

Abstract
Aim
Two unexplored aspects for irinotecan and cisplatin (I&C) combination chemotherapy are (1) actively targeting both drugs to a specific diseased cell type and (2) delivering both drugs on the same vehicle to ensure their synchronized entry into the cell at a well-defined ratio. In this work we report the use of targeted polymeric nanoparticles (NPs) to co-encapsulate and deliver I&C to cancer cells expressing the Prostate Specific Membrane Antigen (PSMA).
Method
We prepared targeted NPs in a single-step by mixing four different precursors inside microfluidic devices.
Results
I&C were encapsulated in 55-nm NPs and showed an 8-fold increase in internalization by PSMA-expressing LNCaP cells compared to non-targeted NPs. NPs co-encapsulating both drugs exhibited strong synergism in LNCaP cells with a combination index of 0.2.
Conclusion
The strategy of co-encapsulating both irinotecan and cisplatin in a single NP targeted to a specific cell type could potentially be used to treat different types of cancer.
Keywords: Nanoparticles, Combination Chemotherapy, Cisplatin, Irinotecan, PSMA
INTRODUCTION
Combination chemotherapy has become a cornerstone in treating diverse types of cancer, having proved to be more effective than single agents against both metastatic cancer and in patients at high risk of relapse after primary surgical treatment [1]. Several combination regimens have been approved and adopted in the clinic with the aim of enhancing therapeutic efficacy, decreasing dosage at equal or increased levels of efficacy, and reducing drug resistance [2]. The use of multiple drugs with different molecular targets can raise the molecular barriers that need to be overcome for cancer cell resistance, thereby more effectively mitigating the cancer cell survival. This line of reasoning has led to the development of curative combination chemotherapy regimens (whereas single agent therapy was not) for germ cell tumors, lymphomas and leukemias. In addition, the application of multiple drugs targeting the same cellular pathways can function synergistically for higher therapeutic efficacy and target selectivity [3].
Although the benefits of combination chemotherapy are very attractive, the reality is that for the vast majority of drug combinations still suffer from several disadvantages, including (1) dissimilar pharmacokinetics and tissue distribution of each drug arising from differences in their disparate physicochemical properties, and (2) more serious side effects because each drug is likely to a have different toxicity profile. These disadvantages make dosing and scheduling of each component of the combination a challenging task. As a consequence, special efforts from the scientific and medical communities have been focused on developing strategies to overcome the disadvantages of combination chemotherapy while retaining the benefits to realize its full potential— an example being the introduction into the clinic of the first liposomal formulations containing combinations of different chemotherapeutic agents by Celator Pharamceuticals [4].
Cisplatin and irinotecan are two established drugs routinely used for treating various types of cancer. Cisplatin is a platinum-based drug that binds to and forms adducts with DNA, ultimately triggering apoptosis [5–7]. Irinotecan is semisynthetic drug of camptothecin that inhibits topoisomerase I, ultimately blocking both DNA replication and transcription [8]. The combination of irinotecan and cisplatin (I&C) displays synergy or supra-additive effects when exposed to cultured human tumor cells, human xenograft tumor models and cancer cells freshly isolated from colorectal patients [9]. In addition, an I&C combination in a Japanese based randomized phase III trial in 2002 showed the regimen resulted in increased overall survival in small cell lung cancer (SCLC) patients when compared to historical controls treated with the conventional regimen of etoposide and cisplatin (E&P) [10]. This study was stopped early due to the positive results after an interim analysis of only 154 patients. In 2006, a phase III trial showed that I&C combination (given in a slightly modified schedule with attenuated cisplatin doses to mitigate toxicities) showed this regimen was as effective as E&P in SCLC [11] and was associated with less hematological toxicities but more diarrhea. As such a formulation which can provide the same dose intensity as the Japanese study with less toxicity has great potential to show improved efficacy compared with standard dose E&P. Recently, a preclinical study showed that not only was the combination of irinotecan and cisplatin important for achieving enhanced cytotoxicity, but the ratio in which they were administered also played a key role in determining therapeutic efficacy [9]. An optimal ratio of drugs resulted in greater synergy and that ratio varied depending on cell type [9]. Currently, two important aspects that need to be investigated for I&C combination are (i) actively targeting the drug combination to a specific diseased cell type and (ii) delivering both drugs on the same delivery vehicle to create an over-lapping pharmacological profile mediated by the controlled release properties of the vehicle, while ensuring their entry into the cell at a well-defined ratio. Because targeting combined with synchronized and sustained delivery may potentiate the synergistic toxicity of I&C, an enabling technology is needed to test this hypothesis.
Targeted polymeric nanoparticles (NPs) can encapsulate, traffic, and deliver chemotherapeutic agents in a controlled fashion to specific diseased tissues, thereby providing a tremendous potential in the fight against cancer [12]. In different animal models and in humans, such NPs have demonstrated improved drug pharmacokinetics, biodistribution, cell- or tissue-specific targeting, and tolerability, resulting in an increase of the therapeutic index of several drugs known to have limited efficacy and/or high toxicity [13–15]. These advantages have driven researchers to implement targeted NPs for the co-encapsulation and co-delivery of two or more drugs in a single NP in order to match the pharmacokinetics and biodistribution of drugs having different chemical and physical properties [16]. Recently, we reported the co-encapsulation of cisplatin and docetaxel, two drugs with quite different chemical properties, in PSMA-targeted polymeric NPs and demonstrated controlled release of both drugs to PSMA-expressing prostate cancer cells [17]. This task was accomplished by conjugating a hydrophilic cisplatin prodrug to the backbone of polylactic acid (PLA) to facilitate its incorporation into the hydrophobic NP core [17]. Extension of this technology to the co-encapsulation of cisplatin and irinotecan presents an attractive option for targeting I&C therapy with controlled and synchronized delivery of each drug for improved synergism.
In the present work we report poly(D,L-lactide–co–glycolide)-co-poly(ethylene glycol) (PLGA-PEG)-based targeted NPs with both irinotecan and cisplatin encapsulated in the same targeted NP. Incorporation of cisplatin into the NP core was accomplished by conjugating a cisplatin prodrug to the backbone of PLA (PLA-cisplatin) while irinotecan was encapsulated during the self-assembly process of the NP. These NPs were targeted to prostate cancer cells over-expressing PSMA receptor using the small molecule S,S-2-[3-[5-amino-1-carboxypentyl]-ureido]-pentanedioic acid (PSMA Ligand; LIG), previously reported to bind to PSMA receptors and currently in clinical development as a component of a cancer-imaging agent [18, 19]. The targeted NPs were prepared in a single step by conjugating LIG to the end of PEG in PLGA-PEG (PLGA-PEG-LIG), then mixing it with unmodified PLGA-PEG, PLA-cisplatin, and free irinotecan followed by self-assembly through the nanoprecipitation method in microfluidic devices [20, 21]. Targeted NPs were characterized with respect to size and drug encapsulation, and selective uptake by PSMA-expressing cells was evaluated. Synergistic performance of the dual drug targeted NPs in PSMA-expressing LNCaP cells was determined at a fixed drug ratio by comparing the cytotoxicity of dual drug targeted NPs to that of NPs encapsulating the same amount of single drugs. Finally, the combination index (CI) was used to quantify the extent of synergism under such conditions.
MATERIALS AND METHODS
Materials
Poly(D,L-lactide–co–glycolide)-co-Poly(ethylene glycol) was purchased from Boehringer Ingelheim (Ingelheim am Rhein, Germany). Poly(D,L-lactide–co–glycolide) (50/50) with terminal carboxylate groups (PLGA, inherent viscosity 0.67 dL/g, MW ~ 45 kDa) was purchased from Lactel (Pelham, AL, USA) and tBOC-NH-PEG-NH2 (MW 5000) and tBOC-NH-PEG-NHS, (MW 5000) were purchased from Laysan Bio, Inc (Arab, AL, USA). MTT reagent was procured from Sigma-Aldrich (St. Louis, MO, USA). LNCaP cell line was obtained from American Type Culture Collection (Manassas, VA, USA).
Synthesis of PLGA-PEG-LIG
The synthesis of PLGA-PEG-LIG (PLGA MW 45K and PEG MW 5K) was achieved by first conjugating LIG to PEG followed by conjugation of the resultant LIG-PEG to PLGA. LIG (3.9 mg, 8.9 μmol) was dissolved in 400 μL of dimethylformamide (DMF) and allowed to react with tBOC-NH-PEG-NHS (22 mg, 4.5 μmol) in the presence of N,N-diisopropylethylamine (DIEA, 10 μL) for 12 h. The reaction product was dialyzed for 24 h in water to remove unreacted LIG, then lyophilized, and finally resuspended in 400 μL of trifluoroacetic acid (TFA) to remove tBOC. After 4 h, PEG-LIG was dried and dissolved in 200 μL of DMSO. In parallel, PLGA-COOH (100 mg, 2.2 μmoL) was allowed to react with N-hydroxysuccinimide (NHS) in the presence of 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide (EDC) in dichloromethane (DCM) for 2 h. The resulting PLGA-NHS was dried and dissolved in 300 μL of DMSO. Finally, PEG-LIG and PLGA-NHS in DMSO were mixed with DIEA, allowed to react for 24 h, precipitated in cold methanol, and dried under vacuum. PLGA-PEG-LIG was characterized by gel permeation chromatography (GPC) to confirm the synthesis of PLGA-PEG-LIG.
Synthesis of PLA-Cisplatin
The synthesis of PLA-cisplatin was accomplished as previously described [17]. First, PLA-OBn was synthesized via ring opening polymerization reaction by dissolving a toluene-functionalized cyclic lactide monomer and simple lactide monomer in a toluene solution containing benzyl alcohol (initiator) and Tin catalyst. The solution was refluxed at 120 °C for 10 h. Then, the hydroxyl functionalized biodegradable polylactide polymer (PLA-OH) was obtained by benzyl deprotection using a Pd/C catalyst at 50 psi pressure for 8 h. Finally, succinic acid-derivative platinum (IV) (cisplatin prodrug) was dissolved in anhydrous DMF and added with excess equivalents of N-hydroxybenzotriazole (HOBt), N,N′-dicyclohexylcarbodiimide (DCC) and stirred at room temperature for 30 min. To this reaction mixture a solution of PLA-OH polymer in dichloromethane was added and stirred at room temperature for 24 hours, followed by precipitation in diethyl ether.
Synthesis and Characterization of NPs
NP synthesis was carried out in microfluidic devices made of PDMS as previously reported [21, 22]. The device had one inlet each for water and precursors streams, and one outlet. The water stream was split into two in order to achieve two water streams at the flow focusing junction. The mixing channel was 20 μm wide, 60μm high and 1 cm long and formed nanoparticles by focusing in two dimensions as described earlier [21]. The 2.5 mL and 0.5 mL gastight syringes used for aqueous and organic streams, respectively, were mounted on syringe pumps to control flow through the device. NPs were prepared by nanoprecipitation. Briefly, 250 μL of PLGA-PEG at 10 mg/mL in acetonitrile was mixed with 250 μL of PLGA-PEG-LIG and 500 μL of PLA-cisplatin both at 10 mg/mL in acetonitrile. Irinotecan was mixed with the polymers to reach an initial drug loading of 5%, corresponding to a concentration of 0.5 mg/mL. The organic stream (polymers and drugs) was run at 5 μL/min while maintaining a total aqueous flow rate of 50μL/min. NPs were collected at the outlet stream and washed three times with water using Amicon centrifugation filtration membrane to remove excess drugs and organic solvents. Dynamic light scattering (DLS) was used to determine particle size and size distribution, using a Zetasizer Nano ZS instrument (Malvern Instruments Ltd., U.K.). Particle visualization was carried out through TEM (JEOL 2011 instrument at an acceleration voltage of 200 kV.) The sample was prepared by depositing 10 μL of the NP solution onto a 300-mesh carbon-coated copper grid. After 30 min incubation samples were blotted away and grids were negatively stained for 20 min with 1% (w/v) uranyl acetate aqueous solution. The stability of NPs was evaluated by suspending freshly made NPs in a PBS solution containing 10% serum and measuring the size of the particles at different time points over a period of 24 h. PLGA NPs were used a control since they are known to aggregate over time [20]. Drug loading and encapsulation efficiency (EE) were determined by quantifying the amount of drug in NP. To evaluate drug loading, NPs were dissolved in a 50/50 acetonitrile/water solution immediately after synthesis and vortexed for several hours to induce NP dissociation. The amount of platinum inside the NPs was quantitated by flameless atomic absorption spectroscopy (AAS), from which the load of cisplatin was computed. Irinotecan was detected using spectrophotometry on a TECAN 1000 plate reader at a wavelength of 385 nm. A calibration curve with known concentrations of irinotecan was prepared, and the amount of irinotecan encapsulated in the NPs was calculated accordingly. In vitro release kinetics of irinotecan and cisplatin were determined by dialyzing 100 μL aliquots of NPs in mini dialysis cassettes with a MW cutoff of 10 kDa against a 4-liter bath of PBS at 37°C. Three aliquots were removed at different time points over a period of 72 hours and the amount of irinotecan and cisplatin remaining inside the NPs were measured as described above.
NP Uptake by LNCaP cells
LNCaP cells were cultured in RPMI 1640 medium with 10% fetal bovine serum, 50 units/mL of penicillin, and 50 μg/mL of streptomycin. Cells were seeded at a density of 50,000 cells per well on a 24-well plate for 24 h. Subsequently, the cell medium was replaced with NPs dissolved in the same medium at a concentration of 1 mg/mL and incubated for 4 h followed by three washes with 1% BSA solution in PBS to remove excess NPs. LNCaP cells were treated with trypsin, removed from the plate, centrifuged, and were reconstituted in 250 μL of PBS. Immediately after cells were analyzed through flow cytometry analysis on a BD Biosciences LSR II with high-throughput sampler (HTS) option in a 96-well plate, with 10,000 cells collected for each measurement (n = 4 per formulation).
In vitro NP Cytotoxicity (MTT Assay)
The cytotoxicity of NPs was investigated through the MTT assay. First, cells were seeded on a 96-well plate in 100 μL of cell medium and incubated for 24 h. Then, the medium was replaced with NP suspension at varying concentrations and incubated for 12 h at 37 °C. The medium was changed after 12 h, and the cells were incubated for 48 h. Subsequently, 20 μL of MTT (5 mg mL in PBS) were added to the cells and allowed for incubation for 5 h. After removing the medium the cells were lysed by adding 100 μL of DMSO, and the absorbance of the purple formazan was recorded at 550 nm using plate reader.
RESULTS AND DISCUSSIONS
The binding, uptake and synergistic mechanism of cellular cytotoxicity of targeted I&C NPs is schematically presented (Figure 1). NPs comprise of a PLGA core in which both drugs are incorporated and a PEG shell modified with LIG molecules that target PSMA. As NPs reach the cell surface, multivalent interactions between LIG and the extracellular domain of over-expressed PSMA receptors result in endocytosis [23]. Through competition assays and microscopy, previous studies have shown that endocytosis of LIG-targeted NPs is inhibited in the presence of free LIG and that LIG- (and LIG analogs) targeted NPs localized both in the cytoplasm and cell nucleus after being internalized [14, 23–25]. Once inside, NPs release both drugs in a controlled and sustained fashion [17]. In the nucleus, irinotecan inhibits DNA-topoisomerase I complexes, while cisplatin forms DNA cross-links. The end result of both events is cell cycle arrest, transcription inhibition [7], and apoptosis [28]. Several studies have suggested that irinotecan acts synergistically with cisplatin by inducing an increase in the lifetime of Pt-DNA adducts resulting in enhanced cytotoxicity [28]. However, the exact mechanism has not yet been determined.
Figure 1.
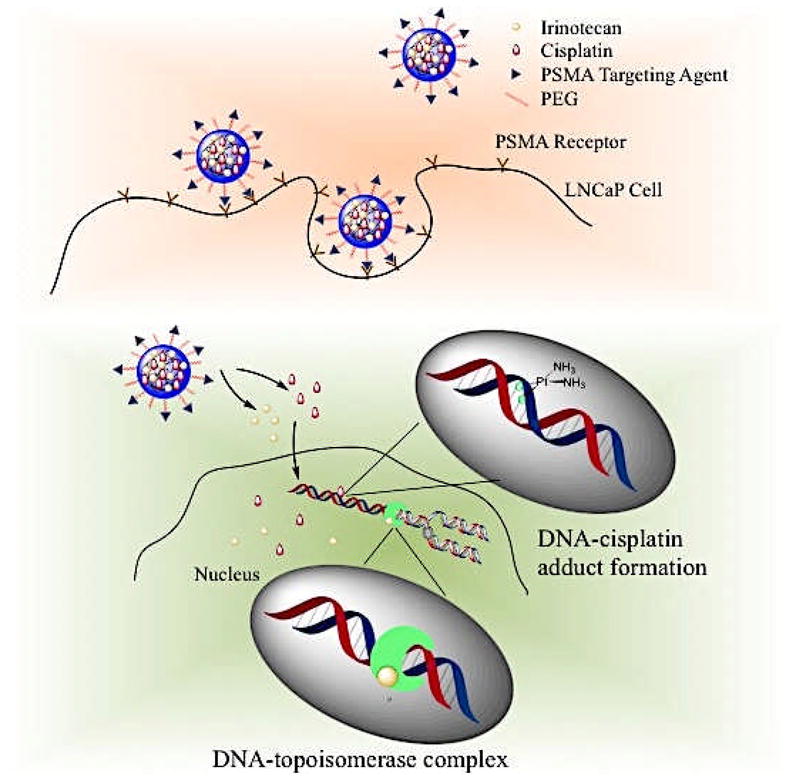
Schematic showing the strategy of co-encapsulation of irinotecan and cisplatin in a single nanoparticle targeted to PSMA receptors in prostate cancer cells. Once internalized, NPs co-deliver irinotecan and cisplatin to enable synergistic toxicity.
To minimize the number of steps in the synthesis of targeted NPs, we prepared a ligand-conjugated PLGA-PEG copolymer (Figure 2A). LIG is a urea-based small molecule that is not subject to supply, stability and analytical challenges associated with macromolecule-based targeting ligands, such as aptamers or antibodies [27]. It contains a free primary amine that can be used to anchor LIG to the NP surface without affecting the targeting capabilities of the molecule. LIG was allowed to react with succinimide-modified tBOC-NH-PEG-NHS, which after deprotection was conjugated to a succinimide-modified PLGA. The functionalized PLGA-PEG-LIG contains necessary components for forming and targeting the NPs, enabling their single step self-assembly and simplifying the optimization and potential scale up [28]. To co-deliver irinotecan and cisplatin, two therapeutic agents with different chemical properties (hydrophobic and hydrophilic, respectively), we synthesized a modified PLA polymer with reactive hydroxyl functional groups to enable conjugation of a platinum prodrug to the polymer backbone (Figure 2B). Previous work demonstrated that this platinum prodrug can be cleaved from the backbone of the polymer and reduced to cisplatin intracellularly, which then forms the cis-diammineplatinum(II) 1,2-d(GpG) cross-links on nuclear DNA, the principal adduct made by cisplatin [17, 29]. AAS of PLA-cisplatin confirmed that ~2.5% by weight of platinum was conjugated to the polymer.
Figure 2.

(A) Synthesis of PLGA-PEG-LIG. (B) Synthesis of PLA-cisplatin
The targeted NPs presented here comprise four different components that self-assemble in a single mixing step into a drug-loaded core-shell nanostructure containing targeting moieties on the surface. These components are: (i) PLGA-PEG block copolymer that forms the core-shell structure, (ii) PLGA-PEG-LIG that self-assembles together with PLGA-PEG and orients LIG toward the NP surface, (iii) PLA-cisplatin prodrug that assembles in the hydrophobic NP core by hydrophobic-hydrophobic interactions of PLGA and PLA, (iv) irinotecan, which partitions into the hydrophobic core based on its physicochemical properties. NPs were prepared in microfluidic channels by a rapid mixing strategy called hydrodynamic flow focusing (HFF) [21, 30] (Figure 3A). In this method, polymers and drugs are mixed in acetonitrile and run in a stream that becomes horizontally focused into a very thin stream when it encounters two streams of water running at a flow rate 10 times higher (Figure 3B). Upon focusing, nanoprecipitation occurs through mixing of the aqueous and organic streams inducing NP self-assembly. This method ensures controlled precipitation and rapid mixing, which results in reproducible, monodisperse NPs with smaller size and higher drug loading than NPs prepared through conventional bulk methods [21]. Figure 3C shows the characterization of the NPs by DLS, resulting in an average size of 55 nm and a polydispersity of 0.04, indicative of relatively monodisperse particles. A TEM image of these NPs reveals an average size of ~55 nm, which matches that obtained from DLS (Figure 3D). By assuming both an average NP density of 1.27 g/mL [31] and that all PLGA-PEG-LIG self-assembles into a NP with LIG molecules on the NP surface [28], we compute that NPs with average diameter of 55 nm and comprising 25% by weight of PLGA-PEG-LIG will have an average of 330 LIG molecules per NP.
Figure 3.
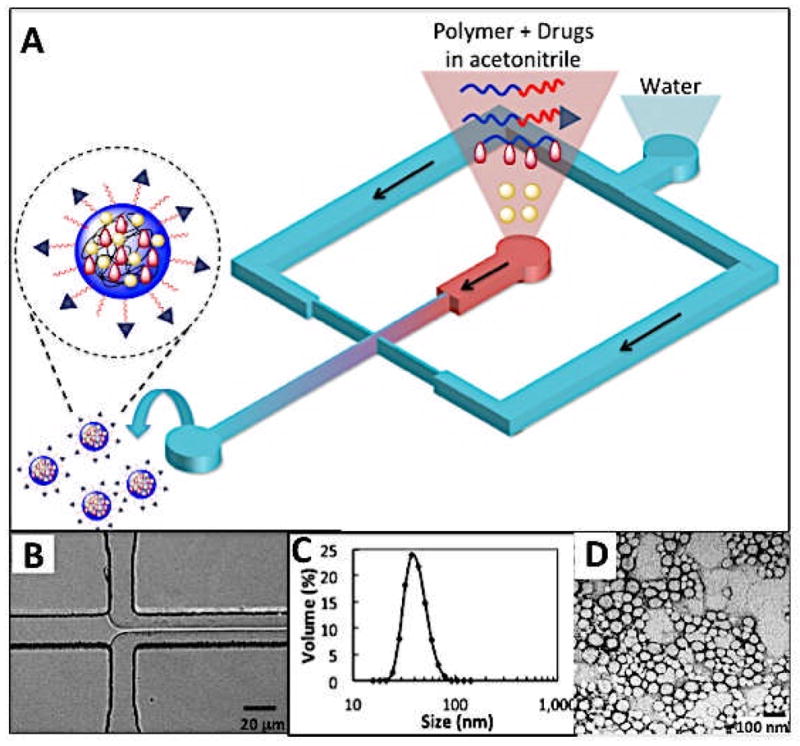
(A) Single-step synthesis of targeted nanoparticles in microfluidic devices using hydrodynamic flow focusing. (B) Visualization of flow focusing in a microfluidic channel during operation. Acetonitrile flow rate = 5 μL/min and water flow rate = 50 μL/min. (C) Size distribution of targeted nanoparticles by volume. Average size = 55 nm. Polydispersity index = 0.04. (D) TEM image of nanoparticles stained with 1% solution of uranyl acetate. Average size = 55 nm.
To evaluate the effect on NP size of incorporating multiple drugs, we measured the size and stability of NPs encapsulating only irinotecan (NPI), only cisplatin (NPC), and both irinotecan and cisplatin (NPI&C) (Figure 4A and 4B). In addition, we determined the drug loading, encapsulation efficiency (EE), and release kinetics of each drug in the NPs (Figure 4C and D). Interestingly, the size of all three NPs was similar with average diameter of approximately 55 ± 4 nm, which remained stable for a period of 24 h. This consistency in NP size is presumably a consequence of the rapid mixing environment enabled by the microfluidic devices, which ensures complete mixing of precursors in water at a time scale smaller than the NP self-assembly and results in the formation of a relatively uniform population of NPs [32]. Cisplatin was encapsulated with very high efficiency (>80%) owing to the hydrophobic-hydrophobic interaction between PLGA and the PLA backbone. Irinotecan exhibited EE and drug loading values of 10% and 0.5%, respectively, after self-assembly with PLGA-PEG. However, when irinotecan was mixed with both PLGA-PEG and PLA-cisplatin, its EE and drug loading increased to 44% and 2.2%, respectively (Figure 4C). This increase may be a consequence of the increased hydrophobicity of the NP core due to the hydrophobic PLA polymer that favors encapsulation of the hydrophobic irinotecan (as shown previously for a similar system of free PLGA and docetaxel [21]). This formulation resulted in an irinotecan to cisplatin mol ratio of 1.5:1 and was used for the all experiments in this study. From the kinetics it was found that encapsulated irinotecan was released at a faster rate than covalently conjugated cisplatin prodrug, which is expected and agrees well with a previous report on a similar system [17]. Through this NP platform the encapsulation and release rate of irinotecan or other encapsulated drugs can be modulated by varying the molecular weight of the PLGA in PLGA-PEG, the concentration of PLGA-PEG in the organic solution and the initial loading wt.% of irinotecan [20, 33], while for cisplatin variation of the MW of PLA in PLA-cisplatin, and the number of cisplatin molecules in PLA-cisplatin allow for controlling its encapsulation and release. Considering different types of cancer may benefit from different drug ratios and release rates [9], the ability to control the encapsulation and release rate is an advantage that would potentially allows for tuning the payload according to the therapeutic need of each type of cancer.
Figure 4.
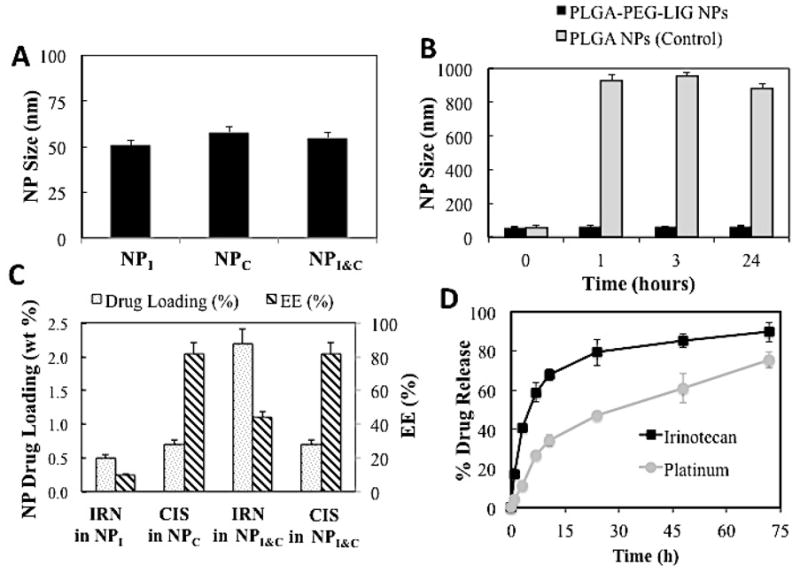
Characterization of nanoparticles containing either irinotecan (NPI) or cisplatin (NPC) prodrug or both (NPI&C). (A) Size (B) Size stability over time in 10% serum (PLGA NPs were used as a positive control), (C) Drug loading and encapsulation efficiency, and (D) drug release kinetics in PBS at 37°C.
Next we tested the targeting capabilities of the PSMA-targeted NPs in vitro. To enable analysis of NP uptake by flow cytometry, 2.5% by weight of the fluorescent probe PLGA-Alexa 488 was added to the organic stream containing both drugs and polymers. In a previous work we showed that this amount was low enough so as not to affect the NP physicochemical properties but high enough to be detected readily by FACS at very low NP concentrations [28]. To test the targeting capabilities of our NPs, we incubated them with PSMA-overexpressing LNCaP cells for 4 h, trypsinized them to remove any bound NPs on the cell surface, and measured the fluorescence intensity of 10,000 cells (Figure 5A). Cells treated with PSMA-targeted NPs exhibited 8-fold more fluorescence compared to those treated with non-targeted NPs. FACS fluorescence histograms show that NP uptake by cells occurred uniformly across all the cells rather than by a few cells engulfing most of NPs (Figure 5B). Previous studies have demonstrated that LIG- (and LIG analogs) targeted nanoparticles enter the cell through endocytosis and localize both in the cytoplasm and cell nucleus [23–25]. It is therefore likely that these NPs are escaping the endosome and reaching other cell compartments, including nucleus. The exact mechanism will be explored in future studies. These results indicate that our dual-drug targeted NPs can selectively target cells over-expressing PSMA receptors and that they can enter the cells more readily than non-targeted NPs.
Figure 5.
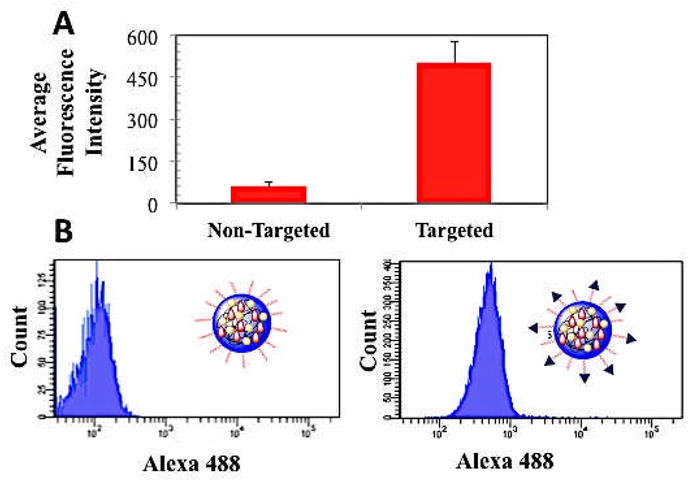
Uptake of PSMA-targeted NPs in LNCaP cells overexpressing PSMA receptors compared to non-targeted NPs. The NPs contain the fluorescent reporter Alexa 488 conjugated to PLGA (PLGA-Alexa 488). (A) Average fluorescence of 10,000 cells obtained by FACS. (B) FACS histograms of fluorescence intensity.
Finally, we carried out cytotoxicity studies in vitro to assess the synergy of cisplatin and irinotecan in LNCaP cells. Targeted NPI, NPC, and NPI&C were exposed to LNCaP cells at different concentrations while keeping the same incubation volumes. The cytotoxicities were evaluated using 3-(4,5-dimethlylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Based on the concentration of drugs in each NP we created dose-response curves and determined the IC50 values for each NP formulation (Figure 6A). NPI&C was 3.6 times and 10.6 more toxic than NPC, and NPI, respectively, after 12 h of exposure to LNCaP cells. Whereas these results suggest that NPI&C is more cytotoxic than the single-drug NPs, it does not assess whether this effect is synergistic or simply additive. To determine synergism of the dual-drug NPs, we used the Talay and Chou method [34] and calculated a combination index (CI) at ED80, as previously reported by others for a similar system [9]. In this method a CI ~1 indicates additivity, CI >1 indicates an antagonism and CI <1 indicates synergism. Remarkably, NPI&C had a CI of 0.20, which falls in the range of strong synergism [34]. These results demonstrate that PSMA-targeted NPI&C exhibit synergistic cytotoxicity against prostate cancer cells under the investigated experimental conditions. All previous work on cisplatin/irinotecan combinations was carried out with either free drugs or each drug encapsulated in a separate liposome, the present investigation being the first for this combination that has been evaluated (1) by encapsulating both agents in the same vehicle and (2) with a nanoparticle targeted to a specific cell type. Finally, although the initial drug ratio of irinotecan/cisplatin in the targeted NP was fixed at 1.5, differences in release kinetics of each drug may alter the actual ratio that reaches the cell nucleus. In fact, investigating the performance of targeted NPs containing different initial drug ratios and/or varying drug release kinetics could potentially result in finding formulations with even greater synergism.
Figure 6.

Cytotoxicity of I&C targeted NPs (NPI&C) in LNCaP cells compared irinotecan targeted NPs (NPI) (A) and cisplatin targeted NPs (NPC) (B). Dual-drug NPs had an IC50 10.6 times and 3.6 times lower than cisplatin and irinotecan NPs, respectively. (C) Combination index determined by Talay and Chou method at ED80. According to the metric 0.20 corresponds to strong synergism. The NPs used had a final irinotecan to cisplatin ratio of 1.5:1 by mole.
SUMMARY AND CONCLUSIONS
In summary, we have devised a novel strategy for trafficking and delivering irinotecan and cisplatin to a specific cell population by encapsulating both drugs in one NP and targeting the NPs to specific cells with a small molecule targeting agent. Although the two drugs have different chemical and physical properties, they were successfully incorporated into the same NP by conjugating the more hydrophilic drug, cisplatin, to the backbone of a PLA-based polymer and encapsulating the more hydrophobic irinotecan in a conventional fashion through nanoprecipitation. NPs prepared with the use of a single-step in microfluidic device exhibited an average size of 55 ± 4 nm that remained essentially unchanged before and after the addition of both drugs. The low polydispersity index of ~0.04 is indicative of a relatively monodisperse population, presumably arising from the rapid mixing environment offered by the microfluidic devices. Specific uptake of targeted NPs by LNCaP cells overexpressing the PSMA receptor was demonstrated by the 8-fold increase in fluorescence associated with targeted NPs compared to non-targeted NPs. Finally synergistic cytotoxicity of irinotecan-cisplatin targeted NPs in LNCaP cells was assessed by a CI of 0.20, which is characteristic of strong synergism. From these results we anticipate that by implementing a two-drugs-in-one-NP strategy together with active targeting to specific cell receptors, combination chemotherapy with irinotecan and cisplatin could potentially be implemented, even in cancers that have traditionally exhibited poor therapy response, such as prostate cancer [35]. In addition, a single-step synthesis of NPs composed of approved drugs and clinically validated biomaterials may accelerate translation of such novel therapeutics to the clinic.
FUTURE PERSPECTIVE
Targeted NPs encapsulating both cisplatin and irinotecan have the potential to enhance the effect of I&C regime for different cancer types. These studies yielded promising results in vitro, and in vivo validation needs to be performed as a next step. Specifically, comparisons of administration of NPs co-encapsulating both drugs, NPs with the drugs encapsulated separately, and free drugs will be important for validating this system. In such experiments, the different in vivo barriers commonly encountered by NPs and small molecule drugs would be monitored. In addition, the present studies were performed at a fixed irinotecan:cisplatin ratio of 1.5:1 (each released at different rate); it is therefore possible that enhanced synergism may occur with targeted NPs having different drug ratios or exhibiting different release rates, both of which ultimately affect the amount of drug that reach the cell nucleus at a given time. The present platform technology could be implemented for different drug combinations with varying physicochemical properties, with the potential of increasing efficacy and reducing toxicity, especially for combinations in which the toxicity profile has resulted in limited or no use.
EXECUTIVE SUMMARY.
Our aim was to develop a targeted polymeric nanoparticle (NP) that can co-encapsulate, traffic, and deliver irinotecan and cisplatin (I&C) in a controlled and synchronized fashion.
We prepared targeted NPs in a single step by mixing the biodegradable polymer PLGA-PEG conjugated to the targeting moiety LIG (PLGA-PEG-LIG), plain PLGA-PEG, PLA-cisplatin, and irinotecan in acetonitrile followed by self-assembly through nanoprecipitation. LIG is a small molecule that binds strongly to PSMA receptors overexpressed in prostate cancer cells as well as the neovasculature of several tumors.
Using PSMA-expressing LNCaP cells, we demonstrated enhanced uptake of targeted NPs compared to non-targeted NPs, and strong synergistic cytotoxicity of irinotecan and cisplatin compared to NPs containing either irinotecan or cisplatin.
This approach could result in the potential use of I&C regime in prostate cancer combination chemotherapy and could be extended to other cancers where I&C has shown to demonstrate efficacy such as small cell lung cancer and colon cancer.
Footnotes
FINANCIAL DISCLSURE
O.C.F and R.L have financial interests in BIND Biosciences and Selecta Biosciences. O.C.F., R.L. and S.J.L. have financial interests in Blend Therapeutics. We thank the Koch Institute Flow Cytometry Core at MIT for the use of FACS and the Center for Material Science and Engineering (CMSE) imaging facility for the electron microscopy image acquisition. This research was supported by the Koch-Prostate Cancer Foundation Award in Nanotherapeutics (R.L. and O.C.F.) and by the NCI Center of Cancer Nanotechnology Excellence at MIT-Harvard (U54-CA151884). P.M.V. is supported by NSF graduate research fellowship. E.MP. is supported by a NDSEG graduate research fellowship. No writing assistance was utilized in the production of this manuscript.
References
*of interest
**of considerable interest
- 1.Chabner BA, Roberts TG., Jr Timeline: Chemotherapy and the war on cancer. Nat Rev Cancer. 2005;5(1):65–72. doi: 10.1038/nrc1529. [DOI] [PubMed] [Google Scholar]
- 2.Jia J, Zhu F, Ma X, Cao Z, Li Y, Chen YZ. Mechanisms of drug combinations: interaction and network perspectives. Nat Rev Drug Discov. 2009;8(2):111–128. doi: 10.1038/nrd2683. [DOI] [PubMed] [Google Scholar]
- 3.Lehar J, Krueger AS, Avery W, et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol. 2009;27(7):659–666. doi: 10.1038/nbt.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayer LD, Harasym TO, Tardi PG, et al. Ratiometric dosing of anticancer drug combinations: controlling drug ratios after systemic administration regulates therapeutic activity in tumor-bearing mice. Mol Cancer Ther. 2006;5(7):1854–1863. doi: 10.1158/1535-7163.MCT-06-0118. [DOI] [PubMed] [Google Scholar]
- 5.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7(8):573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 6.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4(4):307–320. doi: 10.1038/nrd1691. Key publication where it is demonstrated that a specific drug combination of irinotecan and cisplatin could be antagonistic, additive or synergistic depending on the administered drug ratio. [DOI] [PubMed] [Google Scholar]
- 7.Jung Y, Lippard SJ. Direct cellular responses to platinum-induced DNA damage. Chem Rev. 2007;107(5):1387–1407. doi: 10.1021/cr068207j. [DOI] [PubMed] [Google Scholar]
- 8.Garcia-Carbonero R, Supko JG. Current perspectives on the clinical experience, pharmacology, and continued development of the camptothecins. Clin Cancer Res. 2002;8(3):641–661. [PubMed] [Google Scholar]
- 9*.Tardi PG, Dos Santos N, Harasym TO, et al. Drug ratio-dependent antitumor activity of irinotecan and cisplatin combinations in vitro and in vivo. Mol Cancer Ther. 2009;8(8):2266–2275. doi: 10.1158/1535-7163.MCT-09-0243. Excellent review on nanotechnology, specifically nanoparticles for cancer therapy. [DOI] [PubMed] [Google Scholar]
- 10.Noda K, Nishiwaki Y, Kawahara M, et al. Irinotecan plus cisplatin compared with etoposide plus cisplatin for extensive small-cell lung cancer. N Engl J Med. 2002;346(2):85–91. doi: 10.1056/NEJMoa003034. [DOI] [PubMed] [Google Scholar]
- 11.Hanna N, Bunn PA, Jr, Langer C, et al. Randomized phase III trial comparing irinotecan/cisplatin with etoposide/cisplatin in patients with previously untreated extensive-stage disease small-cell lung cancer. J Clin Oncol. 2006;24(13):2038–2043. doi: 10.1200/JCO.2005.04.8595. Excellent review on the use of nanoparticles for combination chemotherapy. It discusses the advantages of using nanoparticles for combination chemotherapy and summarizes previous work done in the field. [DOI] [PubMed] [Google Scholar]
- 12**.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. doi: 10.1038/nnano.2007.387. Key publication showing co-encapsulation and delivery of two drugs with different chemical and physical properties in the same nanoparticle. [DOI] [PubMed] [Google Scholar]
- 13.Shi J, Xiao Z, Kamaly N, Farokhzad OC. Self-Assembled Targeted Nanoparticles: Evolution of Technologies and Bench to Bedside Translation. Acc Chem Res. 2011 doi: 10.1021/ar200054n. [DOI] [PubMed] [Google Scholar]
- 14.Hrkach J, Von Hoff D, Ali M, et al. Preclinical Development and Clinical Translation of a PSMA-Targeted Docetaxel Nanoparticle with a Differentiated Pharmacological Profile. Sci Transl Med. 2012;4 doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 15.Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem Soc Rev. 2012;41(7):2971–3010. doi: 10.1039/c2cs15344k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16**.Hu C, Aryal S, Zhang L. Nanoparticle-assisted combination therapies for effective cancer treatment. Therapeutic Delivery. 2010;1(2):323–334. doi: 10.4155/tde.10.13. First publication where polymeric PLGA-PEG nanoparticles were prepared in microfluidic devices. [DOI] [PubMed] [Google Scholar]
- 17**.Kolishetti N, Dhar S, Valencia PM, et al. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc Natl Acad Sci U S A. 2010;107(42):17939–17944. doi: 10.1073/pnas.1011368107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maresca KP, Hillier SM, Femia FJ, et al. A series of halogenated heterodimeric inhibitors of prostate specific membrane antigen (PSMA) as radiolabeled probes for targeting prostate cancer. J Med Chem. 2009;52(2):347–357. doi: 10.1021/jm800994j. [DOI] [PubMed] [Google Scholar]
- 19.Hillier SM, Maresca KP, Femia FJ, et al. Preclinical evaluation of novel glutamate-urea-lysine analogues that target prostate-specific membrane antigen as molecular imaging pharmaceuticals for prostate cancer. Cancer Res. 2009;69(17):6932–6940. doi: 10.1158/0008-5472.CAN-09-1682. Important publication where several references related to previous investigations on the synergism between irinotecan and cisplatin are provided together with an elucidation of the potential mechanism for their synergistic effect. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng J, Teply BA, Sherifi I, et al. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials. 2007;28(5):869–876. doi: 10.1016/j.biomaterials.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21*.Karnik R, Gu F, Basto P, et al. Microfluidic platform for controlled synthesis of polymeric nanoparticles. Nano Lett. 2008;8(9):2906–2912. doi: 10.1021/nl801736q. [DOI] [PubMed] [Google Scholar]
- 22.Valencia PM, Basto PA, Zhang L, et al. Single-step assembly of homogenous lipid-polymeric and lipid-quantum dot nanoparticles enabled by microfluidic rapid mixing. ACS Nano. 2010;4(3):1671–1679. doi: 10.1021/nn901433u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chandran SS, Banerjee SR, Mease RC, Pomper MG, Denmeade SR. Characterization of a targeted nanoparticle functionalized with a urea-based inhibitor of prostate-specific membrane antigen (PSMA) Cancer Biol Ther. 2008;7(6):974–982. doi: 10.4161/cbt.7.6.5968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas M, Kularatne SA, Qi L, et al. Ligand-targeted delivery of small interfering RNAs to malignant cells and tissues. Ann N Y Acad Sci. 2009;1175:32–39. doi: 10.1111/j.1749-6632.2009.04977.x. [DOI] [PubMed] [Google Scholar]
- 25.Kularatne SA, Wang K, Santhapuram HK, Low PS. Prostate-specific membrane antigen targeted imaging and therapy of prostate cancer using a PSMA inhibitor as a homing ligand. Mol Pharm. 2009;6(3):780–789. doi: 10.1021/mp900069d. [DOI] [PubMed] [Google Scholar]
- 26.Zastre J, Anantha M, Ramsay E, Bally M. Irinotecan-cisplatin interactions assessed in cell-based screening assays: cytotoxicity, drug accumulation and DNA adduct formation in an NSCLC cell line. Cancer Chemother Pharmacol. 2007;60(1):91–102. doi: 10.1007/s00280-006-0353-z. [DOI] [PubMed] [Google Scholar]
- 27*.Gu FX, Karnik R, Wang AZ, et al. Targeted nanoparticles for cancer therapy. Nano Today. 2007;2(3):14–21. [Google Scholar]
- 28.Valencia PM, Hanewich-Hollatz MH, Gao W, et al. Effects of Ligands with Different Water Solubilities on Self-Assembly and Properties of Targeted Nanoparticles. Biomaterials. 2011 doi: 10.1016/j.biomaterials.2011.04.078. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci U S A. 2008;105(45):17356–17361. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rhee M, Valencia PM, Rodriguez MI, Langer R, Farokhzad OC, Karnik R. Synthesis of size-tunable polymeric nanoparticles enabled by 3D hydrodynamic flow focusing in single-layer microchannels. Adv Mater. 2011;23(12):H79–83. doi: 10.1002/adma.201004333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vauthier C, Schmidt C, Couvreur P. Measurement of the density of polymeric nanoparticulate drug carriers by isopycnic centrifugation. J Nanoparticle Res. 1999;1:411–418. [Google Scholar]
- 32.Johnson BK, Prud’homme RK. Mechanism for rapid self-assembly of block copolymer nanoparticles. Phys Rev Lett. 2003;91(11):118302. doi: 10.1103/PhysRevLett.91.118302. [DOI] [PubMed] [Google Scholar]
- 33.Avgoustakis K. Pegylated poly(lactide) and poly(lactide-co-glycolide) nanoparticles. preparation, properties and possible applications in drug delivery. Curr Drug Deliv. 2004;1(4):321–333. doi: 10.2174/1567201043334605. [DOI] [PubMed] [Google Scholar]
- 34.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58(3):621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 35.Reese DM, Tchekmedyian S, Chapman Y, Prager D, Rosen PJ. A phase II trial of irinotecan in hormone-refractory prostate cancer. Invest New Drugs. 1998;16(4):353–359. doi: 10.1023/a:1006120910380. [DOI] [PubMed] [Google Scholar]