

Abstract
The inhibition of viral hemorrhagic septicemia rhabdovirus (VHSV) in vitro infection by pHs of <7 (low pH) has been previously reported. Nevertheless, the details of the mechanism underlying this effect remain obscure. We present evidence showing that low-pH inhibition occurs during a viral postadsorption step. Thus, while VHSV bound, replicated within single cells, and presented its G protein on the membranes of infected cells at both low and physiological pHs, both cell-to-cell spreading of infection (as estimated by the appearance of foci of infected cells) and fusion (as estimated by a syncytium assay) were inhibited by this low pH. The decreased VHSV titers and the inhibition of both cell-to-cell spreading of infection and fusion could be reversed by adjusting the pH to 7.5 at any time during infection. This effect should be taken into account to avoid false negatives in the diagnosis of VHSV by cell culture. On the other hand, the cell-to-cell spreading of infection at pH 7.5 could be stopped at any time by reducing the pH to 6.5. Since at low pH there were changes in the protein G conformation and smaller and imbalanced amounts of N with respect to M1, M2, and G viral proteins, alterations of the assembly and/or budding of VHSV are most probably involved in the absence of newly released infective virions.
In many enveloped viruses, after the virus particle is internalized by a receptor-mediated endocytosis mechanism (6), viral and cell membrane fusions are triggered by the decrease of the endosomal pH (30). Conformational changes are induced by the low pH in the viral glycoproteins (25, 51) to cause fusion. Thus, agents that inhibit the lowering of the pH in the endosomes, such as 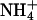 , also inhibit viral fusion and infectivity (11, 44, 49). However, it has been shown that a low-pH environment outside the host cells can also inhibit the in vitro viral replication and/or fusion of viruses such as vesicular stomatitis virus (VSV) (32), rabies virus (RV) (21, 22, 26, 35), viral hemorrhagic septicemia rhabdovirus (VHSV) (10, 50, 52), influenza virus (5), poliovirus (18), or retrovirus (4, 42, 56).
, also inhibit viral fusion and infectivity (11, 44, 49). However, it has been shown that a low-pH environment outside the host cells can also inhibit the in vitro viral replication and/or fusion of viruses such as vesicular stomatitis virus (VSV) (32), rabies virus (RV) (21, 22, 26, 35), viral hemorrhagic septicemia rhabdovirus (VHSV) (10, 50, 52), influenza virus (5), poliovirus (18), or retrovirus (4, 42, 56).
It was proposed earlier that the inhibition of VSV infectivity caused by low pH was due not to inhibition of viral RNA or protein synthesis (18) but to some yet-unidentified alterations in the nucleocapsid replication complex (32). Since then, there have been no further references about the mechanisms causing low-pH inhibition of VSV or any other rhabdoviruses, such as RV or VHSV. However, the study of this aspect of rhabdovirus infection might have important implications for the inactivation of virus in biological fluids (29) and for laboratories responsible for proper surveillance of these diseases, which should correctly diagnose possible false negatives.
To further study this phenomenon, we choose VHSV (a salmonid rhabdovirus) as a model among rhabdoviruses, because VHSV causes cytopathic effects of established fish cell lines only at pH 7.0 or higher (10, 50, 52), and this might cause problems in cell culture detection of this virus. The step of infection which is inhibited by low pH in VHSV remains to be investigated.
In the present work, we show that VHSV binding and replication do occur in the initially infected cells at low pH and that VHSV G protein is expressed on the membranes of the infected cells. Only the release of newly formed viral particles and, therefore, subsequent cell-to-cell spreading of infection require a higher pH. We have also shown that in VHSV, the low pH causes changes in the G protein conformation and an imbalance and reduction of the accumulated levels of M1, M2, and G proteins with respect to the N protein. Furthermore, we have shown that low-pH inhibition of VHSV infection and fusion could be reversed and infection could be resumed when the pH was readjusted to 7.5. This could have important applications in diagnosis.
MATERIALS AND METHODS
EPC cell culture and VHSV.
The VHSV strain VHSV-07.71, isolated in France from rainbow trout (Oncorhynchus mykiss) (33) was used. Supernatants from VHSV-07.71-infected epithelioma papulosum cyprini (EPC) cell cultures (17) were clarified by centrifugation at 2,000 × g for 20 min and kept in aliquots frozen at −70°C until used as the VHSV inoculum. To further purify VHSV, clarified supernatants were centrifuged at 100,000 × g for 2 h over a 10% sucrose cushion. The pellet was used as purified VHSV. EPC cell cultures were maintained as reported previously (1, 9). The cell culture medium contained RPMI 1640 (Dutch modification), 10% fetal calf serum (FCS), 1 mM pyruvate, 2 mM glutamine, 50 μg of gentamicin per ml, and 2 μg of amphotericin B (Fungizone) per ml. Cells were grown at 28°C under a 5% CO2 atmosphere.
Assay of VHSV-infected EPC cells by flow cytometry.
EPC cells in six-well plates, plated with about 300,000 cells/well, were infected with 2 × 10−3 focus-forming units (FFU) of VHSV per cell at pH 6, 6.5, 7, 7.5, 8, and 8.5. After 2 h at 14°C (virus adsorption), the inoculum was removed, cell monolayers were washed, and 1.5 ml of fresh culture medium adjusted to each pH was added. The cells were incubated for a further 22 h at 14°C. The cells were then trypsinized, resuspended in 0.1% bovine serum albumin-0.01% NaN3-5 mM EDTA in PBS (130 mM NaCl, 2 mM KCl, 8 mM Na2HPO4, 1.4 mM KH2PO4 [pH 7.4]), and centrifuged at 2,000 × g for 10 min. The pellets were incubated for 1 h 30 min in 1 ml of a 1:250 dilution of monoclonal antibody (MAb) against N protein of VHSV (2C9) (13, 47) in 0.1% bovine serum albumin-0.01% NaN3-5 mM Na EDTA-2% rabbit serum-10% FCS in PBS. Cells were then washed with PBS and incubated for 45 min with fluorescein-labeled rabbit anti-mouse immunoglobulin G (IgG) Ab (Sigma Chemical Co., St. Louis, Mo.) diluted 1:100. Finally, cells were washed three times, and 25,000 cells at each pH were analyzed with a Coulter Epics XL cytometry apparatus (Becton Dickinson, San Jose, Calif.) by using the Expo 32 software (Becton Dickinson). The percentage of infected cells was calculated as number of Ab-stained cells at each pH/number of Ab-stained cells at pH 7.5 ×100.
Immunostaining focus assay of VHSV.
A previously developed immunostaining focus assay for VHSV infectivity was used (34, 41). Briefly, VHSV diluted in 100 μl of cell culture medium with 2% FCS and adjusted to pH 7.5 was added to EPC cell monolayers, incubated for 2 h at 14°C, and then washed. After addition of fresh medium adjusted to either pH 7.5 or 6.5, the VHSV-infected EPC cell monolayers were incubated at different times at 14°C. After incubation, monolayers were fixed for 10 min in cold methanol and air dried. MAb 2C9 directed toward the N protein of VHSV, diluted 1,000-fold in dilution buffer (0.24 mM merthiolate, 5 g of Tween 20/liter, and 50 mg of phenol red/liter in PBS [pH 6.8]), or a cocktail of anti-G MAbs 3F1A12 and I10 (16), diluted 200-fold, was added to the wells (100 μl/well) and incubated for 1 h at room temperature. After washing with distilled water, 100 μl of peroxidase-labeled (Nordic, Tilburg, The Netherlands) or fluorescein-labeled (Sigma) rabbit anti-mouse IgG Ab was added per well and the incubation was continued for 30 min. After three washings by immersion in distilled water, 50 μl of diaminobenzidine (DAB) (1 mg/ml; Sigma) in PBS containing H2O2 (34, 47) was added per well, and the reaction was allowed to proceed until brown foci were detected with an inverted microscope. Once washed with water and air dried, brown foci (DAB-stained foci) of 15 to 20 DAB-stained cells or brown cells (DAB-stained single cells) were counted with an inverted microscope with a 10× ocular eye grid (34) VHSV titers were expressed as FFU per well. Fluorescence was estimated with an inverted fluorescence microscope (Leica Ltd., Cambridge, United Kingdom) with a digital camera.
Binding of biotin-labeled VHSV to EPC cells.
Purified VHSV was labeled with biotin by using a biotin-labeling kit according to instructions of the manufacturer (Pierce Biotechnology). EPC cell monolayers were incubated with biotin-labeled VHSV at a multiplicity of infection (MOI) of 1 for 2 h at 4°C. Supernatants were then washed, and streptavidin labeled with fluorescein (Nordic) in dilution buffer was added and incubated for 30 min. Fluorescence was detected with a Leica inverted microscope with a digital camera. The percentage of labeled cells at each pH was calculated as number of labeled cells/number of cells × 100.
Determination of binding of Abs to solid-phase VHSV by enzyme-linked immunosorbent assay (ELISA).
Polystyrene plates (Dynatech, Plochingen, Germany) were coated with 5 μg of purified VHSV per well in 100 μl of distilled water, incubated overnight at 37°C to dryness, and kept sealed with blue silica gel at 4°C. The polyclonal antibodies (PAbs) and MAbs were diluted in dilution buffer, adjusted to pH 7.5, and incubated overnight at 4°C. After blocking with dilution buffer and washing, the plates were incubated for 60 min at room temperature with the diluted PAbs or MAbs (100 μl/well) and washed once with distilled water. Peroxidase-labeled rabbit anti-mouse IgG Ab (Nordic) diluted 500-fold in dilution buffer was added (100 μl per well), incubated for 30 min, and then washed three times with distilled water. For color development, 50 μl of substrate buffer (150 mM sodium citrate, 3 mM H2O2, and 1 mg of o-phenylenediamine per liter [pH 4.8]) was pipetted per well, and the reaction was stopped with 50 μl of 4 M H2SO4 per well after 30 min. The absorbance at 620 nm was used to correct for individual nonsignificant differences between wells.
Immunoblotting of VHSV proteins.
The accumulation of VHSV proteins at pH 6.5 or 7.5 was estimated by Western blotting. Flasks of 150 cm2 were infected 2 h at 14°C with 10 FFU of VHSV per cell at pH 7.5 for 2 h, and then 50 ml of fresh medium adjusted to pH 7.5 or 6.5 was added. At 24 h postinfection, the flasks were frozen and thawed, the cells were pelleted by centrifugation at 6,000 × g for 45 min, and the pellets were resuspended in 400 μl of electrophoresis buffer. Sodium dodecyl sulfate-15% polyacrylamide gels were loaded with 20 μl of samples in electrophoresis buffer containing β-mercaptoethanol. To identify the viral proteins, both molecular weight markers (Bio-Rad, Richmond, Calif.) and purified VHSV were analyzed simultaneously. Two parallel gels were run, one for Coomassie blue staining and the other for Western blotting. The proteins in the gel were transferred for 3 h at 125 V in 2.5 mM Tris-9 mM glycine-20% methanol to nitrocellulose membranes (Bio-Rad). The membranes were then blocked with 2% dry milk-0.05% Tween 20-0.3% rabbit serum in PBS and incubated with anti-VHSV PAb (16), anti-M1 protein MAb 1C10 (46), anti-M2 protein MAb 4E4 (46), or anti-N protein MAb 2C9 (1:200 dilution) before incubation with a peroxidase-conjugated rabbit anti-mouse IgG antibody (Nordic) (1:500). Finally, the nitrocellulose membranes were treated with 1 mg of naphthol per ml-0.015% H2O2 (pH 7.5), washed, and dried. The bands were subjected to densitometry with the Scion program. Relative peak areas were calculated as area under protein/area under protein N × 100.
Syncytium formation by infected EPC cells.
Cell-to-cell fusion of VHSV-infected EPC monolayers was estimated after infecting EPC cell monolayers in 96-well plates with different MOIs of VHSV at pH 7.5. After removal of the viral inoculum, fresh medium adjusted to pH 7.5 or 6.5 was added. At 24 h postinfection, the monolayers were incubated with fusion medium (36) at pH 6 for 30 min at 14°C, followed by incubation with fusion medium at pH 7.5 for 2 h. After removal of the medium, EPC cell monolayers were fixed in cold methanol for 10 min, dried, and stained with Giemsa stain. The number of nuclei in syncytia of three or more nuclei was counted among 10,000 nuclei per well. Results were expressed as the percentage of nuclei in syncytia (i.e., number of nuclei in syncytia/total number of nuclei × 100).
Isolation of RNA from VHSV-infected EPC monolayers.
EPC cell monolayers were infected with VHSV at 5 FFU/cell. The virus inoculum was removed 1 h later, and the cells were incubated in RPMI 1640 medium-2% FCS adjusted to pH 6.5 or 7.5. The cells were harvested for RNA isolation at 18 h postinfection. Total RNA was obtained from the cytoplasm of infected EPC cells by using an RNA isolation kit (RNAgents, Promega, Wis.), according to the manufacturer's instructions and adjusted to the number of cells employed (∼4 × 107). The total RNA concentration was determined by optical density at 260 nm and ethidium bromide staining on agarose gels. The same amount of RNA template was used in each reverse transcription-PCR (RT-PCR) assay. The sequences of specific primers for VHSV G and N mRNAs were ATGGAATGGAACACTTTTTTCTTGGTG (forward) and GGTGACTCGATAAGTCACTCTGTGCAG (reverse) for G protein and GAAGATAGGAAGGTGATTGTGG (forward) and GAGTTTCCTGATGGCTGCCTTG (reverse) for N protein.
Determination of G and N mRNAs by RT-PCR.
The RT reaction was performed Moloney murine leukemia virus reverse transcriptase and the reverse specific primers for the VHSV G and N genes given above. Reaction samples were incubated at 42°C for 40 min. PCR amplification was carried out in a GeneAmp PCR system 2700 cycler (Applied Biosystems). Reaction samples (50 μl) contained a 200 μM concentration of each deoxynucleoside triphosphate, a 0.1 μM concentration of the 5′ and 3′ primers, and 5 U of Taq DNA polymerase (Roche, Mannheim, Germany). Temperature profiles of the PCR assays consisted of 30 cycles of denaturation for 1 min at 94°C, annealing for 1 min at 52°C, and elongation for 2 min at 72°C. PCR products were characterized by electrophoresis on ethidium bromide-stained 2% agarose gels.
RESULTS
Number of VHSV-infected EPC cells at different pHs.
The influence of pH on the total number of VHSV-infected EPC cells at different pHs (6 to 8.8) was estimated by flow cytometry by using the anti-N protein MAb 2C9 to stain VHSV-infected cells. At 24 h postinfection, the maximal numbers of VHSV-infected EPC cells (taken as 100%) were stained at pH 7.2 (Fig. 1A). This percentage decreased to 15% at pH 6.2. VHSV-infected cells could not be detected at pHs of lower than 6.2. The number of infected cells at pHs >7.4 also decreased, but ∼55% remained infected at pH 8.8, confirming that infections are more sensitive to acidic than to basic pHs, as previously reported for VHSV (10) and for other rhabdoviruses (18, 32). To further study the influence of pH on VHSV infection, we chose pHs 7.5 and 6.5 as representative of physiological and low pHs, respectively. Figure 1B shows that the total number of VHSV-infected cells at each time was always 102- to 103-fold higher at pH 7.5 than at pH 6.5.
FIG. 1.

Relative number of infected cells 24 h after VHSV infection at different pH values (A) and time course of the number of VHSV-infected cells at pH 7.5 and 6.5 (B). (A) EPC cell monolayers were infected with VHSV (MOI of 0.01) at pHs between 6 and 8.8. At 24 h after infection, the total number of infected cells at each pH was estimated by flow cytometry by using the anti-VHSV N protein MAb 2C9 to stain the cells. The percentage of VHSV-infected cells was calculated as the number of MAb-stained cells at each pH/number of MAb-stained cells at pH 7.5 × 100. (B) EPC cell monolayers were infected with VHSV (MOI of 0.01) for 2 h in medium at pH 7.5, and after washing, medium at pH 7.5 (•) or 6.5 (○) was added to the monolayers. The total number of infected cells at each pH was estimated by staining with anti-VHSV N protein MAb 2C9, peroxidase-conjugated rabbit anti-mouse IgG antibody, and DAB. The number of DAB-stained cells was then counted at different times after infection.
Influence of pH on binding of VHSV to EPC cells.
Binding of VHSV to EPC cells was estimated either indirectly by assaying the FFU in the supernatants after viral adsorption or directly by counting the number of fluorescent cells after adsorption with biotin-labeled VHSV. EPC cell monolayers were incubated with different numbers of FFU of VHSV at pH 7.5 and 6.5. After 2 h, supernatants were separated from the cells in the monolayer and then titrated for VHSV. Figure 2A shows that the percentages of VHSV recovered in the supernatants with respect to the initial inoculum were ∼53% at pH 7.5 and ∼40% at pH 6.5. These percentages were similar in all the inoculum sizes tested (from 25 to 250 FFU/well). The binding of biotin-labeled VHSV to EPC cells produced characteristic images of cells with fluorescent caps (Fig. 2C and D), and its quantification showed that the percentage of fluorescent EPC cells at pH 6.5 was 1.4-fold higher than that at pH 7.5 (Fig. 2B). Both of these results suggest that the small differences found in the binding of VHSV to the EPC cell monolayers did not explain the 102- to 103-fold lower titers of VHSV found at pH 6.5 compared to those obtained at pH 7.5 (Fig. 1B).
FIG. 2.

VHSV remaining in the supernatant after adsorption to EPC cell monolayers (A) and capping of biotin-labeled VHSV at the surface of infected EPC cells (B, C, and D) at pH 7.5 and 6.5. (A) EPC cell monolayers were infected with VHSV at different initial numbers of FFU per well at pH 7.5 (•) and 6.5 (○), and the supernatants were removed 2 h later. Percentages of FFU in the supernatant were calculated as FFU in the supernatant per well/initial FFU per well × 100. Averages and standard deviations from two different experiments, each performed in triplicate, are given. (B) Biotin-labeled VHSV was added to EPC cell monolayers at 4°C and left for 2 h at pH 7.5 and 6.5. After washing with medium, fluorescein-labeled streptavidin was added. The percentage of fluorescent cells was calculated as number of fluorescent cells at each pH per well/number of cells per well × 100. Averages and standard deviations from two different experiments, each performed in duplicate, are given. (C) An infected EPC cell under the normal light microscope. (D) The same infected EPC cell under UV light. Similar capping images were obtained at pH 7.5 and 6.5.
Kinetics of spreading of VHSV infection at pH 7.5 and 6.5.
To investigate cell-to-cell spreading of the infection, VHSV-infected EPC cell monolayers were stained with anti-VHSV Abs at different times during infection. In cultures infected at pH 7.5, DAB-stained foci containing one to two cells per foci appeared 12 h after infection. DAB-stained foci of 10 to 20 cells per foci appeared 24 h after infection (Fig. 3A and B), showing that the primary infection of single cells spread to neighboring cells within 24 h. Larger foci of 50 to 70 cells were observed after 36 h (Fig. 3A and B), and the entire cell monolayer was stained after 48 h and destroyed 60 h after infection (not shown). In contrast to those results, only single DAB-stained cells appeared at pH 6.5 (Fig. 3E and F), and their numbers remained constant from 12 to 60 h after infection, showing that at low pH, the infection did not spread from initially infected single cells to other cells. VHSV-infected RTG-2 (a cell line derived from trout) cell monolayers showed a staining profile (Fig. 3D and H) and kinetics of infection (not shown) similar to those for the VHSV-infected EPC cell monolayers (not shown) at the two pHs studied, thus showing that the low-pH inhibition of infection and its kinetics were independent of the cell line used for the in vitro VHSV infections. All of these experiments indicated that infection at pH 6.5 resulted in a single round of VHSV infection with new VHSV particles being unable to bud and/or to infect any other cells.
FIG. 3.

Staining of VHSV-infected cell monolayers with anti-VHSV G protein MAbs at pH 7.5 and 6.5. EPC (A, B, C, E, F, and G) or RTG-2 (D and H) cell monolayers were infected with VHSV (MOI of 0.01) for 2 h with medium at pH 7.5. After washing, medium at pH 7.5 (A, B, C, and D) or 6.5 (E, F, G, and H) was added to the cells. The cells were then fixed and stained with a cocktail of anti-G protein MAbs (3F1A12 and I10). (A and E) Distribution of focus size (number of infected cells per stained foci) at 12 h (□), 24 h (○), and 36 h (8) after VHSV infection. (B and F) EPC cell monolayers fixed with cold methanol and treated with peroxidase-labeled rabbit anti-mouse IgG Abs 24 h after infection. (C and G) EPC cell monolayers fixed with 4% paraformaldehyde and treated with fluorescein-labeled anti-mouse IgG Abs 24 h after infection. (D and H) RTG-2 cell monolayers fixed with cold methanol and treated with fluorescein-labeled anti-mouse IgG Abs 24 h after infection. Bars, 10 μm.
Expression and function of protein G at the membranes of VHSV-infected cells at pH 7.5 and 6.5.
To investigate whether the inhibition of VHSV infection at low pH was due to the absence of the G protein in the membranes of the infected cells, the presence of G protein was estimated by fluorescence of paraformaldehyde-fixed, VHSV-infected EPC cell monolayers at pH 7.5 and 6.5. Figure 3C and G show that membrane fluorescence was visible in EPC cells at both pHs. Whereas at pH 7.5 the membrane fluorescence was present in foci of stained cells, at pH 6.5 the membrane fluorescence was detected only in single cells. These results, although qualitative, showed that the inhibition of cell-to-cell spreading of VHSV at pH 6.5 is not caused by the absence of G protein in the membranes of the VHSV-infected cells.
Since the protein G expressed in the membranes could be functionally altered by low pH, its fusion properties were assayed after infection at pH 6.5 and 7.5 by the syncytium-forming assay (14). No syncytia were formed in EPC cell monolayers infected at pH 6.5 (Fig. 4C), while syncytia were abundant in those infected at pH 7.5, suggesting that the G protein present in the membranes of VHSV-infected cells at low pH might have some alteration in its fusion functionality. This alteration could be due to conformational changes, as it occurs in other rhabdoviruses with protein G at low pH.
FIG. 4.

(A and B) Spreading of VHSV infection at low pH after adjustment of pH to 7.5. VHSV-infected EPC cell monolayers (MOI of 0.01) were incubated for 24 h. The medium pH was then changed, and the cells were incubated for a further 24 h and stained 48 h after the beginning of the infection. When the medium was changed, monolayers at pH 7.5 were adjusted to pH 6.5 (A) and monolayers at pH 6.5 were adjusted to pH 7.5 (B). The number of foci at each focus size (number of stained cells per foci) was counted and expressed as number of foci per each focus size/total number of foci counted × 100. ○, 24 h at pH 7.5; •, 24 h + 24 h at pH 7.5; ◑, 24 h at pH 7.5 + 24 h at pH 6.5; □, 24 h at pH 6.5; ▪, 24 h + 24 h at pH 6.5; ◨, 24 h at pH 6.5 + 24 h at pH 7.5. (C) Syncytium formation of VHSV-infected EPC cells at pH 7.5 and 6.5. VHSV-infected EPC cell monolayers (MOI of 0.01) were incubated for 12 h. The medium pH was changed, and cells were incubated 12 h more and stained 24 h after the beginning of the infection. When the medium was changed, monolayers at pH 6.5 were adjusted to pH 7.5 and those at 7.5 were adjusted to pH 7.5 or 6.5. To assay for syncytium formation, the cultures were exposed to fusion medium at pH 6 for 30 min at 14°C, and the medium was changed to pH 7.5 and incubated for a further 2 h at 20°C. Results are expressed as the percentage of nuclei in syncytia as determined by the formula number of nuclei in syncytia/total number of nuclei × 100. Averages and standard deviations from two different experiments, each performed in triplicate, are given. •, 12 h + 12 h at pH 7.5; ○, 12 h + 12 h at pH 6.5; ◐, 12 h at pH 6.5 + 12 h at pH 7.5.
Reversibility of the inhibition of VHSV infectivity and fusion at pH 6.5 by pH 7.5.
Further experiments were performed to determine whether the VHSV infectivity inhibited at pH 6.5 could be activated by adjusting the pH to 7.5. Experiments were carried out with an initial adsorption step of 2 h at pH 7.5 or 6.5. The pH of the culture medium was then changed to 6.5 or 7.5, respectively, and maintained for 7 days more before harvesting. The titers of VHSV produced at pH 6.5 + 6.5 (pH at the adsorption step + pH at the infection step), including both cell-associated virus (cell lysates from VHSV-infected cells remaining as a monolayer) and cell-free virus (supernatants from VHSV-infected cell monolayers), were reduced about 104-fold compared to the titers of VHSV obtained at pH 7.5 + 7.5 (Table 1). No cytopathic effects were observed at pH 6.5 + 6.5 while all of the cells in the monolayer were lysed at pH 7.5 + 7.5. While 52.9% of the VHSV infectious particles produced were cell associated at pH 6.5 + 6.5, only 0.05% were cell associated and 108 FFU/ml were in the supernatant at pH 7.5 + 7.5 On the other hand, the titers of VHSV produced at pH 7.5 + 6.5 were also reduced more than 104-fold compared to the titers of VHSV obtained at pH 6.5 + 7.5, thus showing that it is the pH at the infection step rather than the pH at the adsorption step which inhibited the VHSV infectivity and confirming the conclusions drawn from the results of the binding and capping experiments.
TABLE 1.
Titers of supernatant and cell-associated VHSV at different adsorption and infection pHsa
| pH, adsorption + infection | VHSV titer,b FFU (106)/ml
|
Cell-associated virus, % | Cytopathic effect | |
|---|---|---|---|---|
| Supernatant | Cell lysate | |||
| 7.5 + 7.5 | 250.0000 | ≤0.1000 | ≤0.05 | Yes |
| 7.5 + 6.5 | 0.0067 | 0.0059 | 45.38 | No |
| 6.5 + 6.5 | 0.0025 | 0.0027 | 52.90 | No |
| 6.5 + 7.5 | 300.0000 | ≤0.0800 | ≤0.02 | Yes |
EPC cell monolayers in 25-cm2 flasks seeded with 9 × 106 cells per flask were incubated for 2 h with VHSV at an MOI of 0.01 at pH 7.5 or 6.5 (adsorption step). After washing, 7 ml of cell culture medium at pH 7.5 or 6.5 was added per flask, and the cultures were incubated for 7 days at 14°C (infection step). The supernatants (extracellular viral particles) and cell lysates (intracellular viral particles) were then titrated. Cell-associated virus was calculated as VHSV titer in cell lysate/VHSV titer in supernatant × 100.
Mean from two experiments.
Further experiments were carried out with an initial step of 24 h at pH 7.5 or 6.5 and then a second step reversing the pH of the culture medium to pH 6.5 or 7.5, respectively (Fig. 4). Foci of 2 to 4 stained cells obtained after 24 h at pH 7.5 increased to only 4 to 8 cells per 24 h after the pH was changed to 6.5, while the number of cells increased to 20 to 30 stained cells per foci 24 h after to medium was changed with the same initial pH (Fig. 4A). In contrast, foci of 1 to 2 stained cells obtained after 24 h at pH 6.5 increased to 3 to 10 cells 24 h after the pH was changed to 7.5 but remained at 1 to 2 stained cells 24 h after the medium was changed with the same initial pH (Fig. 4B). Both results showed that VHSV infection was stopped at pH 6.5 and that the VHSV infection spread from the initial infected cells to neighboring cells at pH 7.5, whatever the initial pH was.
The possible conformational changes of the G protein at pH 7.5 and 6.5 determined by using PAbs and MAbs of known epitope specificity were then studied by using binding of Abs to solid-phase VHSV by ELISA (Table 2). An increase of 2.5-fold in absorbance at pH 6.5 relative to pH 7.5 was obtained with PAb against frg11 (one of the regions of protein G involved in fusion). Furthermore, all of the MAbs tested had ∼1.4-fold-increased reactivity at pH 6.5 relative to pH 7.5, thus suggesting conformational changes in the respective regions defined by those MAbs (amino acids 140 and 433, 253, 139 to 153, and 399 to 413 for MAbs C10, 3F1A12, I10, and 1P1H3, respectively). The increase in Ab binding to VHSV at pH 6.5 was reported earlier (12) and appears to be due to the increase in the exposition of the epitopes on the surface of the G protein.
TABLE 2.
Binding of Abs to solid-phase VHSV at pH 6.5 relative to binding at pH 7.5a
| Ab | Epitopeb | Relative bindingc |
|---|---|---|
| Anti-frg 11 PAb | 56-110 | 2.5 ± 0.25 |
| Neutralizing MAb C10 | 140 and 433 | 1.3 ± 0.10 |
| Neutralizing MAb 3F1A12 | 253 | 1.4 ± 0.09 |
| MAb 110 | 139-153 | 1.4 ± 0.20 |
| MAb 1P1H3 | 399-413 | 1.4 ± 0.25 |
The binding of different Abs to solid-phase VHSV was estimated by ELISA at pH 7.5 and 6.5. The Abs were diluted 50-fold from 1/20 to 1/2,500 and those dilutions giving absorbances of between 0.5 to 1.5 were chosen for the calculations.
Epitope mapping is indicated by the corresponding amino acid number in protein G as previously reported (2, 14, 16).
The relative binding was calculated as absorbance at pH 6.5/absorbance at pH 7.5. Averages and standard deviations were calculated from two experiments, each performed in duplicate. Relative binding of the anti-N MAb 2C9 was 0.9 ± 0.1.
Other experiments were similarly designed to assay for the possible reversibility of fusion. The 1 to 2% of nuclei in syncytia obtained when the EPC cell monolayers were infected for 12 h + 12 h at pH 6.5 and then assayed for fusion activity increased to 20 to 40% when the pH was changed to 7.5 for 12 h more (Fig. 4C). In parallel cultures, EPC cell monolayers infected for 12 h + 12 h at pH 7.5 and then assayed for fusion could reach 100% of nuclei in syncytia, depending on the initial MOI (Fig. 4C).
VHSV proteins and mRNA accumulation in VHSV-infected cell monolayers at pH 7.5 and 6.5.
Protein extracts were prepared from VHSV-infected EPC cell monolayers (MOI of 10) at 24 h postinfection at pH 7.5 and 6.5. These protein extracts were electrophoresed, electrotransferred to nylon membranes, and then stained with a PAb against VHSV (Fig. 5A, inset). Figure 5A shows the densitometry of the bands corresponding to immunoblotted N, M1, M2, and G proteins relative to the N protein band at pH 7.5. Reductions of 1.6-, 2.2-, and 3.6-fold in the accumulated amounts of M1, M2, and G proteins, respectively, at pH 6.5 relative to pH 7.5 were found. In contrast, the N protein remained at similar concentrations at both pHs, indicating that the low pH caused not only a reduction but also an imbalance in the accumulation of VHSV proteins. Similar results were found when specific MAbs for the N (2C9), M1 (1C10), and M2 (4E4) proteins (46) were used for the immunoblotting (data not shown).
FIG. 5.

Western blotting analysis of N, M1, M2, and G proteins (A) and RT-PCR of G and N cDNAs (B) present in VHSV-infected cells at pH 7.5 or 6.5. (A) VHSV-infected EPC cell monolayers (MOI of 10) were harvested at 24 h postinfection, concentrated by centrifugation, lysed, and separated by polyacrylamide gel electrophoresis. After transfer to nitrocellulose membranes, they were treated with a PAb against VHSV. Inset, Western blotting of VHSV proteins at pH 6.5 and 7.5. Bands and Western blotting were measured with a densitometer, and the relative peak areas were calculated as the area under each protein band at each pH/area under protein N band at pH 7.5 × 100. Averages and standard deviations from two different experiments are given. White bars, densitometry of proteins at pH 6.5. Black bars, densitometry of proteins at pH 7.5. (B). An RT-PCR was performed with total RNA isolated from VHSV-infected EPC cells at pH 7.5 or 6.5 at 24 h postinfection. Inset, agarose gel of PCR products obtained by RT and amplification of the same amount of RNA, using specific primers for VHSV G and N proteins. cDNA quantification (arbitrary units) was performed by densitometry of the DNA bands in the agarose gel. Relative peak areas were calculated as the area under each cDNA band at each pH/area under cDNA from N at pH 7.5 × 100. Averages and standard deviations from two different experiments are given. White bars, densitometry of cDNAs at pH 6.5. Black bars, densitometry of cDNAs at pH 7.5.
An RT-PCR was performed with total RNA isolated from VHSV-infected EPC cells at pH 7.5 or 6.5 at 18 h postinfection, as described in Materials and Methods. Primers specific for N and G protein cDNAs generated fragments of the expected size, i.e., 408- and 317-bp DNA products, respectively (Fig. 5B). However, while the intensity of the N band was similar at pH 7.5 and 6.5, the intensity of the G band was 2.8-fold lower at pH 6.5 (Fig. 5B), confirming an imbalance and a reduction of N and G protein accumulation profiles at low pH.
DISCUSSION
Incubation of VHSV-infected cells at pH 6.5 resulted in more than 99% inhibition of viral titers compared to those at pH 7.5, confirming well-known observations described by several authors years ago (10, 50, 53). However, because of the importance of this phenomenon for current diagnostic procedures based on cell culture techniques and since an explanation was not offered and the step of infection inhibited was not identified, this work focused on these questions. Evidence was gathered to show that the low-pH inhibition of VHSV infection was not due to alterations in virus binding, to alterations in the first round of viral replication, or to the absence of G protein in the membranes of the infected cells. Low-pH inhibition of VHSV infection was accompanied by inhibition of cell-to-cell spreading of infection and fusion, inactivation of the G protein fusion activity, changes in G protein conformation, and imbalance and reduction of accumulation of the M1, M2, and G proteins.
The similarity of the results of binding and capping of VHSV at pH 7.5 and 6.5 indicated that the low-pH inhibition of infectivity operated on a postadsorption step. The results even showed a slightly higher binding of VHSV to EPC cells at low pH, thus confirming earlier reported results of binding of VHSV to model phospholipid liposomes (8). Attachment of RV (54) was also more efficient at low pH than at higher pHs, and similar results were described for murine retrovirus (42) and for salmon anemia virus, an orthomyxovirus-like virus infecting fish (11). In contrast, differences in binding at low pH were described for VSV, which could account for at least some of the observed inhibition of infectivity (32).
Although the absence of G protein in the membranes of infected cells could have explained the inhibition of VHSV infectivity at low pH, as happens in VSV or RV lacking the corresponding G protein (15, 37), the presence of G protein was detected in VHSV-infected EPC cell membranes at both pH 6.5 and 7.5. However, the G protein produced at pH 6.5 lacked fusogenic activity, suggesting that the viral G protein at pH 6.5 is exposed at the cell surface in an inactive fusion state. For RV it has been demonstrated that the G protein can assume at least three different states and that there is a pH-dependent equilibrium among these states (26, 27). To avoid undesirable fusion, the G protein of RV is transported in an inactive conformation because the pH of the Golgi compartments is sufficiently low (27). However, G protein must acquire its native structure at the cell surface to shift towards an activated state at fusion pH (around pH 6). At a pH above 7 at the cell surface, it needs to acquire the native structure (26, 27). In VSV, these same three states have been postulated (7, 43, 55).
Inactivation of fusion activity of the G protein observed in the case of RV is due to conformational changes in the G protein (23, 25, 27). Conformational changes in the G protein of VHSV seem also to occur by exposure to low pH, as shown by alterations in the binding of several MAbs (Table 1) and PAbs against frg11, a region involved in fusion (12, 36, 40, 45). Recently, a domain which regulates the low-pH-induced conformational changes has been identified both RV and VSV (19, 24, 35, 48). This domain extends from amino acid 366 to 415 of the G protein of RV (24). In VHSV, the alteration in the binding of MAb IPH13, mapping from amino acid 399 to 413 of the G protein sequence (Table 1), to solid-phase VHSV at pH 6.5 suggests that, as in mammalian rhabdovirus, the carboxyl terminus of the G protein of VHSV also plays a role in the regulation of the low-pH conformational changes.
The inhibitory effects of low pH were active at any time during the infection process from 2 h to weeks. However, the spreading of cytopathic effects (not shown), the VHSV titers (Table 2), the cell-to-cell spreading of VHSV infection (Fig. 4A and B), and the fusion (Fig. 4C), which were inhibited at low pH, resumed when the pH was adjusted to physiological. On the other hand, VHSV infection and cell-to-cell spreading at physiological pH could be stopped at any time during infection by adjustment to pH 6.5. Therefore, all of the detected inhibitory effects of low pH upon the VHSV infection process seem to be reversible, thus suggesting a simple confirmatory procedure to apply to diagnostics.
Immunostaining of single cells or foci at low or at physiological pH, respectively, provided evidence that the observed low-pH inhibition of infectivity is localized at the step of cell-to-cell spreading of VHSV particles. VHSV replication inside the initially infected cells occurred between pH 6.6 and 8.2; however, cell-to-cell spreading of newly released virus was restricted to pHs higher than 6.5. Thus, DAB-stained single cells but no DAB-stained foci were observed at pH 6.5, and significant numbers of DAB-stained foci were observed only at pHs >7.0, either because a lower number of newly virions were released, because the new virus released was inactivated, or both. It did not seem possible that new viruses were being released at low pH, because the titers were always low even when the assay was performed at physiological pH (Table 1). Since it has been reported that highly efficient budding of rhabdovirus is accomplished by a concerted action of both G protein and other core proteins (38), the above-mentioned observations probably reflect an inefficient assembly and/or budding of rhabdovirus particles at pH 6.5, indicating that the reduced amount of new virus released is determined not only by the change in conformation of the G protein (detected at low pH [Table 1]) but also by changes in other proteins.
Confirming these hypotheses, an imbalance in the levels of M1, M2, and G proteins relative to N protein at physiological pH was observed at low pH. In addition, at low pH, the amounts of M1, M2, and G proteins were lower than those at physiological pH relative to the amounts of N protein. This effect was specific for the viral proteins, since expression of green fluorescent protein in transfected EPC cells at low pH did not significantly change with respect to the expression at physiological pH (data not shown). The induction of an imbalance of the viral proteins observed in the infected cells at low pH could explain the lower yield of VHSV particles detected in the supernatants at pH 6.5 (Table 1), due to difficulties in the achievement of the optimal protein molar ratios for efficient virus assembly and/or budding (20, 38, 39). In contrast, no significant effect of low pH on mRNA or protein synthesis was reported for VSV (18, 32).
In addition to the imbalance and lower accumulation profiles of the M1, M2, and G proteins, the conformational changes detected in the G protein at pH 6.5 (Table 2) could also reduce the required interactions between the G and M1 proteins for budding and thus increase the difficulty for new particles to get outside the cell. It is known that low pH alters the maturation, association, assembly, and/or structure of one or more viral VSV proteins on nucleocapsids (32) and that an optimal molar ratio between the N and P proteins is required for VSV replication (28). Other interactions among the M1, M2, and G proteins could also be altered by low pH; for instance, in influenza virus the analogous M1 protein dissociates from newly synthesized viral ribonucleocapsids when exposed to low pH (5).
The imbalance and reduction of the accumulated levels of the M1, M2, and G proteins with respect to the N protein, which remained at similar concentrations at both pHs, might be due to the effect of L protein attenuation and/or alteration of the L-M1 complex interactions that are necessary for functional activity. The amount of VHSV proteins accumulated at pH 6.5 decreased as the distance of their genes increased from the 3′ termini of the viral RNA (M1 > M2 > G). Since it is accepted that the level of gene transcription in rhabdoviruses is inversely proportional to the gene distance from the 3′ viral termini (3, 31), due to an attenuation of the polymerase at each gene junction (28), this could be one explanation for the reduced levels of VHSV proteins accumulated at low pH. However, there is no evidence for any of the hypotheses, and they remain to be further investigated.
In conclusion, the reduction of VHSV cytopathic effects and titers found at low pH in this and related earlier work (10, 50, 53) was accompanied by an inhibition of cell-to-cell spreading of infection, fusion inhibition, and imbalance of the optimal viral protein ratios in the infected cells. Since the low-pH inhibition could be reversed as the pH was returned to physiological values, it might be used to determine the correct diagnosis of otherwise negative samples. Further reconstitution studies with isolated proteins could shed more light on the mechanism of inhibition of VHSV infectivity at low pH.
Acknowledgments
This work was supported by the INIA ACU01-03 and SC0046, Spain.
Thanks are due to Beatriz Bonmati for technical assistance and to Carolina Tafalla for reviewing the English of the manuscript.
REFERENCES
- 1.Basurco, B., and J. M. Coll. 1989. Spanish isolates and reference strains of viral haemorrhagic septicaemia virus shown similar protein size patterns. Bull. Assoc. Fish Pathol. 9:92-95. [Google Scholar]
- 2.Bearzotti, M., A. F. Monnier, P. Vende, J. Grosclaude, P. DeKinkelin, and A. Benmansour. 1995. The glycoprotein of viral hemorrhagic septicemia virus (VHSV): antigenicity and role in virulence. Vet. Res. 26:413-422. [PubMed] [Google Scholar]
- 3.Benmansour, A., G. Paubert, J. Bernard, and P. De Kinkelin. 1994. The polymerase-associated protein (M1) and the matrix protein (M2) from a virulent and avirulent strain of viral hemorrhagic septicemia virus (VHSV), a fish rhabdovirus. Virology 198:602-612. [DOI] [PubMed] [Google Scholar]
- 4.Brorson, K., S. Krejci, K. Lee, E. Hamilton, K. Stein, and Y. Xu. 2003. Bracketed generic inactivation of rodent retroviruses by low pH treatment for monoclonal antibodies and recombinant proteins. Biotechnol. Bioeng. 82:321-329. [DOI] [PubMed] [Google Scholar]
- 5.Bui, M., G. Whittaker, and A. Helenius. 1996. Effect of M1 protein and low pH on nuclear transport of influenza virus ribonucleoproteins. J. Virol. 70:8391-8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carneiro, F. A., M. L. Bianconi, G. Weissmuller, F. Stauffer, and A. T. Da Poian. 2002. Membrane recognition by vesicular stomatitis virus involves enthalpy-driven protein-lipid interactions. J. Virol. 76:3756-3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clague, M. J., C. Schoch, L. Zech, and R. Blumenthal. 1990. Gating kinetics of pH-activated membrane fusion of vesicular stomatitis virus with cells: stopped-flow measurements by dequenching of octadecylrhodamine fluorescence. Biochemistry 29:1303-1308. [DOI] [PubMed] [Google Scholar]
- 8.Coll, J. M. 1995. Synthetic peptides reveal a phospholipid binding domain in the glycoprotein of VHSV, a salmonid rhabdovirus. Vet. Res. 26:399-407. [PubMed] [Google Scholar]
- 9.DeKinkelin, P. 1972. Le virus D'Egtved. II. Purification. Ann. Rech. Vet. 3:199-208. [Google Scholar]
- 10.DeKinkelin, P., and R. Scherrer. 1970. Le virus Egtved. I. Stabilité, dévelopment et structure du virus de la souche dainose FÇ1. Ann. Rech. Vet. 1:17-30. [Google Scholar]
- 11.Eliassen, T. M., M. K. Froystad, B. H. Dannevig, M. Jankowska, A. Brech, K. Falk, K. Romoren, and T. Gjoen. 2000. Initial events in infectious salmon anemia virus infection: evidence for the requirement of a low-pH step. J. Virol. 74:218-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Estepa, A., and J. M. Coll. 1996. Pepscan mapping and fusion related properties of the major phosphatidylserine-binding domain of the glycoprotein of viral hemorrhagic septicemia virus, a salmonid rhabdovirus. Virology 216:60-70. [DOI] [PubMed] [Google Scholar]
- 13.Estepa, A., D. Frias, and J. M. Coll. 1992. Susceptibility of trout kidney macrophages to viral haemorrhagic septicaemia virus. Viral Immunol. 5:283-292. [DOI] [PubMed] [Google Scholar]
- 14.Estepa, A. M., A. I. Rocha, V. Mas, L. Perez, J. A. Encinar, E. Nunez, A. Fernandez, J. M. Gonzalez Ros, F. Gavilanes, and J. M. Coll. 2001. A protein G fragment from the salmonid viral hemorrhagic septicemia rhabdovirus induces cell-to-cell fusion and membrane phosphatidylserine translocation at low pH. J. Biol. Chem. 276:46268-46275. [DOI] [PubMed] [Google Scholar]
- 15.Etessami, R., K.-K. Conzelmann, B. Fadai-Ghotbi, B. Natelson, H. Tsiang, and P.-E. Ceccaldi. 2000. Spread and pathogenic characteristics of a G-deficient rabies virus recombinant: an in vitro and in vivo study. J. Gen. Virol. 81:2147-2153. [DOI] [PubMed] [Google Scholar]
- 16.Fernandez-Alonso, M., G. Lorenzo, L. Perez, R. Bullido, A. Estepa, N. Lorenzen, and J. M. Coll. 1998. Mapping of linear antibody epitopes of the glycoprotein of VHSV, a salmonid rhabdovirus. Dis. Aquat. Organ. 34:167-176. [DOI] [PubMed] [Google Scholar]
- 17.Fijan, N., D. Sulimanovic, M. Bearzotti, D. Mizinic, L. O. Zwillenberg, S. Chilmonczyk, J. F. Vautherot, and P. de Kinkelin. 1983. Some properties of the Epithelioma papulosum cyprini (EPC) cell line from carp Cyprinus carpio. Ann. Virol. 134:207-220. [Google Scholar]
- 18.Fiszman, M., J. B. Leaute, C. Chany, and M. Girard. 1974. Mode of action of acid pH values on the development of vesicular stomatitis virus. J. Virol. 13:801-808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galbrath, D. W., C. A. Zether, K. R. Harkins, and C. L. Alonso. 1992. Biosynthesis, processing and targeting of the G-protein of vesicular stomatitis virus in tobacco protoplast. Planta 186:324-336. [DOI] [PubMed] [Google Scholar]
- 20.Gao, Y., N. J. Greenfield, D. Z. Cleverley, and J. Lenard. 1996. The transcriptional form of the phosphoprotein of vesicular stomatitis virus is a trimer: structure and stability. Biochemistry 35:14569-14573. [DOI] [PubMed] [Google Scholar]
- 21.Gaudier, M., Y. Gaudin, and M. Knossow. 2002. Crystal structure of vesicular stomatitis virus matrix protein. EMBO J. 21:2886-2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaudin, Y. 2000. Reversibility in fusion protein conformational changes. The intriguing case of rhabdovirus-induced membrane fusion. Subcell. Biochem. 34:379-408. [DOI] [PubMed] [Google Scholar]
- 23.Gaudin, Y., P. de Kinkelin, and A. Benmansour. 1999. Mutations in the glycoprotein of viral haemorrhagic septicaemia virus that affect virulence for fish and the pH threshold for membrane fusion. J. Gen. Virol. 80:1221-1229. [DOI] [PubMed] [Google Scholar]
- 24.Gaudin, Y., H. Raux, A. Flamand, and R. Ruigrok. 1996. Identification of amino acids controlling the low-pH-induced conformational change of rabies virus glycoprotein. J. Virol. 70:7371-7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaudin, Y., R. W. H. Ruigrok, and J. Brunner. 1995. Low-pH induced conformational changes in viral fusion proteins: implications for the fusion mechanism. J. Gen. Virol. 76:1541-1556. [DOI] [PubMed] [Google Scholar]
- 26.Gaudin, Y., R. W. H. Ruigrok, M. Knossow, and A. Flamand. 1993. Low-pH conformational changes of rabies virus glycoprotein and their role in membrane fusion. J. Virol. 67:1365-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaudin, Y., C. Tuffereau, P. Durrer, A. Flamand, and R. W. H. Ruigrok. 1995. Biological function of the low-pH, fusion-inactive conformation of rabies virus glycoprotein (G): G is transported in a fusion-inactive state-like conformation. J. Virol. 69:5528-5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Green, T. J., S. Macpherson, S. Qiu, J. Lebowitz, G. W. Wertz, and M. Luo. 2000. Study of the assembly of vesicular stomatitis virus N protein: role of the P protein. J. Virol. 74:9515-9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kasermann, F., K. Wyss, and C. Kempf. 2001. Virus inactivation and protein modifications by ethyleneimines. Antiviral Res. 52:33-41. [DOI] [PubMed] [Google Scholar]
- 30.Kielian, M., and S. Jungerwirth. 1990. Mechanisms of enveloped virus entry into cells. Mol. Biol. Med. 7:17-31. [PubMed] [Google Scholar]
- 31.Kurath, G., and J. C. Leong. 1985. Characterization of infectious hematopoietic necrosis virus mRNA species reveals a nonvirion rhabdovirus protein. J. Virol. 53:462-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.La Ferla, F. M., and R. W. Peluso. 1989. The 1:1 N-NS protein complex of vesicular stomatitis virus is essential for efficient genome replication. J. Virol. 63:3852-3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.LeBerre, M., P. De Kinkelin, and A. Metzger. 1977. Identification sérologique des rhabdovirus des salmonidés. Bull. Office Int. Epizooties 87:391-393. [Google Scholar]
- 34.Lorenzo, G., A. Estepa, and J. M. Coll. 1996. Fast neutralization/immunoperoxidase assay for viral haemorrhagic septicemia with anti-nucleoprotein monoclonal antibody. J. Virol. Methods 58:1-6. [DOI] [PubMed] [Google Scholar]
- 35.Maillard, A., M. Domanski, P. Brunet, A. Chaffotte, E. Guittet, and Y. Gaudin. 2003. Spectroscopic characterization of two peptides derived from the stem of rabies virus glycoprotein. Virus Res. 93:151-158. [DOI] [PubMed] [Google Scholar]
- 36.Mas, V., L. Perez, J. A. Encinar, M. T. Pastor, A. Rocha, E. Perez-Paya, A. Ferrer-Montiel, J. M. Gonzalez Ros, A. Estepa, and J. M. Coll. 2002. Salmonid viral haemorrhagic septicaemia virus: fusion-related enhancement of virus infectivity by peptides derived from viral glycoprotein G or a combinatorial library. J. Gen. Virol. 83:2671-2681. [DOI] [PubMed] [Google Scholar]
- 37.Mebatsion, T., M. Konig, and K. K. Conzelmann. 1996. Budding of rabies virus particles in the absence of the spike protein. Cell 84:941-951. [DOI] [PubMed] [Google Scholar]
- 38.Mebatsion, T., F. Weiland, and K.-K. Conzelmann. 1999. Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. J. Virol. 73:242-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morimoto, K., H. D. Foley, J. P. McGettigan, M. J. Schnell, and B. Dietzschold. 2000. Reinvestigation of the role of the rabies virus glycoprotein in viral pathogenesis using a reverse genetics approach. J. Neurovirol. 6:373-381. [DOI] [PubMed] [Google Scholar]
- 40.Nunez, E., A. M. Fernandez, A. Estepa, J. M. Gonzalez-Ros, F. Gavilanes, and J. M. Coll. 1998. Phospholipid interactions of a peptide from the fusion-related domain of the glycoprotein of VHSV, a fish rhabdovirus. Virology 243:322-330. [DOI] [PubMed] [Google Scholar]
- 41.Perez, L., V. Mas, J. Coll, and A. Estepa. 2002. Enhanced detection of viral hemorrhagic septicemia virus (a salmonid rhabdovirus) by pretreatment of the virus with a combinatorial library-selected peptide. J. Virol. Methods 106:17-23. [DOI] [PubMed] [Google Scholar]
- 42.Portis, J. L., F. J. McAtee, and L. H. Evans. 1985. Infectious entry of murine retroviruses into mouse cells: evidence of a postadsorption step inhibited by acidic pH. J. Virol. 55:806-812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Puri, A., J. Winick, R. J. Lowy, D. Covell, O. Eidelman, A. Walter, and R. Blumenthal. 1988. Activation of vesicular stomatitis virus fusion with cells by pretreatment at low pH. J. Biol. Chem. 263:4749-4753. [PubMed] [Google Scholar]
- 44.Rigaut, K. D., D. E. Birk, and J. Lenard. 1991. Intracellular distribution of input vesicular stomatitis virus proteins after uncoating. J. Virol. 65:2622-2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rocha, A., M. Fernandez-Alonso, V. Mas, L. Perez, A. Estepa, and J. M. Coll. 2002. Antibody response to a fragment of the protein G of VHS rhabdovirus in immunised trout. Vet. Immunol. Immunopathol. 86:89-99. [DOI] [PubMed] [Google Scholar]
- 46.Sanz, F., B. Basurco, M. Babin, J. Dominguez, and J. M. Coll. 1993. Monoclonal antibodies against the structural proteins of viral haemorrhagic septicaemia virus isolates. J. Fish Dis. 16:53-63. [Google Scholar]
- 47.Sanz, F., and J. M. Coll. 1992. Detection of viral haemorrhagic septicemia virus by direct immunoperoxidase with selected anti-nucleoprotein monoclonal antibody. Bull. Eur. Assoc. Fish Pathol. 12:116-119. [Google Scholar]
- 48.Shokralla, S., Y. He, E. Wanas, and H. P. Ghosh. 1998. Mutations in a carboxy-terminal region of vesicular stomatitis virus glycoprotein G that affect membrane fusion activity. Virology 242:39-50. [DOI] [PubMed] [Google Scholar]
- 49.Superti, F., M. Derer, and H. Tsiang. 1984. Mechanism of rabies virus entry into CER cells. J. Gen. Virol. 65:781-789. [DOI] [PubMed] [Google Scholar]
- 50.Vestergaard-Jorgensen, P. E. 1972. Egtved virus: antigenic variation in 76 virus isolates examined in neutralization test and by means of the fluorescent antibody technique. Symp. Zool. Soc. 30:333-339. [Google Scholar]
- 51.Weissenhorn, W., A. Dessen, L. J. Calder, S. C. Harrison, J. J. Skehel, and D. C. Wiley. 1999. Structural basis for membrane fusion by enveloped viruses. Mol. Membr. Biol. 16:3-9. [DOI] [PubMed] [Google Scholar]
- 52.Wolf, K., and M. C. Quimby. 1973. Fish viruses: buffers and methods for plaquing eight agents under normal atmosphere. Appl. Microbiol. 25:659-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolf, K., M. C. Quimby, L. L. Pettijohn, and M. L. Landolt. 1973. Fish viruses: isolation and identification of infectious hematopoietic necrosis virus in Eastern North America. J. Fish Res. 30:1625-1627. [Google Scholar]
- 54.Wunner, W. H., K. J. Reagan, and H. Koprowski. 1984. Characterization of saturable binding sites for rabies virus. J. Virol. 50:691-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yao, Y., K. Ghosh, R. F. Epand, R. M. Epand, and H. P. Ghosh. 2003. Membrane fusion activity of vesicular stomatitis virus glycoprotein G is induced by low pH but not by heat or denaturant. Virology 310:319-332. [DOI] [PubMed] [Google Scholar]
- 56.Ye, K., H. K. Dhiman, J. Suhan, and J. S. Schultz. 2003. Effect of pH on infectivity and morphology of ecotropic Moloney murine leukemia virus. Biotechnol. Prog. 19:538-543. [DOI] [PubMed] [Google Scholar]