

Protein kinase TORC2 is regulated by Ras response to distinct stimulatory ligands. Cells insensitive to one chemoattractant for TORC2 activation remain fully responsive to other ligands. Receptor-specific inhibitory circuits in Dictyostelium are found upstream and independent of GEF/Ras and downstream, feedback, or feedforward responses.
Abstract
Global stimulation of Dictyostelium with different chemoattractants elicits multiple transient signaling responses, including synthesis of cAMP and cGMP, actin polymerization, activation of kinases ERK2, TORC2, and phosphatidylinositide 3-kinase, and Ras-GTP accumulation. Mechanisms that down-regulate these responses are poorly understood. Here we examine transient activation of TORC2 in response to chemically distinct chemoattractants, cAMP and folate, and suggest that TORC2 is regulated by adaptive, desensitizing responses to stimulatory ligands that are independent of downstream, feedback, or feedforward circuits. Cells with acquired insensitivity to either folate or cAMP remain fully responsive to TORC2 activation if stimulated with the other ligand. Thus TORC2 responses to cAMP or folate are not cross-inhibitory. Using a series of signaling mutants, we show that folate and cAMP activate TORC2 through an identical GEF/Ras pathway but separate receptors and G protein couplings. Because the common GEF/Ras pathway also remains fully responsive to one chemoattractant after desensitization to the other, GEF/Ras must act downstream and independent of adaptation to persistent ligand stimulation. When initial chemoattractant concentrations are immediately diluted, cells rapidly regain full responsiveness. We suggest that ligand adaptation functions in upstream inhibitory pathways that involve chemoattractant-specific receptor/G protein complexes and regulate multiple response pathways.
INTRODUCTION
The seven-transmembrane receptors (7-TMRs) activate multiple downstream signaling cascades via heterotrimeric G protein–dependent and –independent pathways in all Eukarya (Ferguson and Caron, 1998; Rosenbaum et al., 2009). When activated, however, these receptors often elicit only transient responses. Downstream pathways are rapidly deactivated, and cells may become insensitive to the stimulating ligand concentration (Ferguson and Caron, 1998). Cells remain unresponsive if the stimulus is maintained persistently but regain sensitivity as ligand concentrations decline.
Sensory adaptation to a persistent stimulus (e.g., odorant, visual) is essential to discern input directionality and enable detection of diverse stimuli within a mixture of varying amplitudes. More broadly, such responses enable homeostatic balance to rapid hormonal perturbations.
Loss of sensitivity to chemoattractants is essential during many phases of the Dictyostelium life cycle. Dictyostelium grow as single-celled organisms and use folate as a nutrient-sensing chemoattractant during growth. On depletion of food sources, however, Dictyostelium are induced to enter multicellular development (McMains et al., 2008). During early development, Dictyostelium secrete cAMP, which functions as a chemoattractant. Cells respond to the extracellular cAMP signal by moving inward toward the source of cAMP synthesis and secreting additional cAMP (McMains et al., 2008; Cai and Devreotes, 2011). Thus cells coalesce at signaling centers and aggregate, but the cAMP signal is also relayed outward to recruit and synchronize additional cells. Still, Dictyostelium respond only transiently to a cAMP stimulus and then enter an insensitive phase characterized by arrested cellular movement and attenuated cAMP synthesis. Once the extracellular cAMP is degraded by secreted phosphodiesterase (PDE), cells regain sensitivity to cAMP and reinitiate a cycle of sensitization/desensitization (McMains et al., 2008; Cai and Devreotes, 2011), which ensures inward directional movement toward the center of cAMP synthesis but an outwardly relayed cAMP signal (Wessels et al., 1992).
Many molecular pathways in Dictyostelium respond transiently to cAMP receptor stimulation. These include activation of adenylyl and guanylyl cyclases (Tomchik and Devreotes, 1981; Van Haastert and Van der Heijden, 1983), actin polymerization (Hall et al., 1988), activation of ERK2 kinase (Maeda et al., 1996, 2004; Brzostowski and Kimmel, 2006) and phosphatidylinositide 3-kinase (PI3K; Huang et al., 2003, Brzostowski et al., 2004), ion influx (Milne and Devreotes, 1993), and Ras GDP-GTP cycling (Kae et al., 2004; Sasaki et al., 2004; Charest et al., 2010). However, the mechanisms that regulate the multiple deactivating pathways are very poorly understood. Here we dissect the regulation of the TOR kinase complex 2 (TORC2), which also exhibits activating/deactivating responses to cAMP (Lee et al., 2005; Kamimura et al., 2008; Cai et al., 2010; Charest et al., 2010; Liao et al., 2010).
TORC2 phosphorylates a C-terminal HM regulatory motif within two target substrates of Dictyostelium, AGC kinases PKBR1 and AKT (Kamimura and Devreotes, 2010; Liao et al., 2010). Relative in vivo TORC2 kinase activity can thus be quantified by immunoblot assay using a specific phospho-motif antibody. Stimulation of Dictyostelium with the chemoattractants cAMP and folate leads to very rapid but transient activation of TORC2 (Liao et al., 2010). TORC2 phosphorylation of AKT and PKBR1 is independent of the lipid kinase PI3K (Liao et al., 2010); however, data from several groups indicate a dependence on RasC activation (Lim et al., 2001; Cai et al., 2010; Charest et al., 2010).
By treating cells with cAMP and/or folate in various combinations, we show that cells that developed insensitivity to one chemoattractant can still activate TORC2 in response to the other ligand. TORC2 responses to cAMP or folate thus are not cross-inhibited. Furthermore, using a series of signaling mutants, we show that folate and cAMP responses require the identical GEF/RasC pathway but separate upstream receptor/G protein couplings. This common GEF/Ras pathway is also insensitive to cross-inhibition; cells that have become desensitized to RasC activation by one chemoattractant remain responsive to the other ligand.
Whereas downstream TORC2 kinase deactivation may be regulated by a negative feedback loop or a delayed inhibitory feedforward pathway centered on modulating RasC-GTP levels (Zhang et al., 2008; Charest et al., 2010; Takeda et al., 2012), our data indicate that acquired insensitivity to persistent chemoattractant stimulation must function independently of a common GEF/RasC pathway and more likely occurs via an upstream inhibitory pathway. We suggest that desensitization to continuous chemoattractant stimulation involves an adaptive response mediated by the chemoattractant-specific receptor/G protein complexes, thus insulating the cAMP and folate pathways and preventing their cross-adaptation.
RESULTS
Transient TORC2 activation by chemoattractants cAMP and folate
During development, Dictyostelium secrete cAMP with highly regulated periodicities (McMains et al., 2008). These cAMP pulses elicit corresponding periodic responses in a series of downstream pathways, including ERK2 phosphorylation, adenylyl and guanylyl cyclase activation, and cell shape change (McMains et al., 2008). Because TORC2 phosphorylation of the AKT and PKBR1 kinases of Dictyostelium is also cAMP stimulated (Kamimura and Devreotes, 2010; Liao et al., 2010), we were interested to determine whether TORC2 kinase activity also oscillated during development. Cells were differentiated in shaking culture and allowed to establish endogenous cAMP oscillations (Kimmel, 1987). Cell aliquots were assayed by immunoblot (Liao et al., 2010) using antibodies specific to either the phospho-form of ERK2 or to the TORC2 phosphorylated C-terminal sequence identical in both AKT and PKBR1 (FEGFpTYVA [pT435 for AKT and pT470 for PKBR1]).
Phospho-ERK2 showed characteristic maxima at ∼6-min intervals, which parallels endogenous extracellular cAMP signaling (Kimmel, 1987; Maeda et al., 2004); TORC2 phosphorylation of PKBR1 and AKT followed similar temporal kinetics to that of phospho-ERK2 (Figure 1A). We also looked at relative TORC2 phosphorylation levels of PKBR1 during normal develop on solid substrata in the absence of exogenous cAMP stimulation. TORC2 phosphorylation of PKBR1 is maximal at 5–15 h of development (Figure 1B), approximating times of maximal in vivo cAMP signaling (Kimmel, 1987; Brzostowski and Kimmel, 2006).
FIGURE 1:

Chemoattractant-mediated TORC2 phosphorylation of AKT and PKBR1. (A) Spontaneous oscillations of AKT, PKBR1, and ERK2 phosphorylation in Dictyostelium. Cells were pulsed with a 75 nM final concentration of cAMP every 6 min for 6 h. Cells were washed, resuspended in fresh buffer, and incubated without exogenous cAMP to allow spontaneous oscillations. Aliquots were collected at 1-min intervals and AKT, PKBR1, and ERK2 phosphorylations assayed by immunoblot. For p-ERK2 we used an antibody specific to the phospho-form of ERK2. For TORC2, we used an antibody that recognizes the TORC2-phosphorylated C-terminal sequence identical in both AKT and PKBR1 (FEGFpTYVA [pT435 for AKT and pT470 for PKBR1]). Relative phosphorylation changes were quantified. (B) Phosphorylation of PKBR1 during development. PKBR1 phosphorylation and actin levels were assayed by immunoblot during development at the times indicated. (C) Response of AKT and PKBR1 phosphorylation to cAMP and folate at different developmental stages. Cells were collected at times of differentiation in shaking culture, as indicated, washed of endogenous ligands, treated with caffeine to inhibit adenylyl cyclase, and washed. Cells were then stimulated with 10 μM cAMP or 50 μM folate. TORC2 phosphorylation of AKT and PKBR1 was assayed by immunoblot at the times indicated. (D) Cells at 0 h of development are equivalently responsive to folate and cAMP. Cells were stimulated with 10 μM cAMP or 50 μM folate. TORC2 phosphorylation of AKT and PKBR1 was assayed by immunoblot at the times indicated. For quantification, see Supplemental Figure S1. PHAPS, GacQ, and SHAPS indicate substrates phosphorylated by AKT/PKBR1 and assayed by immunoblot.
Because TORC2 is also activated by the chemoattractant folate (Liao et al., 2010), we sought to determine a developmental stage at which cells were responsive to both stimuli and to then assess the contributing effects of the different chemoattractants. Cells were stimulated at different times of development with exogenous saturating levels of either cAMP or folate and TORC2 activity assayed. The cells showed nearly identical response to cAMP at all stages examined (Figure 1C). Quiescent cells had only low levels of TORC2 phosphorylation of PKBR1 and AKT, but phosphorylation levels rose rapidly (∼15 s) but transiently and declined rapidly to basal levels. Thus the TORC2 pathway activation is quick but transitory, indicating a slightly delayed, antagonistic regulatory response to cAMP.
Folate-stimulated phosphorylation of PKBR1/AKT by TORC2 showed similar activating and inhibiting regulatory profiles in undifferentiated cells (Figure 1C). TORC2 phosphorylation of AKT and PKBR1 was very rapid, but dephosphorylation was equally rapid. However, unlike with cAMP, only undifferentiated cells were maximally responsive to folate; folate response diminished significantly as differentiation proceeded (Figure 1C). Nonetheless, the data indicate a stage at which cells are equally responsive to cAMP and folate for TORC2 activation. When cells from the same culture are stimulated with either cAMP or folate, they elicit very similar responses for TORC2 phosphorylation of PKBR1 and AKT (Figure 1D and Supplemental Figure S1) and for phosphorylation of the previously identified PKBR1/AKT substrates PHAPS, GacQ, and SHAPS (Figure 1D; Kamimura and Devreotes, 2010; Liao et al., 2010).
Of interest, folate stimulation also elicited a weak secondary but reproducible activation of TORC2 at ∼120 s (Figure 1D) that was not observed with cAMP. Because folate is able to activate adenylyl cyclase (De Wit et al., 1986), we postulated that the reactivation of TORC2 might result from a secondary cellular response to cAMP. To test this directly, we studied TORC2 response to folate in aca-null cells that lack adenylyl cyclase A. Although wild-type cells are able to mount a secondary TORC2 activation response to folate, the aca-null cells did not (Figure 2 and Supplemental Figure S2). These data indicate that the secondary TORC2 response is cAMP dependent, but, more significantly, suggest that cells deactivated to folate may still be responsive to cAMP.
FIGURE 2:
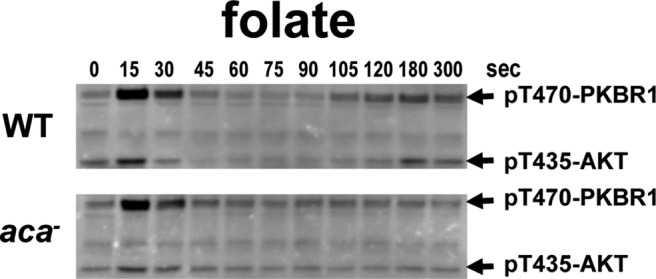
Secondary activation of TORC2 phosphorylation of AKT and PKBR1 after folate stimulation is caused by increases in cAMP. WT and aca-null cells were stimulated with 50 μM folate, and TORC2 phosphorylation of AKT and PKBR1 was assayed by immunoblot at the times indicated. For quantification, see Supplemental Figure S2.
Loss of cellular response to cAMP and folate
The rapid kinetics of activation/deactivation of TORC2 can occur via several mechanistic pathways. TORC2 activation could induce a negative feedback signal that directly suppresses TORC2. In addition, ligand stimulation could elicit a fast, TORC2-activating response but a more slowly functioning inhibitory signal. Furthermore, cellular response to an initial ligand concentration may only be transitory; cells may then develop insensitivity to a persistent, nonvarying stimulus. To analyze the potential for this latter response, we first determined cAMP concentration sensitivity (Figure 3A and Supplemental Figure S3A) for TORC2 phosphorylation of PKBR1 (EC50 ≈ 15 nM) and AKT (EC50 ≈ 45 nM). These stimulation data closely match the G protein–dependent, high-affinity binding sites (Kd ≈ 25 nM) for cAMP receptor CAR1 (Johnson et al., 1992). Next cells were stimulated with various concentrations of cAMP, allowed to deactivate, and then restimulated with a saturating dose (10 μM) of cAMP. Samples were assayed for PKBR1 and AKT phosphorylation 30 s after the initial stimulus and after the secondary stimulus (Figure 3B and Supplemental Figure S3A). Data show that the secondary response to saturating cAMP is inversely proportional to the initial cAMP stimulus. These data are consistent with cells becoming insensitive to a persistent, nonvarying cAMP stimulus.
FIGURE 3:

Adaptation of TORC2 activation to cAMP and folate. (A) cAMP dose–response activation of AKT and PKBR1. Cells were treated with various concentrations of cAMP and samples collected after 30 s. TORC2 phosphorylation of AKT and PKBR1 was assayed by immunoblot at the doses indicated and relative phosphorylation levels quantified. The EC50 for TORC2 phosphorylation of PKBR1 is 15 nM cAMP and is 45 nM cAMP for phosphorylation of AKT. For quantification, see Supplemental Figure S3A. (B) Response to a secondary saturating cAMP stimulus is inversely related to initial cAMP dose. Cells were treated with various concentrations of cAMP and samples collected after 30 s, followed by a second 10 μM cAMP stimulus at 60 s, with samples collected 30 s later, at 90 s. TORC2 phosphorylation of AKT and PKBR1 was assayed by immunoblot. For quantification, see Supplemental Figure S3A. (C) Adaptation of AKT and PKBR1 phosphorylation is not the result of cAMP degradation. Cells treated with or without DTT were stimulated with 10 μM cAMP, and TORC2 phosphorylation of AKT and PKBR1 was assayed by immunoblot at the times indicated. (D) Folate dose–response activation of AKT and PKBR1. Cells were treated with various concentrations of folate and samples collected after 15 s. TORC2 phosphorylation of AKT and PKBR1 was assayed by immunoblot at the doses indicated and relative phosphorylation levels quantified. The EC50 for TORC2 phosphorylation of PKBR1 is 65 nM folate and is 80 nM folate for phosphorylation of AKT. For quantification, see Supplemental Figure S3B. (E) Response to a secondary saturating folate stimulus is inversely related to the initial folate dose. Cells were treated with various concentrations of folate, with samples collected after 15 s, followed by a second 50 μM folate stimulus at 60 s, with samples collected 15 s later, at 75 s. TORC2 phosphorylation of AKT and PKBR1 was assayed by immunoblot. For quantification, see Supplemental Figure S3B.
Because Dictyostelium secrete a cAMP PDE, it is possible that the deactivating responses observed result from ligand clearing, as we previously showed for ERK2 regulation (Brzostowski and Kimmel, 2006), and not simply from ligand insensitivity. To examine this potential effect on TORC2 activity, we stimulated cells with saturating levels of cAMP in the presence or absence of dithiothreitol (DTT), an inhibitor of the secreted cAMP PDE of Dictyostelium (Brzostowski and Kimmel, 2006). DTT preserved input cAMP levels, and we observed no differences in phosphorylation of PKBR1 and AKT between the treated and untreated cell populations (Figure 3C). Cells remained adapted for 10 min. Thus the rapid decline in PKBR1/AKT phosphorylation is not the result of fluctuations in extracellular cAMP levels.
We also determined the dose–response effects of folate (Figure 3D and Supplemental Figure S3B) on TORC2 phosphorylation of PKBR1 (EC50 ≈ 65 nM) and AKT (EC50 ≈ 80 nM). These dose-response data are similar to those for the cellular G protein–dependent, high-affinity binding state (Kd ≈ 60 nM) for folate (De Wit and Bulgakov, 1985). The initial and secondary responses of TORC2 to folate exhibited a desensitizing behavior similar to that observed for cAMP (Figure 3E and Supplemental Figure S3B) and indicate that inhibitory signal strength for both cAMP and folate is proportional to the stimuli concentration; folate is not subject to significant degradation or modification during this brief time course.
Cellular responses to cAMP and folate are not cross-inhibitory
Because cAMP and folate appear to elicit similar responses to TORC2 signaling, we sought to use related assays to determine components that were common or distinct between the two pathways. We stimulated cells with a saturating dose of cAMP, allowed cells to adapt, and restimulated them with either additional cAMP or a saturating dose of folate (Figure 4A and Supplemental Figure S4A). Cells remained unresponsive to the secondary cAMP stimulus but were fully responsive to folate. As a response control, naive cells were also stimulated with saturating folate; these showed identical response to cells previously treated with cAMP. Thus, although cAMP-treated cells became insensitive to cAMP, they were fully responsive to stimulation by folate (Figure 4A and Supplemental Figure S4A).
FIGURE 4:

TORC2 phosphorylation of AKT and PKBR1 does not cross-adapt to different chemoattractants. (A) Cells stimulated with saturating doses of cAMP remain responsive to folate. Cells were stimulated with 10 μM cAMP and then stimulated with either 10 μM cAMP or 50 μM folate at 60 s. As a control, cells were stimulated with 50 μM folate at 0 s. TORC2 phosphorylation of AKT and PKBR1 was assayed by immunoblot at the times indicated. For quantification, see Supplemental Figure S4A. (B) Cells stimulated with saturating doses of folate remain responsive to cAMP. Cells were stimulated with 50 μM folate and then stimulated with either 50 μM folate or 10 μM cAMP at 60 s. As a control, cells were stimulated with 10 μM cAMP at 0 s. TORC2 phosphorylation of AKT and PKBR1 was assayed by immunoblot at the times indicated. For quantification, see Supplemental Figure S4B. (C) Cells stimulated with subsaturating doses of cAMP remain responsive to subsaturating doses of folate. Cells were stimulated with 15 nM cAMP and then stimulated with either 15 nM cAMP or 70 nM folate at 75 s. As a control, cells were stimulated with 70 nM folate at 0 s. TORC2 phosphorylation of AKT and PKBR1 was assayed by immunoblot at the times indicated. For quantification, see Supplemental Figure S4C. (D) Cells stimulated with subsaturating doses of folate remain responsive to subsaturating doses cAMP. Cells were stimulated with 70 nM folate and then stimulated with either 70 nM folate or 15 nM cAMP at 60 s. As a control, cells were stimulated with 15 nM cAMP at 0 s. TORC2 phosphorylation of AKT and PKBR1 was assayed by immunoblot at the times indicated. For quantification, see Supplemental Figure S4D.
In a reciprocal experiment, folate-stimulated cells could not be reactivated with an additional folate stimulus but were responsive for TORC2 activation by saturating cAMP (Figure 4B and Supplemental Figure S4B). These data indicate that the inhibitory pathways for cAMP and folate are ligand specific and do not elicit cross-regulation. Thus the inhibitory targets must lie upstream of TORC2-mediated phosphorylation (Figure 4B and Supplemental Figure S4B).
To further examine the potential for cross-inhibition, we modified our approach by using subsaturating doses of both cAMP and folate. Here, we would predict that cAMP-stimulated cells would be partially responsive to a secondary stimulation with the same subsaturating dose of cAMP but again would be fully responsive to a subsaturating dose of folate. Indeed, that was observed (Figure 4C and Supplemental Figure S4C). Similarly, folate-stimulated cells were partially responsive to a secondary subsaturating dose of folate but were fully responsive to subsaturating doses of cAMP (Figure 4D and Supplemental Figure S4D).
Chemoattractants cAMP and folate use a common GEF/Ras pathway but distinct receptor/G protein couplings
Because cells do not exhibit cross-inhibition to cAMP and folate, we were interested to determine components in the activation pathways that were unique to the individual stimuli and thus may participate in adaptive response. We used previously characterized cells that are deficient for a variety of signaling proteins (Fey et al., 2009). Cells were stimulated with either cAMP or folate and characterized for TORC2 phosphorylation of PKBR1 and AKT.
cAMP stimulation exhibited absolute dependence on cAMP receptor CAR1, the single Gβ, and Gα2 (Figure 5). These are well-characterized components that define cAMP signaling for several other responses (McMains et al., 2008). None of the other Gα proteins studied was required for cAMP-mediated activation of TORC2 or affected kinase inactivation (Figure 5).
FIGURE 5:

cAMP and folate use different receptors and G protein couplings but the same GEF/Ras pathway to mediate AKT and PKBR1 phosphorylation. Strains deficient for different signaling components were stimulated with 50 μM folate in starvation buffer or with 10 μM cAMP after differentiation for 6 h in shaking culture with cAMP pulses. TORC2 phosphorylation of AKT and PKBR1 was assayed by immunoblot at the times indicated. Genotypes in bold indicate cells that are unresponsive to active TORC2 by either cAMP or folate.
The folate receptor has not been identified, but TORC2 response to folate requires the single Gβ and Gα4 (Figure 6), components of the known folate cascade (Hadwiger et al., 1994). Neither CAR1 nor Gα2 is essential for folate signaling. We also examined cells lacking RasC or its specific activating protein, GefA (Kae et al., 2007), a guanine nucleotide exchange factor (GEF). These proteins are suggested to function highly proximal to receptor/G protein complexes and were shown to participate in TORC2 signal response (Lim et al., 2001; Cai et al., 2010; Charest et al., 2010; Kortholt et al., 2011). We further demonstrate the requirement of both GefA and RasC for TORC2 activation by folate and cAMP in the same cell (Figure 6). We suggest that overlapping signaling circuits for cAMP and folate mediate the activation of a common RasC/TORC2 pathway.
FIGURE 6:
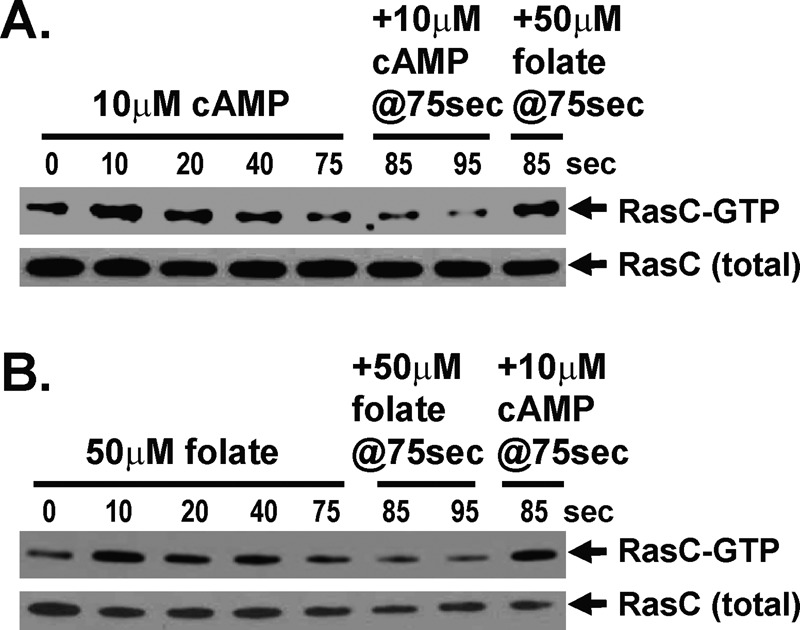
RasC activation does not cross-adapt to different chemoattractants. (A) Cells stimulated with saturating doses of cAMP remain responsive to folate. FLAG-RasC–expressing cells were stimulated with 10 μM cAMP and then stimulated with either 10 μM cAMP or 50 μM folate at 75 s. RasC-GTP levels were determined by interaction-specific affinity and normalized to total RasC by α-FLAG immunoblot assay at the times indicated. For quantification, see Supplemental Figure S5A. (B) Cells stimulated with saturating doses of folate remain responsive to cAMP. FLAG-RasC–expressing cells were stimulated with 50 μM folate and then stimulated with either 50 μM folate or 10 μM cAMP at 75 s. RasC-GTP levels were determined by interaction-specific affinity and normalized to total RasC by α-FLAG immunoblot assay at the times indicated. For quantification, see Supplemental Figure S5B.
RasC responses to cAMP and folate are not cross-inhibitory
Because both cAMP and folate require RasC to elicit TORC2 signaling, we sought to determine whether this pathway was also insensitive to cross-inhibition by the different chemoattractants. RasC-GTP levels rapidly increase in response to a cAMP stimulus and return to basal levels within 60 s (Lim et al., 2001; Cai et al., 2010; Charest et al., 2010). We therefore stimulated cells with saturating cAMP and, after RasC-GTP down-regulation, secondarily stimulated with either additional cAMP or with a saturating dose of folate. Relative RasC activation was monitored in cells expressing FLAG-RasC, normalizing RasC-GTP levels, determined by interaction-specific affinity, to that of total RasC in α-FLAG immunoblot assays. Whereas RasC is initially activated rapidly by cAMP, cells were unresponsive to a secondary cAMP stimulus; however, RasC-GTP rapidly reaccumulated in response to folate (Figure 6A and Supplemental Figure S5A).
In a reciprocal experiment, folate-stimulated cells could also not be reactivated for RasC-GTP signaling by an additional folate stimulus but were fully responsive for RasC-GTP activation by cAMP (Figure 6B and Supplemental Figure S5B). These data indicate that the inhibitory pathways for cAMP and folate are ligand specific, do not cross-regulate RasC/TORC2 signaling, and may require distinct receptor/G protein couplings.
Folate and cAMP responses are nonadditive
The data indicate that neither the RasC nor TORC2 pathways are cross-inhibited by folate and cAMP. Potentially, there may be sequestered or compartmentalized RasC/TORC2 linkages that respond uniquely to either folate or cAMP but not to both. Thus direct inhibition of a cAMP-specific RasC/TORC2 pathway may have no effect on folate activation of a completely independent RasC/TORC2 circuit. A corollary to this predicts that such multiple RasC/TORC2 pathways would each be activated independently of the other and thus must collectively exhibit an additive activation response when both chemoattractant ligands are applied simultaneously.
To address this mechanistically, we examined whether cells responded differently to saturating doses of cAMP and folate, alone or in combination. Cells were stimulated with either cAMP or folate or with both and assayed for TORC2 phosphorylation of PKBR1 and AKT. Only small comparative differences are seen among the various assays (Figure 7A and Supplemental Figure S6A). Cells were not hyperactivated by combining folate and cAMP in a single stimulus. Similarly, maximal RasC activation (Figure 7B and Supplemental Figure S6B) is observed regardless of whether cells are treated with folate and cAMP, singularly or in combination. These data indicate that the folate and cAMP stimulatory pathways are functionally independent but converge on a common downstream circuit comprising RasC/TORC2 components.
FIGURE 7:
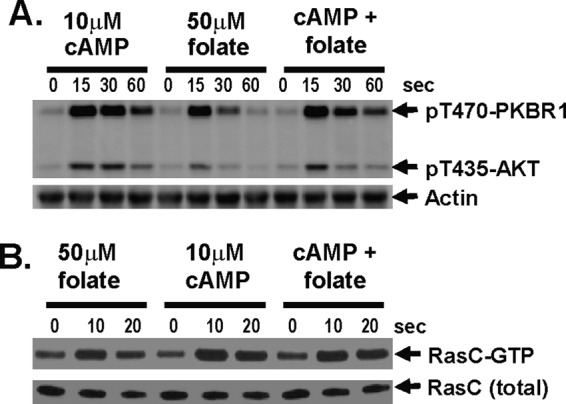
TORC2 and RasC responses to folate and cAMP are nonadditive. (A) AKT and PKBR1 phosphorylations are nonadditive in response to a mixture of saturated cAMP and folate. Cells were stimulated either with 10 μM cAMP plus 50 μM folate or 10 μM cAMP plus 50 μM folate. TORC2 phosphorylation of AKT and PKBR1 was assayed by immunoblot at the times indicated. For quantification, see Supplemental Figure S6A. (B) RasC activation is nonadditive in response to a mixture of saturated cAMP and folate. FLAG-RasC–expressing cells were stimulated either with 50 μM folate plus 10 μM cAMP, or 50 μM folate plus 10 μM cAMP. RasC-GTP levels were determined by interaction-specific affinity and normalized to total RasC by α-FLAG immunoblot assay at the times indicated. For quantification, see Supplemental Figure S6B.
Rapid cellular resensitization to cAMP and folate
Because TORC2 activity oscillates in response to endogenous cAMP fluctuations during normal Dictyostelium development (see Figure 1A), TORC2 must be subject to both desensitizing and resensitizing regulation. We suggest that re-response must require ligand clearance. To follow the kinetics for resensitization in culture, we mimicked ligand degradation by instantaneous dilution of cellular samples by 10-fold.
Cells were stimulated with subsaturating 100 nM cAMP, allowed to deactivate, and diluted 10-fold into fresh buffer, immediately reducing cAMP levels from 100 to 10 nM. The diluted cells were either left untreated or restimulated at various times with 100 nM cAMP and then assayed for TORC2 reactivation (Figure 8A). The untreated cells showed no TORC2 reactivation. The diluted cells were initially unresponsive to secondary stimulation but showed weak activation within 15 s and half-maximal reactivation within ∼30 s (Figure 8A and Supplemental Figure S7A). Thus once cAMP concentrations decline, cells undergo extremely rapid resensitization.
FIGURE 8:
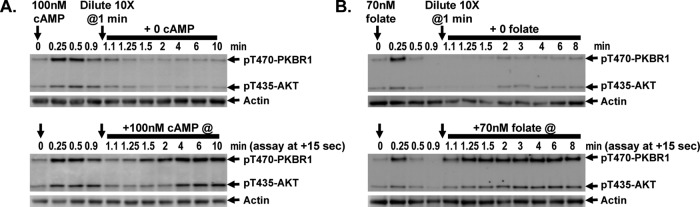
Very rapid deadaptation of TORC2 to cAMP and folate. (A) Cells become rapidly (<2 min) reresponsive to cAMP. Cells were stimulated with 100 nM cAMP, and TORC2 phosphorylation of AKT and PKBR1 was assayed by immunoblot at the times indicated. At 1 min, cells were diluted 10× into buffer to reduce cAMP to ∼10 nM. Cells were either maintained without additional cAMP or stimulated one time with 100 nM cAMP at each of the times indicated and samples removed after an additional 15 s, and TORC2 phosphorylation of AKT and PKBR1 was assayed by immunoblot. For quantification, see Supplemental Figure S7A. (B) Cells become rapidly (<1 min) reresponsive to folate. Cells were stimulated with 70 nM folate, and TORC2 phosphorylation of AKT and PKBR1 was assayed by immunoblot at the times indicated. At 1 min, cells were diluted 10× into buffer to reduce folate to ∼7 nM. Cells were either maintained without additional folate or stimulated one time with 70 nM folate at each of the times indicated and samples removed after an additional 15 s, and TORC2 phosphorylation of AKT and PKBR1 was assayed by immunoblot. For quantification, see Supplemental Figure S7B.
Similar results are seen with folate; after stimulation with 70 μM folate, cells were diluted 10-fold into fresh buffer, immediately reducing folate levels to 7 μM. The diluted cells were either left untreated or restimulated at various times with 70 μM folate and then assayed for TORC2 reactivation (Figure 8B). Resensitization to folate may be more rapid than with cAMP, with half-maximal reactivation at <30 s (Figure 8B and Supplemental Figure S7B).
DISCUSSION
During aggregation, Dictyostelium encounter oscillatory waves of cAMP that emanate and propagate outward from centers of cellular aggregation. As the cAMP wave approaches, Dictyostelium cells orient toward and migrate “up” the cAMP gradient and continue to respond to increasing cAMP concentrations until ligand binding reaches saturation. Simultaneously, cells secrete additional cAMP, which relays the oscillatory cAMP wave and recruits additional outlying cells. As the cells move through the concentration peak, however, the perceived cAMP gradient becomes spatially inverted, and cells deactivate most cAMP-regulated responses and become insensitive to further cAMP stimulation. Deactivation ensures that the propagated cAMP wave is relayed outward and also that cells do not alter their inward directional movement by reorienting toward the reversed cAMP gradient. With time, the extracellular cAMP signal is hydrolyzed by secreted PDE, and cells regain responsiveness to the next oncoming wave of cAMP (Wessels et al., 1992; McMains et al., 2008; Cai and Devreotes, 2011).
Many intracellular signaling pathways in Dictyostelium undergo activated/deactivated cycling in response to cAMP oscillation (McMains et al., 2008). These include ion flux, protein and lipid kinase regulation, cAMP and cGMP synthesis, and actin polymerization (McMains et al., 2008). Their coordinated regulations are required for both chemotactic movement and cAMP signal relay. Response down-regulation is essential for chemotactic aggregation during development, but many of these pathways also show similar on-off regulatory responses to the chemoattractant folate, a bacterial byproduct, during growth phase.
We dissected the sensitivity of Dictyostelium TORC2 kinase activation to the chemically distinct chemoattractants folate and cAMP (Liao et al., 2010). When cells are continually exposed to a nonvarying stimulus, the immediate response to either ligand is transient. The TORC2 substrates AKT and PKBR1 are rapidly (15 s) phosphorylated and almost equally rapidly dephosphorylated. TORC2 can be reactivated in response to a secondary stimulus of the same ligand but only if the initial stimulus is subsaturating. Nonetheless, the secondary activation of TORC2 is never maximal but is inversely proportional to the initial activation. Proposed mechanisms for chemoattractant regulation of TORC2 must integrate inhibition, persistence, and differential sensitivity to varying ligand concentrations within the chemical gradient.
Using a series of specific Dictyostelium signaling mutants, we determined that folate and cAMP activate TORC2 in the same cell, but through interaction with separate receptors and different G protein complexes (Figure 9A). Nonetheless, both chemoattractants use an identical GefA/RasC pathway to mediate TORC2 activation. TORC2 may be deactivated through depletion of RasC-GTP levels via negative feedback loops or incoherent feedforward circuits (Figure 9B; see Ma et al., 2009). TORC2-activated AKT/PKBR1 phosphorylates ScaA, a GefA scaffolding protein (Charest et al., 2010), and phosphorylated Sca is suggested to suppress GefA stimulation of RasC-GTP (Charest et al., 2010). Alternatively, receptor activation may generate rapid activation of GefA and RasC-GTP production (Kae et al., 2007) but a delayed RasGAP (i.e., GTPase-activating protein) signal (Zhang et al., 2008; Takeda et al., 2012), which more slowly returns RasC to its GDP-bound basal state.
FIGURE 9:
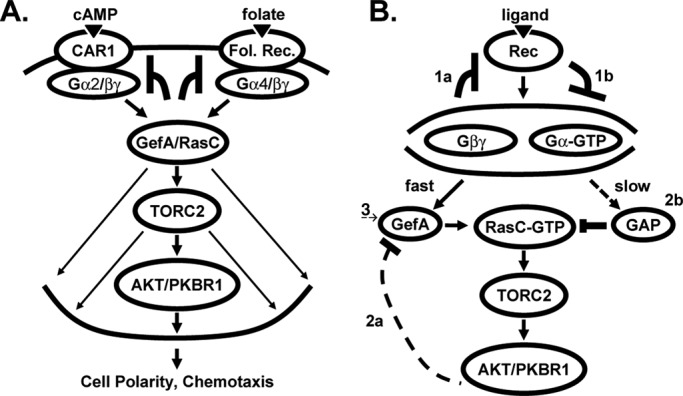
RasC/TORC2 regulation by chemokine signaling. (A) cAMP binds receptor CAR1 and induces dissociation of its coupled heterotrimeric G protein complex Gα2/βγ. Folate binds its receptor and dissociates its coupled heterotrimeric G protein complex Gα4/βγ. Both cAMP and folate use the same GefA-RasC axis to mediate AKT and PKBR1 phosphorylation by TORC2. Because cAMP and folate do not exhibit cross-inhibition, we suggest that adaptation must occur upstream and independently of GefA/RasC, potentially via the cAMP and folate receptors and their respective G protein complexes. (B) Receptor stimulation leads to G protein activation and dissociation. Many downstream pathways in Dictyostelium require signaling via Gβγ, although involvement of Gα-GTP is not excluded. RasC-GTP is required to activate TORC2. Subsequently, TORC2 activity may be suppressed through 1) adaptive responses that are specific to individual receptor/G protein complexes or 2) inhibitory down-regulation of RasC-GTP levels. Adaptation: 1a) Negative feedback regulation after G protein activation; 1b) delayed feedforward activation of an inhibitory signal that can sequester or inactivate specific G protein subunits. RasC down-regulation: 2a) Negative feedback regulation of the GefA activator (Charest et al., 2010); 2b) slow, feedforward activation of an inhibitory rasGAP (Zhang et al., 2008; Takeda et al., 2012). 3) Because folate and cAMP are not cross-adaptive (A), we propose that any inhibitory effects that are mediated via GefA (2a) or rasGAP (2b) must be transient, allowing reversal of GefA/rasGAP (or PPase) activity to an initial basal state (Figure 8 and Supplemental Figure S7). The adaptive receptor/G protein circuits (1) are thus the primary pathways that maintain ligand-specific desensitization of the RasC/TORC2 pathway but coordinate with downstream signaling (2, 3).
Nonetheless, we showed that cells that are unable to respond to one chemoattractant can fully activate TORC2 when stimulated with the other ligand. Accordingly, neither RasC nor its GEF or GAP regulators are targets for persistent ligand-mediated down-regulation. We therefore propose that downstream inhibitory effects (Figure 9B) mediated via GefA (Charest et al., 2010) or rasGAP (Zhang et al., 2008; Takeda et al., 2012) must be transient (Figure 8 and Supplemental Figure S7) and subject to reversion to an initial basal state (Figure 9B; Charest et al., 2010). Thus ligand response inhibition would occur upstream of RasC through an independent adaptive pathway involving desensitization of ligand-specific receptor/G protein complexes (Figure 9A).
In sensory and hormonal networks, adaptation or stimulus desensitization expands the range of signal strength detection. In these G protein–coupled receptor systems, desensitization may be mediated by receptor phosphorylation and interaction with arrestin (Ferguson and Caron, 1998; Reiter and Lefkowitz, 2006). The mechanistic targets for adaptation in Dictyostelium, however, are not clear. We suggest that inhibitory signaling must function upstream of RasC regulation and involve specific receptor/G protein complexes. However, the rapidity of TORC2 adaptive response in Dictyostelium limits the potential involvement of receptor modification. Phosphorylation of cytosolic serines in the C-terminus of CAR1 can be detected within 10 s of cAMP stimulation, but phosphorylation half-time requires ∼2 and >10 min to reach a plateau (Vaughan and Devreotes, 1988). In addition, dephosphorylation of CAR1 is significantly delayed (t1/2 ≈ 6 min; Vaughan and Devreotes, 1988) in comparison to deadaptive kinetics for TORC2 (t1/2 < 30 s). Furthermore, cells that only express nonphosphorylatable CAR1 variants (Kim et al., 1997) exhibit WT TORC2 adaptive responses (unpublished observations). Although the folate receptor has not been identified and studied biochemically, the data seem to preclude receptor modification as a defining motif for adaptive regulation.
The temporal kinetics for cAMP-stimulated Gα2/Gβγ dissociation (Janetopoulos et al., 2001) is very rapid and anticipates RasC-GTP accumulation (Sasaki et al., 2004) and TORC2 activation (Figures 3, 4, and 6). The kinetics for Gα2/Gβγ reassociation (Xu et al., 2007) and TORC2 deadaption (Figure 8A) upon removal of cAMP are also very similar (t1/2 < 30 s). Although comparable studies on Gα4 do not exist, the activation of TORC2 appears very tightly coordinated with that of G proteins. Nonetheless, Gα2/Gβγ is constitutively dissociated in the presence of persistent cAMP stimulation (Janetopoulos et al., 2001), whereas TORC2 is continuously down-regulated under identical conditions (Figure 3). Thus the Gα2/Gβγ assembly state is not sufficient to determine TORC2 activity; adaptation must function independently.
It is generally considered that released Gβγ is the activation module for heterotrimeric G protein signaling in Dictyostelium (Okaichi et al., 1992; Lilly and Devreotes, 1995). These data primarily derive from genetically based experiments, however, rather than from direct biochemical interactive proof. Thus a role for Gα in cAMP or folate signaling cannot be excluded. Indeed, data suggest both positive and inhibitory roles for Gα in Dictyostelium (Okaichi et al., 1992; Srinivasan et al., 1999; Brzostowski et al., 2004). Potentially, dissociated Gα and/or Gβγ convey an adaptive signal (Levine et al., 2006). For Gα, this might involve modification, sequestration, or the regulation of a specific regulator of G protein signaling, as suggested in other systems (Sethakorn et al., 2010). It is also interesting that Gα2 is rapidly (<20 s) phosphorylated upon cAMP stimulation (Gundersen and Devreotes, 1990). Gβ function may be regulated by modification (Chakrabarti et al., 2005) or phosducin-mediated assembly with Gγ (Knol et al., 2005). Regardless, G protein alterations that mediate adaptation must be rapidly reversible upon ligand removal and only target ligand-dissociated Gα/Gβγ subunits without affecting the additional Gα/Gβγ complexes.
We also suggest that adaptive mechanisms that regulate RasC-GTP cycling and TORC2 activity may have broad effect on cellular response and globally regulate other rapid cAMP- and folate-stimulated processes, including guanylyl cyclase and PI3K activity. Indeed, guanylyl cyclase responses to folate and cAMP are also not cross-inhibitory (Van Haastert, 1983). It is less apparent whether the more slowly responding (e.g., adenylyl cyclase) or G protein–independent (e.g., ERK2 and Ca2+) cAMP-regulated pathways are similarly affected. The time scale for RasC-GTP cycling and TORC2 deadaptation (t1/2 < 30 s) is significantly more rapid than that previously observed for adenylyl cyclase (t1/2 = 2–3 min) and other processes (Dinauer et al., 1980; Xiong et al., 2010).
Although we propose that adaptive mechanisms must function upstream (Figure 9, A and B), our data do not exclude the transitory roles for negative feedback loops or feedforward circuits that inhibit the downstream excitable RasC network (Figure 9B; Zhang et al., 2008; Charest et al., 2010; Takeda et al., 2012). Although both upstream and downstream events may act in concert, any downstream inhibitory effects must be transitory (Figures 8 and 9 and Supplemental Figure S7), allowing the excitable RasC network to rapidly respond to heterologous ligand stimulation. This posits another signaling mechanism that resets the initial basal state (Charest et al., 2010).
Several models have been proposed to explain how chemotaxing cells can strongly polarize their intracellular components within extremely shallow extracellular chemokine gradients (Δ < 5%). Most incorporate a local activation pathway that is proportional to ligand stimulation, and a global, diffusible inhibitory circuit that is equally active at all loci in the cell (Xiong et al., 2010). Our data suggest additional pathways that may intersect with such proposals and support a mechanism for insulating cAMP and folate pathways to prevent cross-inhibition.
MATERIALS AND METHODS
Dictyostelium strains
Dictyostelium were grown and differentiated in shaking culture as previously described (Liao et al., 2010). Spontaneous oscillations were in shaking culture (Kimmel, 1987; Maeda et al., 2004). Log-phase cells were developed on preboiled black filters and cells removed for assay at different developmental times. Mutant strains used in this study are for genes Gα2, Gα3, Gα4, Gα5, Gα7, Gα8, Gα9, CAR1, Gβ, ACA, gefA, and rasC and were previously described (Fey et al., 2009).
ERK2, AKT, and PKBR1 phosphorylation
cAMP and folate stimulations were previously described (Liao et al., 2010). Phosphorylation immunoblot assays of ERK2, AKT and PKBR1 were previously described (Maeda et al., 2004; Brzostowski and Kimmel, 2006; Liao et al., 2010). Antibodies were anti–phospho-PDK2/HM site (#9206; Cell Signaling Technology, Beverly, MA; phospho-p70 S6 kinase [Thr-389; 1A5] mouse monoclonal antibody); anti–phospho-AKT substrate (#9611; Cell Signaling Technology; phospho-(Ser/Thr) Akt substrate antibody); anti–phospho-ERK2 (#9101; Cell Signaling Technology; phospho-p44/42 MAPK [Erk1/2; Thr-202/Tyr-204] antibody); and anti-actin (#1616; Santa Cruz Biotechnology, Santa Cruz, CA; I-19; horseradish peroxidase).
RasC-GTP assay
RasC-GTP levels were monitored using previously described and optimized methods (Sasaki and Firtel, 2009). Briefly, Ras-GTP was isolated from cells by interaction with a purified glutathione S-transferase–Ras-binding domain component. Specific RasC activation was monitored in cells expressing FLAG-RasC, normalizing RasC-GTP levels to that of total RasC.
Supplementary Material
Acknowledgments
We thank dictyBase (Fey et al., 2009) and colleagues for various strains. We thank Leung Kim, Pascale Charest, Rick Firtel, and Shi Shu for providing the Ras-related reagents. This research was supported by the Intramural Research Program of the National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases.
Abbreviations used:
- AKT
also called protein kinase B
- cAMP
3′-5′-cyclic adenosine monophosphate
- ERK
extracellular signal–regulated kinase
- GEF
guanine nucleotide exchange factor
- PKBR1
protein kinase B–related protein 1
- TORC
target of rapamycin complex 2
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-03-0130) on May 8, 2013.
Present addresses: *Baylor College of Medicine, Houston, TX 77030;
†School of Medicine, Georgetown University, Washington, DC 20057.
REFERENCES
- Brzostowski JA, Kimmel AR. Nonadaptive regulation of ERK2 in Dictyostelium: implications for mechanisms of cAMP relay. Mol Biol Cell. 2006;17:4220–4227. doi: 10.1091/mbc.E06-05-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzostowski JA, Parent CA, Kimmel AR. A G alpha-dependent pathway that antagonizes multiple chemoattractant responses that regulate directional cell movement. Genes Dev. 2004;18:805–815. doi: 10.1101/gad.1173404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Das S, Kamimura Y, Long Y, Parent CA, Devreotes PN. Ras-mediated activation of the TORC2-PKB pathway is critical for chemotaxis. J Cell Biol. 2010;190:233–245. doi: 10.1083/jcb.201001129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Devreotes PN. Moving in the right direction: how eukaryotic cells migrate along chemical gradients. Semin Cell Dev Biol. 2011;22:834–841. doi: 10.1016/j.semcdb.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S, Regec A, Gintzler AR. Chronic morphine acts via a protein kinase C(gamma)-G(beta)-adenylyl cyclase complex to augment phosphorylation of G(beta) and G(betagamma) stimulatory adenylyl cyclase signaling. Brain Res Mol Brain Res. 2005;138:94–103. doi: 10.1016/j.molbrainres.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Charest PG, Shen Z, Lakoduk A, Sasaki AT, Briggs SP, Firtel RA. A Ras signaling complex controls the RasC-TORC2 pathway and directed cell migration. Dev Cell. 2010;18:737–749. doi: 10.1016/j.devcel.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wit RJ, Bulgakov R. Guanine nucleotides modulate the ligand binding properties of cell surface folate receptors in Dictyostelium discoideum. FEBS Lett. 1985;179:257–261. doi: 10.1016/0014-5793(85)80530-5. [DOI] [PubMed] [Google Scholar]
- De Wit RJ, Bulgakov R, Rinke de Wit TF, Konijn TM. Developmental regulation of the pathways of folate-receptor-mediated stimulation of cAMP and cGMP synthesis in Dictyostelium discoideum. Differentiation. 1986;32:192–199. doi: 10.1111/j.1432-0436.1986.tb00573.x. [DOI] [PubMed] [Google Scholar]
- Dinauer MC, Steck TL, Devreotes PN. Cyclic 3’,5’-AMP relay in Dictyostelium discoideum. IV. Recovery of the cAMP signaling response after adaptation to cAMP. J Cell Biol. 1980;86:545–553. doi: 10.1083/jcb.86.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SS, Caron MG. G protein-coupled receptor adaptation mechanisms. Semin Cell Dev Biol. 1998;9:119–127. doi: 10.1006/scdb.1997.0216. [DOI] [PubMed] [Google Scholar]
- Fey P, et al. dictyBase—a Dictyostelium bioinformatics resource update. Nucleic Acids Res. 2009;37:D515–D519. doi: 10.1093/nar/gkn844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen RE, Devreotes PN. In vivo receptor-mediated phosphorylation of a G protein in Dictyostelium. Science. 1990;248:591–593. doi: 10.1126/science.2110382. [DOI] [PubMed] [Google Scholar]
- Hadwiger JA, Lee S, Firtel RA. The G alpha subunit G alpha 4 couples to pterin receptors and identifies a signaling pathway that is essential for multicellular development in Dictyostelium. Proc Natl Acad Sci USA. 1994;91:10566–10570. doi: 10.1073/pnas.91.22.10566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AL, Schlein A, Condeelis J. Relationship of pseudopod extension to chemotactic hormone-induced actin polymerization in amoeboid cells. J Cell Biochem. 1988;37:285–299. doi: 10.1002/jcb.240370304. [DOI] [PubMed] [Google Scholar]
- Huang YE, Iijima M, Parent CA, Funamoto S, Firtel RA, Devreotes P. Receptor-mediated regulation of PI3Ks confines PI(3,4,5)P3 to the leading edge of chemotaxing cells. Mol Biol Cell. 2003;14:1913–1922. doi: 10.1091/mbc.E02-10-0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janetopoulos C, Jin T, Devreotes P. Receptor-mediated activation of heterotrimeric G-proteins in living cells. Science. 2001;291:2408–2411. doi: 10.1126/science.1055835. [DOI] [PubMed] [Google Scholar]
- Johnson RL, Van Haastert PJ, Kimmel AR, Saxe CL 3rd, Jastorff B, Devreotes PN. The cyclic nucleotide specificity of three cAMP receptors in Dictyostelium. J Biol Chem. 1992;267:4600–4607. [PubMed] [Google Scholar]
- Kae H, Kortholt A, Rehmann H, Insall RH, Van Haastert PJ, Spiegelman GB, Weeks G. Cyclic AMP signalling in Dictyostelium: G-proteins activate separate Ras pathways using specific RasGEFs. EMBO Rep. 2007;8:477–482. doi: 10.1038/sj.embor.7400936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kae H, Lim CJ, Spiegelman GB, Weeks G. Chemoattractant-induced Ras activation during Dictyostelium aggregation. EMBO Rep. 2004;5:602–606. doi: 10.1038/sj.embor.7400151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura Y, Devreotes PN. Phosphoinositide-dependent protein kinase (PDK) activity regulates phosphatidylinositol 3,4,5-trisphosphate-dependent and -independent protein kinase B activation and chemotaxis. J Biol Chem. 2010;285:7938–7946. doi: 10.1074/jbc.M109.089235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura Y, Xiong Y, Iglesias PA, Hoeller O, Bolourani P, Devreotes PN. PIP3-independent activation of TorC2 and PKB at the cell's leading edge mediates chemotaxis. Curr Biol. 2008;18:1034–1043. doi: 10.1016/j.cub.2008.06.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Soede RD, Schaap P, Valkema R, Borleis JA, Van Haastert PJ, Devreotes PN, Hereld D. Phosphorylation of chemoattractant receptors is not essential for chemotaxis or termination of G-protein-mediated responses. J Biol Chem. 1997;272:27313–27318. doi: 10.1074/jbc.272.43.27313. [DOI] [PubMed] [Google Scholar]
- Kimmel AR. Different molecular mechanisms for cAMP regulation of gene expression during Dictyostelium development. Dev Biol. 1987;122:163–171. doi: 10.1016/0012-1606(87)90342-3. [DOI] [PubMed] [Google Scholar]
- Knol JC, Engel R, Blaauw M, Visser AJ, van Haastert PJ. The phosducin-like protein PhLP1 is essential for G{beta}{gamma} dimer formation in Dictyostelium discoideum. Mol Cell Biol. 2005;25:8393–8400. doi: 10.1128/MCB.25.18.8393-8400.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortholt A, Kataria R, Keizer-Gunnink I, Van Egmond WN, Khanna A, Van Haastert PJ. Dictyostelium chemotaxis: essential Ras activation and accessory signalling pathways for amplification. EMBO Rep. 2011;12:1273–1279. doi: 10.1038/embor.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Comer FI, Sasaki A, McLeod IX, Duong Y, Okumura K, Yates JR 3rd, Parent CA, Firtel RA. TOR complex 2 integrates cell movement during chemotaxis and signal relay in Dictyostelium. Mol Biol Cell. 2005;16:4572–4583. doi: 10.1091/mbc.E05-04-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine H, Kessler DA, Rappel WJ. Directional sensing in eukaryotic chemotaxis: a balanced inactivation model. Proc Natl Acad Sci USA. 2006;103:9761–9766. doi: 10.1073/pnas.0601302103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao XH, Buggey J, Kimmel AR. Chemotactic activation of Dictyostelium AGC-family kinases AKT and PKBR1 requires separate but coordinated functions of PDK1 and TORC2. J Cell Sci. 2010;123:983–992. doi: 10.1242/jcs.064022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilly PJ, Devreotes PN. Chemoattractant and GTP gamma S-mediated stimulation of adenylyl cyclase in Dictyostelium requires translocation of CRAC to membranes. J Cell Biol. 1995;129:1659–1665. doi: 10.1083/jcb.129.6.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CJ, Spiegelman GB, Weeks G. RasC is required for optimal activation of adenylyl cyclase and Akt/PKB during aggregation. EMBO J. 2001;20:4490–4499. doi: 10.1093/emboj/20.16.4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Trusina A, El-Samad H, Lim WA, Tang C. Defining network topologies that can achieve biochemical adaptation. Cell. 2009;138:760–773. doi: 10.1016/j.cell.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda M, Aubry L, Insall R, Gaskins C, Devreotes PN, Firtel RA. Seven helix chemoattractant receptors transiently stimulate mitogen-activated protein kinase in Dictyostelium. Role of heterotrimeric G proteins. J Biol Chem. 1996;271:3351–3354. doi: 10.1074/jbc.271.7.3351. [DOI] [PubMed] [Google Scholar]
- Maeda M, Lu S, Shaulsky G, Miyazaki Y, Kuwayama H, Tanaka Y, Kuspa A, Loomis WF. Periodic signaling controlled by an oscillatory circuit that includes protein kinases ERK2 and PKA. Science. 2004;304:875–878. doi: 10.1126/science.1094647. [DOI] [PubMed] [Google Scholar]
- McMains VC, Liao XH, Kimmel AR. Oscillatory signaling and network responses during the development of Dictyostelium discoideum. Ageing Res Rev. 2008;7:234–248. doi: 10.1016/j.arr.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne JL, Devreotes PN. The surface cyclic AMP receptors, cAR1, cAR2, and cAR3, promote Ca2 +influx in Dictyostelium discoideum by a G alpha 2-independent mechanism. Mol Biol Cell. 1993;4:283–292. doi: 10.1091/mbc.4.3.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okaichi K, Cubitt AB, Pitt GS, Firtel RA. Amino acid substitutions in the Dictyostelium G alpha subunit G alpha 2 produce dominant negative phenotypes and inhibit the activation of adenylyl cyclase, guanylyl cyclase, and phospholipase C. Mol Biol Cell. 1992;3:735–747. doi: 10.1091/mbc.3.7.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter E, Lefkowitz RJ. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab. 2006;17:159–165. doi: 10.1016/j.tem.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki AT, Chun C, Takeda K, Firtel RA. Localized Ras signaling at the leading edge regulates PI3K, cell polarity, and directional cell movement. J Cell Biol. 2004;167:505–518. doi: 10.1083/jcb.200406177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki AT, Firtel RA. Spatiotemporal regulation of Ras-GTPases during chemotaxis. Methods Mol Biol. 2009;571:333–348. doi: 10.1007/978-1-60761-198-1_23. [DOI] [PubMed] [Google Scholar]
- Sethakorn N, Yau DM, Dulin NO. Non-canonical functions of RGS proteins. Cell Signal. 2010;22:1274–1281. doi: 10.1016/j.cellsig.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan J, Gundersen RE, Hadwiger JA. Activated Galpha subunits can inhibit multiple signal transduction pathways during Dictyostelium development. Dev Biol. 1999;215:443–452. doi: 10.1006/dbio.1999.9474. [DOI] [PubMed] [Google Scholar]
- Takeda K, Shao D, Adler M, Charest PG, Loomis WF, Levine H, Groisman A, Rappel WJ, Firtel RA. Incoherent feedforward control governs adaptation of activated ras in a eukaryotic chemotaxis pathway. Sci Signal. 2012;5:ra2. doi: 10.1126/scisignal.2002413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomchik KJ, Devreotes PN. Adenosine 3’,5’-monophosphate waves in Dictyostelium discoideum: a demonstration by isotope dilution–fluorography. Science. 1981;212:443–446. doi: 10.1126/science.6259734. [DOI] [PubMed] [Google Scholar]
- Van Haastert PJ. Relationship between adaptation of the folic acid and the cAMP mediated cGMP response in Dictyostelium. Biochem Biophys Res Commun. 1983;115:130–136. doi: 10.1016/0006-291x(83)90979-8. [DOI] [PubMed] [Google Scholar]
- Van Haastert PJ, Van der Heijden PR. Excitation, adaptation, and deadaptation of the cAMP-mediated cGMP response in Dictyostelium discoideum. J Cell Biol. 1983;96:347–353. doi: 10.1083/jcb.96.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan RA, Devreotes PN. Ligand-induced phosphorylation of the cAMP receptor from Dictyostelium discoideum. J Biol Chem. 1988;263:14538–14543. [PubMed] [Google Scholar]
- Wessels D, Murray J, Soll DR. Behavior of Dictyostelium amoebae is regulated primarily by the temporal dynamic of the natural cAMP wave. Cell Motil Cytoskeleton. 1992;23:145–156. doi: 10.1002/cm.970230207. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Huang CH, Iglesias PA, Devreotes PN. Cells navigate with a local-excitation, global-inhibition-biased excitable network. Proc Natl Acad Sci USA. 2010;107:17079–17086. doi: 10.1073/pnas.1011271107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Meier-Schellersheim M, Yan J, Jin T. Locally controlled inhibitory mechanisms are involved in eukaryotic GPCR-mediated chemosensing. J Cell Biol. 2007;178:141–153. doi: 10.1083/jcb.200611096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Charest PG, Firtel RA. Spatiotemporal regulation of Ras activity provides directional sensing. Curr Biol. 2008;18:1587–1593. doi: 10.1016/j.cub.2008.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.