

Abstract
Objective
Dietary fructose and copper interaction may play an important role in the pathogenesis of non-alcoholic fatty liver disease (NAFLD). In this study, we investigate whether or not modest fructose consumption (3% fructose, w/v) (which is more closely related to the American lifestyle with regard to sugar beverage consumption) affects copper status, and causes liver injury and fat accumulation in marginal copper deficient rats.
Design and Methods
Male weanling Sprague-Dawley rats were fed either an adequate copper (6ppm) or a marginally copper deficient (1.6ppm) diet for 4 weeks. Deionized water or deionized water containing 3% fructose (w/v) was given ad lib.
Results
Modest fructose consumption further impaired copper status in the marginal copper deficient rats and increased hepatic iron accumulation. Liver injury and fat accumulation were significantly induced in the marginal copper deficient rats exposed to fructose.
Conclusions
Our data suggest that modest fructose consumption can impair copper status and lead to hepatic iron overload, which in turn, may lead to liver injury and fatty liver in marginal copper deficient rats. This study provides important information on dietary fructose and copper interaction, suggesting that dietary fructose induced low copper availability might be an important mechanism underlying fructose induced fatty liver.
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is the hepatic manifestation of the metabolic syndrome. NAFLD ranges from simple steatosis to steatohepatitis and cirrhosis, is the major cause of abnormal liver enzymes in the United States, affecting more than 20% of Americans. Nonalcoholic steatohepatitis (NASH), a more serious form of NAFLD, is present in at least 2–3% of adults (1). The mechanisms underlying NASH are not fully understood. A large part of our knowledge on this disease has been derived from animal studies. Previously used animal models of NASH include genetic models, such as OB/OB or DB/DB mouse, or nutritional models, such as methionine and choline-deficient (MCD) diet (2), but these are actually not the real reflections of human disease. The typical characteristics of the Western-style diet include dietary high fat and high fructose corn syrup (HFCS) sweetened beverages (3). The increased consumption of fructose temporally parallels the increased prevalence of obesity and the metabolic syndrome in the United States and worldwide (4, 5). Emerging evidence has shown that high fructose intake may play an important role in the development of NAFLD (6, 7).
The currently used fructose levels in animal studies range from 10% to 30% (w/v) in the drinking water (8–10), or 50% to 66% of calories derived from dietary fructose (11, 12), which are the dietary extremes used for mechanistic studies. However, these levels do not reflect the real life style or current nutritional status of Americans. Recently, the American Lifestyle-Induced Obesity Syndrome (ALIOS) diet, which is a combination of high fat and HFCS in the drinking water, was introduced to mimic the characteristics of the American fast-food diet (13). The drink used in ALIOS model is HFCS-55 equivalent (which simulates the 60% of HFCS used in the US food supply), containing 55% fructose and 45% glucose or sucrose at a concentration of 42g/L, which equals to 2.3–3.3% (w/v) fructose in the drinking water (13, 14).
The pathogenesis of NASH likely involves multiple factors. Copper availability is one such factor which was identified very recently (15, 16). Both human and animal studies showed that decreased copper availability or inadequate dietary copper intake might play an important role in the pathogenesis of NAFLD. Decreased copper levels were found in a significant proportion of NAFLD patients, and a copper deficient diet induced hepatic steatosis in rats (15, 16). Decreased copper availability can be either primary (the average American diet contains only marginal amounts of copper) (17), or secondary. One of the important factors leading to decreased copper availability is high dietary fructose, which has been well-documented by Fields and colleagues and by our group (18–21). Moreover, dietary fructose interacts with copper deficiency and markedly enhances the metabolic complications of copper deficiency in rodents, possibly by disrupting copper absorption from gut (18–21). Our group recently showed that high fructose in the drinking water further impaired the copper status of marginal copper deficient rats, and exacerbated liver injury and fat accumulation (21), suggesting that marginal copper deficiency might be an initial component of the “two hit” model of NASH(22). However, the fructose used in the previous studies, including ours (30% fructose in the drinking water), are all high fructose concentration, which is much higher than that consumed by humans. As mentioned above, 3% fructose (w/v) in the drinking water as added sugar sweetener is more relevant to the American lifestyle.
Actually, fructose absorption is saturated at fairly low levels in both human and rat. In rats, the capacity to absorb fructose is 1.5–2.0g/kg body weight, which is higher than in humans (0.5g/kg body weight) (23, 24). Thus, the estimated fructose absorption is already saturated even with 3% fructose (w/v). Given that the upper gastrointestinal tract is the common site for the absorption of both copper and fructose (24, 25), and the average American diet contains only marginal amounts of copper (17), we hypothesize that even modest fructose (3%, w/v) intake may interfere with copper absorption and impair copper status in marginal copper deficient rats and lead to liver injury and fat accumulation.
METHODS AND PROCEDURES
Animals
Male weanling Sprague-Dawley rats (35–45g) from the Harlan Laboratories (Indianapolis, IN) were fed (AD LIB) a purified AIN-76 diet for laboratory rodents with defined copper content. The copper adequate rats received 6 mg/kg copper, and the marginal copper rats were fed with 1.6 mg/kg of copper for 4 weeks to achieve marginal copper deficiency. The animals were housed in stainless steel cages in a temperature and humidity controlled room with a 12:12h light-dark cycle. Animals had free access to either deionized water or deionized water containing 3% fructose (w/v). Fructose enriched drinking water was changed twice a week. All studies were approved by the University of Louisville Institutional Animal Care and Use Committee, which is certified by the American Association of Accreditation of Laboratory Animal Care. At the end of the experiment, all the animals were sacrificed under the anesthesia with pentobarbital at 50mg/kg I.P. injection. Blood was collected from the inferior vena cava, and citrated plasma was stored at −80°C for further analysis. Portions of liver tissue were fixed with 10% formalin for subsequent sectioning, while others were snap-frozen with liquid nitrogen.
Assessment of Copper and Iron Status
Plasma ceruloplasmin was measured on the basis of its oxidase activity (26). Plasma copper and liver copper were measured by Varian SpectrAA 880/GTA-100 graphite furnace atomic absorption spectrometer (AAS) (Worcester Polytechnic Institute, Worcester, MA). Liver iron were measured by iCE3000 series flame AAS (Thermo Fisher Scientific, Waltham, MA). Plasma ferritin was determined by commercially available kit (ALPCO Diagnostics, Salem, NH).
Hepatic Copper, zinc- superoxide dismutase (SOD1) Enzyme Activity Assay
Briefly, liver tissues (less than 9mg) were homogenized with 10mM Tris-HCl (PH 7.4). After Mn-SOD (SOD2) activity was inhibited by 2% sodium dodecyl sulfate (38), SOD activity was measured using SOD determination kit (Sigma-aldrich, St. Louis, MO). SOD activity was determined by measuring the inhibition rate of formazan dye production.
Liver Enzyme and Plasma MCP-1 Assay
Plasma alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured using commercially available kits (Infinity; Thermo Electron Corporation, Melbourne, Australia) based on a colorimetric method. Plasma monocyte chemoattractant protein-1 (MCP-1) was determined by ELISA kit (R&D Systems, Minneapolis, MN).
GSH and GSSG Assay
Reduced glutathione (GSH) and oxidized glutathione (GSSG) were determined by HPLC as described previously (27).
Histology and Immunohistochemistry
Formalin-fixed, paraffin-embedded liver sections were cut at 5 μm thickness, and stained with hematoxylin and eosin (H&E). Apoptotic hepatocytes were detected by the Terminal Deoxynucleotidyl Transferase Biotin-dUTP Nick End Labeling (TUNEL) assay with an In Situ Apoptosis Detection Kit (Millipore, Billerica, MA).
For immunohistochemical analysis, paraffin-embedded liver sections were incubated with anti-4-hydroxynonenal (HNE) (Alpha Diagnostic International Inc., San Antonio, TX), for 30 min. Staining was visualized using the horseradish peroxidase-conjugated DAKO staining system (DAKO InVision, Carpenteria, CA).
Hepatic Triglyceride Assay
Liver tissues were homogenized in ice-cold phosphate buffered saline. Hepatic total lipids were extracted with chloroform/methanol (2:1) according to the method described by Bligh and Dyer (28). Hepatic triglyceride content was determined by commercially available kits (Infinity, Thermo Electron).
Statistical Analysis
All data were expressed as mean ± SD (Standard Deviation) and analyzed by analysis of variance (ANOVA) followed by Newman Keuls’ Multiple Comparison Test and Student’s T- test. The interactions between copper and fructose were examined by two-way ANOVA. Differences at P < 0.05 were considered to be statistically significant.
RESULTS
Effects of low fructose feeding on copper, iron status and hepatic SOD1 activity
Plasma ceruloplasmin activity and plasma copper were significantly decreased in the rats fed with the marginal copper deficient diet, and they were further decreased in marginal copper deficient rats fed fructose (Figure 1a, b). Similarly, liver copper was significantly decreased in marginal copper deficient rats. However, no significant decrease was observed in those animals additionally fed with fructose (Figure 1c). Hepatic SOD1 activity was markedly decreased in marginal copper deficient rats, as well as in adequate copper rats fed with fructose. However, no further decrease was observed in marginal copper deficient rats additionally fed with fructose (Figure 1d). Liver iron was significantly increased in both adequate copper and marginal copper deficient rats fed with fructose, and it was further increased in marginal copper deficient rats compared to adequate copper rats. However, there was no significant difference in iron levels between adequate copper and marginal copper deficient rats without fructose (Figure 2a). Consistent with the liver iron, plasma ferritin, a marker of total body iron stores, was also significantly increased in marginal copper deficient rats fed with fructose compared to adequate copper rats (Figure 2b).
Figure 1.

Effect of low fructose feeding on copper status. (a) Plasma ceruloplasmin. (b) Plasma copper. (c) Liver copper. (d) Hepatic SOD1 activity. Data represent means ± SD (n=5–10). *p<0.05. A, adequate copper diet; M, marginal copper deficient diet; AF, adequate copper diet+3% fructose drinking; MF, marginal copper deficient diet+3% fructose drinking. SOD1, copper, zinc- superoxide dismutase.
Figure 2.
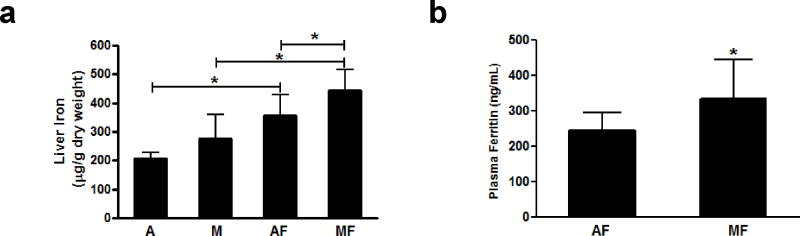
Effect of low fructose feeding on iron status. (a) Liver iron. (b) Plasma ferritin. Data represent means ± SD (n=5–10). *p<0.05. A, adequate copper diet; M, marginal copper deficient diet; AF, adequate copper diet+3% fructose drinking; MF, marginal copper deficient diet+3% fructose drinking.
Dietary marginal copper deficiency and low fructose drinking interact to exacerbate liver injury
Liver injury was assessed by plasma levels of liver enzymes (ALT and AST). Plasma ALT level was not significantly changed by either marginal copper deficiency or fructose feeding. However, marginal copper deficiency and fructose drinking together significantly increased plasma ALT as compared to controls. While there is no significant difference in plasma AST level among the adequate copper rats fed with or without fructose, or in marginally copper deficient rats without fructose, whereas marginal copper deficiency and fructose feeding synergistically increased plasma AST level compared to other groups (Figure 3a). Consistent with the biochemical findings, obvious hepatocyte apoptosis was observed by TUNEL staining in marginal copper deficient rats fed fructose, as seen in representative photomicrographs of TUNEL staining (Figure 3b). In addition, fructose feeding plus marginal copper deficiency increased plasma MCP-1 level compared to marginal copper deficient rats and adequate copper rats fed with fructose (Figure 3c).
Figure 3.

Effect of marginal copper deficiency and low fructose feeding on liver injury. (a) Plasma ALT and AST level. (b) Representative photomicrographs of TUNEL staining of liver section (400×). (c) Plasma MCP-1. Data represent means ± SD (n=5–10). *p<0.05; #, interaction between copper and fructose is significant (p<0.05). A, adequate copper diet; M, marginal copper deficient diet; AF, adequate copper diet+3% fructose drinking; MF, marginal copper deficient diet+3% fructose drinking. ALT, alanine aminotransferase; AST, aspartate aminotransferase; TUNEL, Terminal Deoxynucleotidyl Transferase Biotin-dUTP Nick End Labeling; MCP-1, monocyte chemoattractant protein-1.
Dietary marginal copper deficiency and low fructose ingestion synergistically accelerate hepatic fat accumulation
Neither adequate copper with fructose in the drinking water nor marginal copper deficiency alone significantly increased hepatic triglyceride content compared to adequate copper rats after 4 weeks. However, fructose drinking combined with marginal copper deficiency significantly and synergistically induced hepatic triglyceride accumulation (Figure 4b). Those changes were also consistent with histology as shown by H&E staining (Figure 4a).
Figure 4.

Effect of marginal copper deficiency and low fructose feeding on hepatic lipid accumulation. (a) Representative photomicrographs of the H&E staining of liver section (200×). (b) Hepatic triglyceride. Data represent means ± SD (n=5–10). *p<0.05. #, interaction between copper and fructose is significant (p<0.05). A, adequate copper diet; M, marginal copper deficient diet; AF, adequate copper diet+3% fructose drinking; MF, marginal copper deficient diet+3% fructose drinking. H&E, hematoxylin and eosin;
Decreased antioxidant defense system and increased oxidative stress by dietary marginal copper deficiency and low fructose ingestion
We next examined the effects of marginal copper deficiency and fructose ingestion on oxidative stress, including GSH/GSSG and 4-HNE. Hepatic GSH and GSSG levels were significantly decreased by fructose drinking in either marginal copper deficiency or adequate copper rats. The GSH/GSSG ratio, which is an indicator of oxidative stress, was significantly increased in both adequate and marginal copper deficient rats when fed with fructose compared to controls. Fructose feeding led to a further increased GSH/GSSG ratio in marginal copper deficient rats compared to adequate copper rats (Figure 5a). As shown in Figure 5b, the representative photomicrographs of liver 4-HNE immunohistochemistry staining, immunoreactivity of 4-HNE was slightly increased in the livers of adequate copper rats fed with fructose, and it was significantly enhanced in marginal copper deficient rats fed with fructose compared with controls, suggesting increased lipid peroxidation and oxidative stress in marginal copper deficient rats fed with fructose.
Figure 5.

Effect of marginal copper deficiency and low fructose feeding on hepatic GSH and GSSG. Hepatic (a) GSH, GSSG, and GSH/GSSG ratio measured by HPLC. (b) Representative photomicrographs of the immunohistochemistry staining for 4-HNE in liver section (100×). Data represent means ± SD (n=5–8). *p<0.05. A, adequate copper diet; M, marginal copper deficient diet; AF, adequate copper diet+3% fructose drinking; MF, marginal copper deficient diet+3% fructose drinking. GSH, reduced glutathione; GSSG, oxidized glutathione; 4-HNE, 4-hydroxynonenal.
DISCUSSION
In our current animal study, we used 3% fructose (w/v) as the modest fructose feeding which is much lower than the 30% fructose (w/v) we used previously (21). Actually, 3% fructose (w/v) in the drinking water is very close to the typical HFCS-55 consumption as sweetened beverages in the current American diet (29, 30).
It is well known that the etiology of NAFLD is complex, and multiple factors are likely involved, including nutritional and genetic factors. Recently, decreased copper availability was shown in NAFLD patients (15, 16). Interestingly, decreased copper availability may not only be due to inadequate intake from the food, but may also be due to dietary high fructose, which might impair copper absorption from the intestine (17–21). In the current study, we report for the first that modest fructose intake and marginal copper deficiency together induced liver injury and fat accumulation in rats. The fructose concentration in current study is only about 10% of that in the high fructose drinking, as was used in our previous study. While there is a 10-fold difference in the amount of fructose in the drinking water, the estimated fructose absorption is already saturated even at 3% fructose (23, 24). Therefore, it is plausible that 3% and 30% fructose (w/v) interfere with copper absorption and impair copper status to a similar level, suggesting that marginal copper deficiency may play an important role in the pathogenesis of NAFLD. Further, decreased copper availability might be a critical factor underlying fructose induced NAFLD.
Modest fructose consumption did not significantly increase hepatic triglyceride content in adequate copper rats in 4 weeks. However, it significantly increased hepatic triglyceride content in marginal copper deficient rats by approximately 3-fold during that same time. In our previous study, high fructose feeding (30% fructose, w/v) increased hepatic triglyceride content by 4-fold in marginal copper deficient rats (21). Plasma AST was increased by 5-fold in marginal copper deficient rats when feeding 30% fructose (21), and it increased by 2-fold when animals were fed with 3% fructose. Compared to our previous data from 30% fructose drinking, it appears that increased fat accumulation and liver injury do not parallel the amount of fructose consumption, but seems more to relate to copper status. In fact, previous studies have reported that dietary fructose impaired copper absorption from the intestine (31, 32), potentially by blocking copper transport through copper transporter-1(Ctr-1) (21).
Hepatic iron overload is a common feature associated with copper deficiency (33, 34). Our data clearly showed increased liver iron content in marginal copper deficient rats even with modest fructose consumption (Figure 2a). Basically, excess iron was diffusely distributed throughout hepatocytes and a large portion within Kupffer cells (resident macrophages in liver) (35). Macrophages play a crucial role in iron recycling by phagocytosing senescent erythrocytes and releasing accumulated iron back into the circulation in a regulated manner, thus enabling iron recycling. One important mechanism underlying copper deficiency associated hepatic iron overload is decreased plasma ceruloplasmin, which regulates iron efflux from reticuloendothelial cells, including liver Kupffer cells via ferroxidase (33). Down-regulation of the iron exporter, ferroportin-1 (Fp-1) (36), or some other unknown mechanism may also contribute to the iron accumulation.
One mechanism underlying fructose induced liver injury in marginal copper deficient rats is decreased antioxidant defenses caused by copper deficiency and increased oxidative stress. In the current study, decreased antioxidant defense is characterized by decreased GSSG in animals fed with fructose (which has already been observed previously by our group), possibly due to the down-regulated glutathione peroxidase (GPx) (21). Therefore, GSH cannot be fully oxidized to remove hydrogen peroxide and cannot perform its antioxidant function. However, fructose feeding did not lead to liver injury in adequate copper rats, while it did in the marginal copper deficient animals, suggesting other factors associated with copper deficiency might play a role. In fact, decreased copper, zinc- superoxide dismutase (SOD1) due to copper deficiency is well-documented in previous studies, including from our group, and may be another important factor contributing to decreased antioxidant defense (21, 37). Thus, it is likely that decreased antioxidant defense as a result of dysregulation of the GSH/GSSG system and suppressed SOD1 due to copper deficiency may led to increased accumulation of superoxide and hydrogen peroxide. On the other hand, hepatic iron overload is a common feature associated with copper deficiency (33, 34), and hepatocytes are important cells for iron accumulation (35). Iron is a catalyst to convert hydrogen peroxide to hydroxyl radicals through the Haber-Weiss and Fenton reactions. Therefore, ROS generated from the mitochondrial respiratory chain may be further increased by iron overload, subsequently leading to hepatocyte cell death/liver injury.
In conclusion, our data clearly showed that 3% fructose as the added sugar beverage further impaired copper status and led to significant hepatic fat accumulation and liver injury in marginal copper deficient rats, suggesting that dietary fructose and copper interaction may play an important role in the pathogenesis of fructose induced NAFLD, and marginal copper deficiency might be an important priming factor in the “two hit” model of NASH. One potential mechanism underlying copper deficiency-induced fatty liver is likely due to hepatic iron overload. The pathogenesis of NAFLD is a complicated process and likely involves multiple factors. Our data provide strong evidence in understanding the pathogenesis of NAFLD and potential new therapeutic targets for the future.
Acknowledgments
This study was supported in part by National Institute on Alcohol Abuse and Alcoholism Grants RO1AA015970, PO1AA017103, R37AA010762, RO1AA018869, RC2AA019385, P30AA019360, R01AA018016(C.J.M.), RO1AA014623, RO1AA016013, RO1AA018844 (Z.Z.); the Veterans Administration (C.J.M.); National Institute of Diabetes and Digestive and Kidney Diseases DK-055030 (D.A.S.), RO1DK071765 (C.J.M.), and UofL Clinical and Translational Sciences Pilots, “High fructose feeding induces copper deficiency: A novel mechanism for obesity related hyperlipidemia and fatty liver” (C.J.M.).
Footnotes
DISCLOSURE
The authors declared no conflict of interest.
References
- 1.Cave M, Deaciuc I, Mendez C, Song Z, Joshi-Barve S, Barve S, McClain C. Nonalcoholic fatty liver disease: predisposing factors and the role of nutrition. J Nutr Biochem. 2007;18:184–195. doi: 10.1016/j.jnutbio.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 2.London RM, George J. Pathogenesis of NASH: animal models. Clin Liver Dis. 2007;11:55–74. viii. doi: 10.1016/j.cld.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 3.Odermatt A. The Western-style diet: a major risk factor for impaired kidney function and chronic kidney disease. Am J Physiol Renal Physiol. 2011;301:F919–931. doi: 10.1152/ajprenal.00068.2011. [DOI] [PubMed] [Google Scholar]
- 4.Hallfrisch J. Metabolic effects of dietary fructose. FASEB J. 1990;4:2652–2660. doi: 10.1096/fasebj.4.9.2189777. [DOI] [PubMed] [Google Scholar]
- 5.Spruss A, Bergheim I. Dietary fructose and intestinal barrier: potential risk factor in the pathogenesis of nonalcoholic fatty liver disease. J Nutr Biochem. 2009;20:657–662. doi: 10.1016/j.jnutbio.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 6.Assy N, Nasser G, Kamayse I, Nseir W, Beniashvili Z, Djibre A, Grosovski M. Soft drink consumption linked with fatty liver in the absence of traditional risk factors. Can J Gastroenterol. 2008;22:811–816. doi: 10.1155/2008/810961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ouyang X, Cirillo P, Sautin Y, McCall S, Bruchette JL, Diehl AM, Johnson RJ, Abdelmalek MF. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J Hepatol. 2008;48:993–999. doi: 10.1016/j.jhep.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li JM, Li YC, Kong LD, Hu QH. Curcumin inhibits hepatic protein-tyrosine phosphatase 1B and prevents hypertriglyceridemia and hepatic steatosis in fructose-fed rats. Hepatology. 2010;51:1555–1566. doi: 10.1002/hep.23524. [DOI] [PubMed] [Google Scholar]
- 9.Castro MC, Massa ML, Del Zotto H, Gagliardino JJ, Francini F. Rat liver uncoupling protein 2: changes induced by a fructose-rich diet. Life Sci. 2011;89:609–614. doi: 10.1016/j.lfs.2011.07.024. [DOI] [PubMed] [Google Scholar]
- 10.Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology. 2009;50:1094–1104. doi: 10.1002/hep.23122. [DOI] [PubMed] [Google Scholar]
- 11.Rodríguez-Calvo R, Barroso E, Serrano L, Coll T, Sánchez RM, Merlos M, Palomer X, Laguna JC, Vázquez-Carrera M. Atorvastatin prevents carbohydrate response element binding protein activation in the fructose-fed rat by activating protein kinase A. Hepatology. 2009;49:106–115. doi: 10.1002/hep.22570. [DOI] [PubMed] [Google Scholar]
- 12.Zavaroni I, Chen YD, Reaven GM. Studies of the mechanism of fructose-induced hypertriglyceridemia in the rat. Metabolism. 1982;31:1077–1083. doi: 10.1016/0026-0495(82)90155-x. [DOI] [PubMed] [Google Scholar]
- 13.Tetri LH, Basaranoglu M, Brunt EM, Yerian LM, Neuschwander-Tetri BA. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am J Physiol Gastrointest Liver Physiol. 2008;295:G987–995. doi: 10.1152/ajpgi.90272.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kohli R, Kirby M, Xanthakos SA, Softic S, Feldstein AE, Saxena V, Tang PH, Miles L, Miles MV, et al. High-fructose, medium chain trans-fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology. 2010;52:934–944. doi: 10.1002/hep.23797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aigner E, Theurl I, Haufe H, Seifert M, Hohla F, Scharinger L, Stickel F, Mourlane F, Weiss G, et al. Copper availability contributes to iron perturbations in human nonalcoholic fatty liver disease. Gastroenterology. 2008;135:680–688. doi: 10.1053/j.gastro.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 16.Aigner E, Strasser M, Haufe H, Sonnweber T, Hohla F, Stadlmayr A, Solioz M, Tilg H, Patsch W, et al. A role for low hepatic copper concentrations in nonalcoholic Fatty liver disease. Am J Gastroenterol. 2010;105:1978–1985. doi: 10.1038/ajg.2010.170. [DOI] [PubMed] [Google Scholar]
- 17.Holden JM, Wolf WR, Mertz W. Zinc and copper in self-selected diets. J Am Diet Assoc. 1979;75:23–28. [PubMed] [Google Scholar]
- 18.Fields M, Ferretti RJ, Smith JC, Jr, Reiser S. The interaction of type of dietary carbohydrates with copper deficiency. Am J Clin Nutr. 1984;39:289–295. doi: 10.1093/ajcn/39.2.289. [DOI] [PubMed] [Google Scholar]
- 19.Fields M, Ferretti RJ, Smith JC, Jr, Reiser S. Effect of copper deficiency on metabolism and mortality in rats fed sucrose or starch diets. J Nutr. 1983;113:1335–1345. doi: 10.1093/jn/113.7.1335. [DOI] [PubMed] [Google Scholar]
- 20.Fields M, Ferretti RJ, Reiser S, Smith JC., Jr The severity of copper deficiency in rats is determined by the type of dietary carbohydrate. Proc Soc Exp Biol Med. 1984;175:530–537. doi: 10.3181/00379727-175-41832. [DOI] [PubMed] [Google Scholar]
- 21.Song M, Schuschke DA, Zhou Z, Chen T, Pierce WM, Jr, Wang R, Johnson WT, McClain CJ. High fructose feeding induces copper deficiency in Sprague-Dawley rats: A novel mechanism for obesity related fatty liver. J Hepatol. 2012;56:433–440. doi: 10.1016/j.jhep.2011.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Day CP. From fat to inflammation. Gastroenterology. 2006;130:207–210. doi: 10.1053/j.gastro.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 23.Fujisawa T, Riby J, Kretchmer N. Intestinal absorption of fructose in the rat. Gastroenterology. 1991;101:360–367. doi: 10.1016/0016-5085(91)90012-a. [DOI] [PubMed] [Google Scholar]
- 24.Riby JE, Fujisawa T, Kretchmer N. Fructose absorption. Am J Clin Nutr. 1993;58(5 Suppl):748S–753S. doi: 10.1093/ajcn/58.5.748S. [DOI] [PubMed] [Google Scholar]
- 25.van den Berghe PV, Klomp LW. New developments in the regulation of intestinal copper absorption. Nutr Rev. 2009;67:658–672. doi: 10.1111/j.1753-4887.2009.00250.x. [DOI] [PubMed] [Google Scholar]
- 26.Schosinsky KH, Lehmann HP, Beeler MF. Measurement of ceruloplasmin from its oxidase activity in serum by use of o-dianisidine dihydrochloride. Clin Chem. 1974;20:1556–1563. [PubMed] [Google Scholar]
- 27.Richie JP, Jr, Lang CA. The determination of glutathione, cyst(e)ine, and other thiols and disulfides in biological samples using high-performance liquid chromatography with dual electrochemical detection. Anal Biochem. 1987;163:9–15. doi: 10.1016/0003-2697(87)90085-6. [DOI] [PubMed] [Google Scholar]
- 28.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 29.Popkin BM, Armstrong LE, Bray GM, Caballero B, Frei B, Willett WC. A new proposed guidance system for beverage consumption in the United States. Am J Clin Nutr. 2006;83:529–542. doi: 10.1093/ajcn.83.3.529. [DOI] [PubMed] [Google Scholar]
- 30.Bray GA, Nielsen SJ, Popkin BM. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am J Clin Nutr. 2004;79:537–543. doi: 10.1093/ajcn/79.4.537. [DOI] [PubMed] [Google Scholar]
- 31.Holbrook J, Fields M, Smith JC, Jr, Reiser S. Tissue distribution and excretion of copper-67 intraperitoneally administered to rats fed fructose or starch. J Nutr. 1986;16:831–838. doi: 10.1093/jn/116.5.831. [DOI] [PubMed] [Google Scholar]
- 32.Fields M, Holbrook J, Scholfield D, Smith JC, Jr, Reiser S. Effect of fructose or starch on copper-67 absorption and excretion by the rat. J Nutr. 1986;116:625–632. doi: 10.1093/jn/116.4.625. [DOI] [PubMed] [Google Scholar]
- 33.Harris ZL, Durley AP, Man TK, Gitlin JD. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc Natl Acad Sci USA. 1999;96:10812–10817. doi: 10.1073/pnas.96.19.10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fields M, Lewis CG. Hepatic iron overload may contribute to hypertriglyceridemia and hypercholesterolemia in copper-deficient rats. Metabolism. 1997;46:377–381. doi: 10.1016/s0026-0495(97)90051-2. [DOI] [PubMed] [Google Scholar]
- 35.Collins JF, Prohaska JR, Knutson MD. Metabolic crossroads of iron and copper. Nutr Rev. 2010;68:133–147. doi: 10.1111/j.1753-4887.2010.00271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Z, Zhang F, An P, Guo X, Shen Y, Tao Y, Wu Q, Zhang Y, Yu Y, et al. Ferroportin1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood. 2011;118:1912–1922. doi: 10.1182/blood-2011-01-330324. [DOI] [PubMed] [Google Scholar]
- 37.Prohaska JR. Changes in Cu, Zn-superoxide dismutase, cytochrome c oxidase, glutathione peroxidase and glutathione transferase activities in copper-deficient mice and rats. J Nutr. 1991;21:355–363. doi: 10.1093/jn/121.3.355. [DOI] [PubMed] [Google Scholar]
- 38.Geller BL, Winge DR. A method for distinguishing Cu, Zn- and Mn-containing superoxide dismutases. Anal Biochem. 1983 Jan;128(1):86–92. doi: 10.1016/0003-2697(83)90348-2. [DOI] [PubMed] [Google Scholar]