al. Entorhinal cortex entrains epileptiform activity in CA1 in pilocarpine-treated rats. Neurobioloby of Disease. 2005;19(3):451–460. doi: 10.1016/j.nbd.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Xia YF, He L, Whistler JL, Hjelmstad GO. Acute amphetamine exposure selectively desensitizes kappa-opioid receptors in the nucleus accumbens. Neuropsychopharmacology. 2008;33(4):892–900. doi: 10.1038/sj.npp.1301463. [DOI] [PMC free article] [PubMed] [Google Scholar]