

Abstract
A variety of exposure regimens of cigarette smoke have been used in animal models of lung diseases. In this study, we compared biological responses of smoke exposure in rats, using different smoke concentrations (wet total particulate matter [WTPM]), daily exposure durations, and total days of exposure. As a range-finding acute study, we first compared pulmonary responses between SD and F344 strains after a single nose-only exposure to mainstream cigarette smoke or LPS. Secondly, F344 rats were exposed to cigarette smoke for 2 or 13 weeks under the comparable daily exposure dose (WTPM concentration x daily exposure duration; according to Haber’s rule) but at a different WTPM concentration or daily exposure duration. Blood carboxylhemoglobin was increased linearly to the WTPM concentration, while urinary nicotine plus cotinine value was higher for the longer daily exposure than the corresponding shorter exposure groups. Gamma glutamyl transferase activity in bronchoalveolar lavage fluid (BALF) was increased dose dependently after 2 and 13 weeks of cigarette smoke exposure, while the neutrophil content in BALF was not increased notably. Smoke-exposed groups showed reduced body weight gain and increased relative lung and heart weights. While BALF parameters and the relative lung weights suggest pulmonary responses, histopathological examination showed epithelial lesions mainly in the upper respiratory organs (nose and larynx). Collectively, the results indicate that, under the employed study design, the equivalent daily exposure dose (exposure concentration x duration) induces equivalent pulmonary responses in rats.
Keywords: cigarette smoke, inhalation, exposure marker, bronchoalveolar lavage fluid (BALF), pulmonary inflammation
Introduction
The biological effects of cigarette smoke have been investigated since the 1960s. Previous studies including epidemiological analysis suggest prolonged smoking increases the risk for chronic obstructive pulmonary disease (COPD), lung cancer, and cardiovascular disease. However, despite many researches attempt to establish the causative relationship between the impact of cigarette smoking and the pathogenesis of these diseases1,2,3, the definitive mechanisms have not been elucidated.
For in vivo smoke studies, literatures on biological effects of cigarettes smoking report a variety of exposure regimens and animal models4,5,6,7,8,9,10,11,12,13. As part of product stewardship, many 90-day rodent inhalation studies have been conducted to investigate the biological impacts of ingredients added to cigarettes14,15,16 , typically using nose-only exposures to SD rats according to the OECD and/or NTP guidelines17,18. In these tobacco ingredient studies, the exposure regimens are tightly defined and controlled based on the wet total particulate matter (WTPM) and the exposure duration of 1 to 6 h per day, 5 or 7 days per week for 13 weeks13. Because cigarette smoke exposure can induce respiratory suppression in exposed animals16, exposure biomarkers in blood and/or urine are commonly measured to aid the evaluation between the exposure level and biological effects.
Unlike ingredient testing, many in vivo cigarette smoke disease models, for example on COPD 19,20, have been conducted under manually controlled exposure regimens and mainly using mice. Most of these investigational studies have used chronic whole-body exposures that were controlled according to constant numbers of cigarettes per day, rather than a definite WTPM concentration and daily exposure duration. In addition, only a few studies of disease models6,21,22 have used rats. Considering the need to assess a variety of COPD-related biochemical, physiological (e.g., lung function), and histopathological parameters in smoke-exposed animals, rats could be a useful model for measuring these parameters reliably due to their body size compared to mice.
In the area of inhalation toxicology, Haber’s rule23 is commonly used to estimate the relationship between the exposure dosimetry and the biological effect based on the theory that an equivalent integrated dose of concentration (C) × time (t) induces a comparable toxic effect (k). An example of the use of this theory is the study of pesticides24. However, in studies involving highly active chemicals such as chlorine, lung injury was reported depending not only on the total dose but also each of the exposure concentration and time23,25. We previously reported that an equivalent weekly exposure resulted in comparable biological effects in a subchronic inhalation study using SD rats13, however, the application of Haber’s rule at the time was limited to comparison within 13-week subchronic study durations using SD rats.
In this study, we performed a series of different exposure tests (according to Haber’s rule), comparing different daily exposure regimens under acute as well as subchronic smoke inhalation: Potential differences in terms of exposure regimen and study duration were assessed based on exposure markers, bronchoalveolar lavage fluid (BALF) parameters, and histopathological evaluation.
Materials and Methods
Cigarette and smoke generation
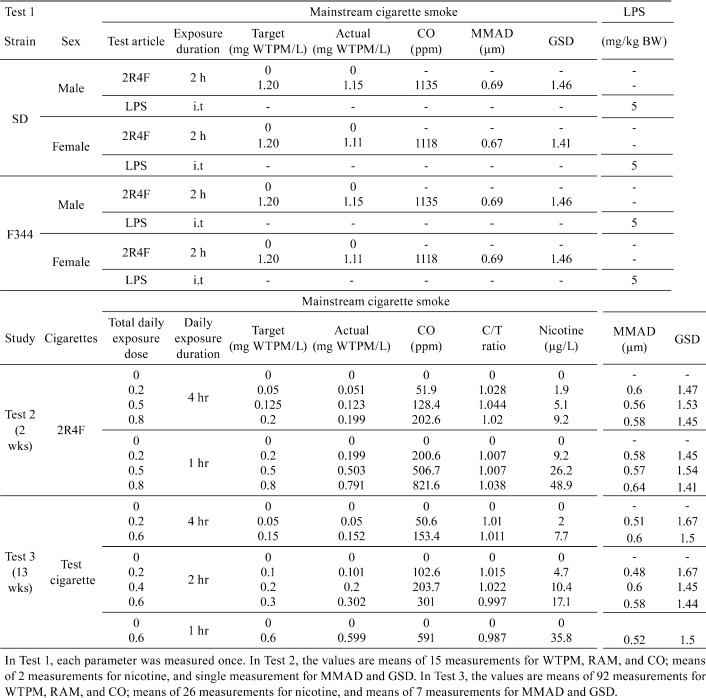
In Test 1 and 2, 2R4F cigarettes from University of Kentucky (Lexington, KY, USA) were used, which had tar, nicotine, and CO values of 11.7, 0.85, and 13 mg/cigarette, respectively. In Test 3, test cigarettes were manufactured by JT with conventional manufacturing equipment. Test 3 cigarette had tar, nicotine and CO values of 5.5, 0.55, and 6.6 mg/cigarette, respectively, according to ISO (International Organization for Standardization) 3402. Cigarettes were smoked in basic conformity with ISO 4387. The cigarettes conditioning and smoke generation methods used were described in our previous studies (Renne et al., 2006).
Characterization of smoke exposure atmosphere
Mainstream cigarette smoke was characterized as described in previous studies16. Briefly, the concentrations of wet total particulate matter (WTPM) and carbon monoxide (CO) were monitored via real-time aerosol monitor (RAM; Microdust, Pro; Casella, Amherst, NH, USA) and CO monitor (California Analytical Instruments, Inc, Orange, CA, USA), respectively. The coefficient of variation (% CV) of exposure concentration (WTPM) was within ± 10% by gravimetric analysis using Cambridge 47-mm glass-fiber filter (Performance Systematix Inc., Grand Rapids, MI, USA). Mean actual exposure concentrations were calculated from the mass collected on the filters and the total volume of air drawn through the filters.
Particle size distribution of smoke was measured using a cascade impactor (In-Tox Products, Moriarty, NM, USA), which has a cut-off diameter in the range of approximately 0.4–2.5 µm. The mass collected on each impactor stage was analyzed gravimetrically for WTPM, and the resulting data were interpreted by probit analysis to obtain the particle size distribution, mass median aerodynamic diameter (MMAD) and geometric standard deviation (GSD). The puff volume produced by the smoking machine pump was measured daily using a soap bubble flow meter. Nose port temperature and RH were measured using Humitter solid state integrated humidity/temperature transmitters (Vaisala Inc., Woburn, MA, USA).
The nicotine concentration in the smoke was analyzed for Test 2 and 3 using gas chromatography. Two filters were collected for nicotine analysis and extracted with isopropanol containing heptadecane. The nicotine samples were analyzed using GC system (Agilent HP-6890) with flame ionization detector (FID) equipped with DB-Wax fused silica capillary column (0.25 mm ID, J&W, Folsom, CA, USA) with a film thickness of 0.25 μm. Temperature and RH of the exposure atmosphere were measured daily from a representative animal exposure port for each exposure group.
LPS instillation
In Test 1, a group of rats were exposed to LPS via a single intratraecheal (i.t.) instillation at 5 mg/kg body weight of LPS (E. coli 055:B5, Sigma-Aldrich; dissolved in saline); this dose was determined in our preliminary study (data not shown).
Animals and animal care
All rats used in this report were purchased from Charles River Japan (Atsugi, Kanagawa, Japan). Animals received certified rodent diet (MF, Oriental Yeast Co., Ltd, Tokyo, Japan) and fresh tap water (City of Kamisu, Ibaragi, Japan) ad libitum, except during exposure periods.
Animals were housed in animal rooms with 12 h light/12 h dark cycles at 23 ± 2 °C with 30–70% relative humidity and air flow with 6 air changes/h: Feed and fresh municipal water were supplied ad libitum except during exposure.
In Test 1, male and female Crlj:CD (SD) and F344/DuCrlCrlj (F344) rats were used at 8 weeks and 9 weeks of age respectively after 2 weeks quarantine. In Test 2 and 3, male F344 rats were used at 7 weeks of age after 2 weeks quarantine.
The studies were carried out in accordance with the Guidelines for Animal Studies of the Ministry of Health, Labuor and Welfare, Japan. The protocols of these studies were approved by the Committee for Ethics in Animal Studies of the test facility prior to commencing the studies.
Study design
In Test 1, 12 male and female SD and F344 rats were divided into sham, LPS, and smoke exposure groups (4 rats/group). Smoke exposure groups were exposed once by nose-only inhalation at 1.2 mg WTPM/L or exposed to filtered air (sham) for 2 h. The LPS group was subjected to a single i.t. instillation of LPS (5 mg/kg) and sacrificed on the following day after smoke exposure and LPS administration.
In Test 2, 128 male F344 rats were divided into 8 groups (16 rats/group; 6 for BALF and 10 for pathology). One-hour daily exposure groups (1-h regimen) were exposed at the 3 WTPM concentrations (0.2, 0.5, and 0.8 mg/L) for 7 days/week. Four-hour daily exposure groups (4-h regimen) were exposed at the corresponding 3 WTPM concentrations (0.05, 0.125, and 0.2 mg/L) for 7 days/week. The Sham group for each regimen was restrained in a similar nose-only exposure unit but exposed to filtered air during 2 weeks of exposure.
In Test 3, 144 male F344 rats were divided into 9 groups (16 rats/group; 6 for BALF and 10 for pathology), and 5 extra rats were assigned for health monitoring. One-hour daily exposure groups (1-h regimen) were exposed at 1 dose (0.6 mg/L) for 7 days/week. Two-hour daily exposed groups (2-h regimen) were exposed at 3 doses (0.1, 0.2, and 0.3 mg/L) for 7 days/week. Four-hour daily exposed groups (4-h regimen) were exposed at 2 doses (0.05 and 0.15 mg/L) for 7 days/week. The Sham group for each regimen was restrained and exposed to filtered air during 13 weeks of exposure.
Rats were observed twice daily prior to and after exposure for mortality and moribundity throughout each test. Individual body weight was measured prior to exposure (all tests) and also weekly and at necropsy (Test 2 and 3).
Respiratory functions during smoke exposure
Tidal volume (TV), respiratory rate (RR), and minute volume (MV), derived from flow signals from spontaneously breathing animals, were measured in 5 rats/group in week 2 (Test 2) and 5 rats/group in week 1, 2 and 12 (Test 3), using a whole-body plethysmography system (BUXCO Electronics Research Systems, Wilmington, NC, USA)26 during exposure. Measurements were performed for 20 min begining 15 min after the start of exposure in all groups, and additionally, 20 min after 1 h had passed in the 2-h exposure groups and 20 min after 3 h had passed in the 4-h exposure groups. The data shown are the averages of the 2 time measurements in the 2-h and 4-h exposure groups.
Exposure markers
Blood carboxyhemoglobin (COHb) and plasma nicotine and cotinine concentrations were determined in 5 rats/group at week 2 (Test 2) and at weeks 2 and 12 (Test 3). Blood samples were collected at the end of daily exposure periods after about 50 min for the 1-h regimen groups. For the 4-h regimen groups, sampling was performed after approximately 50 min, 2.5 h, and 4 h of the exposure period. All animals were bled within 8 minutes after removal from exposure. Blood samples were drawn from the retro-orbital plexus, under carbon dioxide (CO2) anesthesia, into tubes containing EDTA. Blood COHb concentrations were determined using an OSM Hemoximeter (Radiometer, Copenhagen, Denmark). The remaining blood was centrifuged, and obtained plasma samples were stored at –70°C until nicotine and cotinine analysis. The plasma samples were extracted with toluene containing methyl-D3 nicotine and methyl-D3 cotinine (Cambridge Isotope Laboratories, Andover, MA, USA) as internal standards and quantitatively determined using gas chromatography/mass spectrometry (GC/MS) with selected ion monitoring.
Urine was collected in a metabolic cage for overnight (approximately 15 h) after the last exposure. Urinary nicotine and cotinine were analyzed GC/MS system in the same manner same as the plasma samples.
Bronchoalveolar lavage fluid (BALF) collection and analyses
The day following the last exposure, designated rats [4/group (Test 1); 6/group (Test 2 and 3)] were exsanguinated and subjected to bronchoalveolar lavage (BAL) at terminal sacrifice under pentobarbital anesthesia. A 23-gauge catheter was inserted into the left bronchi, and the left lungs were washed with 3 mL of saline by repeating filling and aspiration 5 times. BALF was centrifuged at approximately 400 g for 10 min at 4°C.
The supernatant was analyzed for lactate dehydrogenase (LDH), N-acetyl-beta-D-glucosaminidase (NAG), protein, and gamma glutamyl transferase (γGT) in Test 2 and 3 using an automated clinical chemistry analyzer (TBA-200FR; Toshiba, Tokyo, Japan). Additionally, a commercially available cytokine measurement system (R&D Systems) was used to determine the following cytokine concentrations in Test 1: IL-1β (RLB00), CINC-3 (RCN300), Fractalkine (ELR-Fractalkine-001), and TNF-α (RTA00). In Test 2 and 3, the following cytokines were measured using the Rat Cytokine 9-Plex A Panel (Bio-Rad Laboratories) according to the manufacture’s instructions: IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-10, GM-CSF, INF-γ, and TNF-α.
Cell pellets were resuspended and evaluated for viability, cell count, and cell typing. Total cell numbers were counted using a hemocytometer by the trypan blue exclusion method. Differential cell counts were analyzed using an automated hematology analyzer (XT-2000i; Sysmex Corp., Kobe, Hyogo, Japan) to calculate the percentage of alveolar macrophage, neutrophil, lymphocyte, and eosinophil.
Clinical pathology
At terminal sacrifice in Test 2 and 3, the rats were anesthetized with CO2, and blood samples were obtained from the retro-orbital plexus. Standard hematology and clinical chemistry parameters were measured, according to the methods described in our previous study16.
Necropsy and histopathology
For Test 2 and 3, 10 animals per group were euthanized with approximately 70% CO2 in air and exsanguinated. The respiratory tract, lungs, hearts, and other selected organs (shown in Table 3) were harvested, weighed and fixed in 10% neutral buffered formalin (NBF). Lung was perfused with 10% NBF at 25 cm hydrostatic pressure after being weighed.
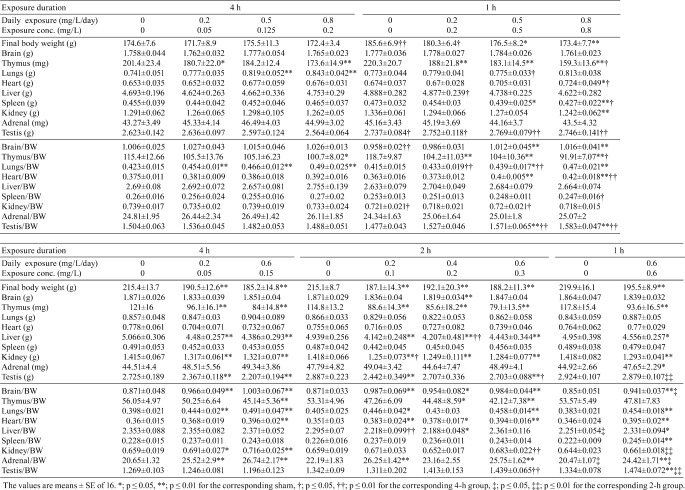
Table 3. Organ Weights in Test 2 and 3.
After a minimum 48 h of fixation, tissues were trimmed and embedded in paraffin and sliced into sections at 4–5 μm thickness. Microscopic slides were prepared according to previous studies13,16. In short, the nasal tissues were cut at 4 different locations to obtain representative sections of the different epithelia27. Three transverse sections of larynx were prepared from the base of the epiglottis, ventral pouch, and through the caudal larynx at the level of the vocal folds28. The lungs were trimmed to provide a section along the mainstem bronchi. Three transverse sections of trachea were prepared. The heart was trimmed longitudinally to contain both left and right atria and ventricles. All sections were stained with hematoxylin and eosin (H&E) stains; slides of nasal tissues and lung were stained with Alcian blue/ periodic acid-Schiff (AB/PAS) stain for evaluation of goblet cells.
The lungs, nasal cavity (four sections), nasopharynx, larynx (three cross sections), trachea (three transverse sections), tracheobronchial lymph nodes, mediastinal (thymic) lymph nodes and, heart, and all gross lesions were examined microscopically.
Statistics
The data were analyzed using SAS system (Ver. 6.12; SAS Institute Japan, Tokyo, Japan) or MiTOX (Mitsui Zosen System Research Inc., Chiba, Japan).
Differences between the treatment groups and their corresponding sham groups were analyzed for (if applicable): body weight, blood COHb concentration, plasma and urinary nicotine and cotinine concentrations, respiratory functions, hematology, blood biochemistry, organ weight, and BALF parameters. Homogeneity of variance was first analyzed using Bartlett’s test. For homogeneous data, one-way analysis of variance was performed. In case difference was detected between groups (for Test 2 and 3), the two-tailed Dunnett’s multiple comparison test was applied. For unequal variances, the Kruskal-Wallis test was performed, and if a difference was shown among groups, the two-tailed Steel’s multiple comparison test was applied. For the histopathological data, the grades were converted to numerical values, and the two-tailed Steel’s rank sum test was used. Comparison between 2 groups was performed by two-tailed Wilcoxon’s test.
In Test 1, difference between strain or sex for the corresponding exposure condition was also evaluated. Homogeneity of data in each rat strain of each sex was analyzed by F-test. Homogeneous data were analyzed by two-tailed Student’s-t test and nonhomogeneous data were analyzed by two-tailed Aspin-Welch t-test.
In Test 2 and 3, difference between exposure regimens was also compared with each test. Homogeneity of data was evaluated using the F-test. For homogeneous data, one-way ANOVA was applied, and then a 2-tailed Tukey’s multiple comparison test was performed if difference between groups was shown. For unequal variances, the Kruskal-Wallis test was applied and the non-parametric Tukey’s multiple comparison test was performed if difference between groups was shown. In comparison among three groups (Test 3), variances were analyzed by Bartlett’s test. If difference among groups was shown, the 2-tailed Tukey’s multiple test was applied.
Results
Test 1 (range-finding study)
In-life exposure and clinical observations: The smoke exposure atmosphere (WTPM, particle size and CO; Table 1) was well controlled in the study. There was no unscheduled removal due to early death or moribund condition following either smoke or LPS exposure.
Table 1. Study Design and Characterization of Exposure Atmosphere.
Immediately following the single smoke exposure, all smoke-exposed SD and F344 groups displayed decreased locomotor activity, ataxic gait, irregular respiration, nasal noise, and salivation. These signs were disappeared in SD rats by the next morning; however, F344 rats still displayed decreased locomotor activity and nasal noise. The mean body weight was decreased in all the smoke-exposed and LPS-instilled groups. Compared with the pre-exposure body weights, the SD rats lost about 8–11% of their body weights and the F344 rats lost similarly at 8-13% to their body weights in all groups including the sham controls. The greatest weight reduction was observed in the smoke-exposed male rats (SD, 11%; F344, 13%).
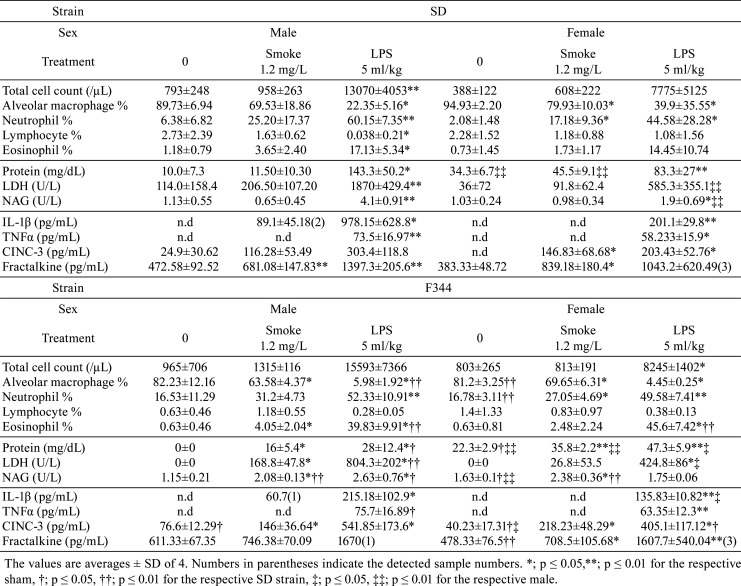
Bronchoalveolar lavage fluid: The total cell and granulocyte counts in BALF were increased with the exposure, most notably after the LPS instillation in both strains and sexes (Table 2). Between the strains, the total cell count in the F344 rats tended to be higher than that of SD rats in both sexes, however, none of differences were statistically significant. LPS instillation induced stronger extravasation of granulocytes compared to cigarette smoke exposure. LDH was increased by LPS and smoke exposure in both strains and sexes, while male F344 rats showed the most marked changes from smoke exposure compared with the sham. Consistent with the differential data, BALF IL-1β, TNF-α, CINC-3, and Fractalkine were markedly induced by LPS stimulation, while cigarette smoke exposure slightly induced CINC-3 and Fractalkine. IL-1β and TNF-α were not detected in any of sham groups and most smoke-exposed groups in both strains and sexes.
Table 2. Cell Typing, Deviation Enzymes, and Cytokines in BALF (Test 1).
Based on these results, male F344 rats were selected to proceed to 2-week (Test 2) and 13-week (Test 3) cigarette smoke studies. Considering the in-life data (body weight loss and acute clinical signs post exposure), the daily exposure dose was reduced up to 0.8 mg/L/day (Test 2) and 0.6 mg/L/day (Test 3).
Test 2 and 3
In-life exposure and clinical observations: The smoke exposure atmosphere for Test 2 and 3 (WTPM, nicotine, CO, and particle size; Table 1) was well controlled throughout the study. The butt-length, temperature, and relative humidity were also well controlled (data not shown).
There was no serological evidence of health problems throughout the study, and no unscheduled removal of animals due to death or moribund condition. Several clinical signs were observed from all smoke-exposed groups throughout the study; however, they were generally minor in consequence and low in incidence, and signs were associated with daily tube constraint during exposure or sample collection. There were no clear exposure-related adverse clinical signs from either test.
Respiratory physiology: Both in Test 2 and 3, the RR in the smoke-exposed group was decreased overall compared with the corresponding sham, while the TV in the smoke-exposed groups was increased (data not shown). As a result, the MVs for the exposed groups were suppressed, generally in a WTPM-dependent manner, with the reduction ranging from 8 to 35% reduction relative to the respective sham controls. This was similarly observed in our previous rat study13,16 and other studies14,29.
Exposure markers: Blood COHb and plasma and urinary (nicotine plus cotinine) values are shown in Fig. 1. In Test 2, the COHb concentration of the 0.8 mg/L group was very high (46%), which could induce risk for CO intoxication in a longer-term study. When the maximal WTPM concentration was lowered to 0.6 mg/L in Test 3, the COHb concentration was 34%. In both tests, the COHb values increased in direct relation with the WTPM concentrations, resulting in higher levels for 1-h groups compared to the corresponding 2- or 4-h groups. The plasma nicotine plus cotinine values were increased dependently with the total daily exposure dose (c x t). Notably, urinary nicotine plus cotinine values tended to be higher in 4-h groups compared with 1-h groups among the equivalent daily exposure groups.
Fig. 1.

Exposure markers. Blood COHb in Test 2 (a) and Test 3 (b), plasma nicotine plus cotinine in Test 2 (c) and Test 3 (d), and urinary nicotine plus cotinine in Test 2 (e) and Test 3 (f). The values are mean ± SD (n=16). *; p ≤ 0.05, **; p ≤ 0.01 for the respective sham, †; p ≤ 0.05, ††; p ≤ 0.01 for the corresponding 4 h group, ‡; p ≤ 0.05, ‡‡; p ≤ 0.01 for the corresponding 2 h group.
Bronchoalveolar lavage fluid: BALF parameters of cell typing and γGT analysis are shown in Fig. 2. In Test 2 and 3, total cell counts were generally increased in a dose-dependent manner (Fig. 2-a and b). However, the increase above the sham groups was minimal and the regimen difference between the equivalent daily exposure groups was not significant in either test. Similarly, the neutrophil counts increased in smoke-exposed groups in Test 3 (Fig. 2-b), but the regimen difference was insignificant. In contrast, γGT in BALF was increased in a dose-dependent manner in both Test 2 (Fig. 2-c) and Test 3 (Fig. 2-d), and the increase above sham was statistically significant. LDH was significantly increased only in the high-dose groups (the 4-h 0.15 mg/L group and the 2-h 0.3 mg/L group) in Test 3 (data not shown). The BAL cytokine data did not show consistent changes over the exposure regimen in either test (data not shown).
Fig. 2.

BALF total cell counts, cell typing, and γGT activity. BALF cell counts and cell typing in Test 2 (a) and Test 3 (b), and BALF γGT activity in Test 2 (c) and Test 3 (d). AM, alveolar macrophage; Lymp, lymphocyte; Neut, neutrophil. The values are average ± S.D (n=5–6). *; p ≤ 0.05, **; p≤ 0.01 for the respective Sham group.
Body and organ weights: Body weight and organ weight at terminal sacrifice are shown in Table 3. The mean body weights of all groups in Test 2 and 3 increased steadily throughout the exposure period, although the degree of weight gain was suppressed depending on the smoke concentration (WTPM) and daily restraint during exposure. In Test 2, 1-h exposure groups showed statistically significant reduction in body weight, while the 4-h smoke-exposed groups had comparable weights to the sham. Between the sham groups, the 4-h sham showed significantly lower body weight compared with the 1-h sham. In Test 3, the body weight was decreased in all smoke-exposed groups similarly regardless of exposure regimen and no significant difference was observed among daily exposure durations. This suggests that by 13 weeks of exposure, the equivalent daily exposure dose induces a comparable effect on body weight gain.
The thymus weight was decreased in all smoke-exposed groups in both Test 2 and 3. The relative lung weight (lung weight/body weight) was significantly higher than that of the corresponding sham in some smoke-exposed groups in Test 2, and most smoke-exposed groups in Test 3. Those changes were comparable among the equivalent exposure groups.
Hematology and blood chemistry: In hematological measurements in Test 2 (data not shown) and 3 (Table 4), red blood cell counts changed sporadically in some smoke-exposed groups after 2 week and 13 week of exposure. Hemoglobin was increased in higher-dose groups in all regimens in Test 3 except for 1-h 0.6 mg/L group. In both tests, hematocrit was commonly increased, whereas platelet counts were generally decreased by smoke exposure. White blood cell counts and lymphocyte percentage were decreased by smoke exposure, while neutrophil percent was increased. MCV and MCH were significantly increased by cigarette smoke exposure without clear dose-dependency. These changes were similar both in Test 2 and 3 and, in comparison of exposure regimen, the daily equivalent dose groups showed similar responses.
Table 4. Hematology and Blood Chemistry in Test 3.
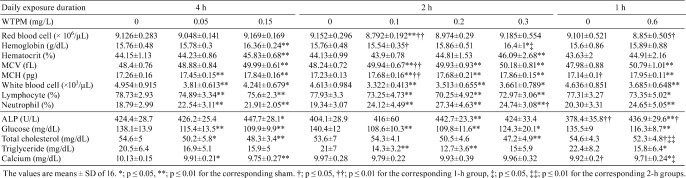
Blood chemistry results (Table 4, Test 3) also showed sporadic changes, possibly except for the following observations: ALP was generally increased by smoke exposure after both 2-week (Test 2) and 13-week (Test 3) exposures. Triglyceride and calcium tended to be decreased by smoke exposure. In both tests, the glucose concentration was decreased by smoke exposure in a dose-dependent manner with some statistical significance. This may be from the combined effect of restraint stress and smoke exposure. Total cholesterol was decreased, and statistical difference was mainly observed in the longer daily exposure groups.
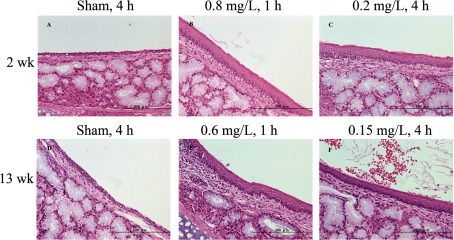
Histopathological examination: Histopathological examination is summarized in Table 5, and representative histological observations are shown in Fig. 3 (nose), Fig. 4 (larynx), and Fig. 5 (lungs), respectively. The numbers in the table represent the incidence of lesions, and the severity is graded as minimal, mild, moderate, and severe.
Table 5. Histopathological Changes in Test 2 and 3.
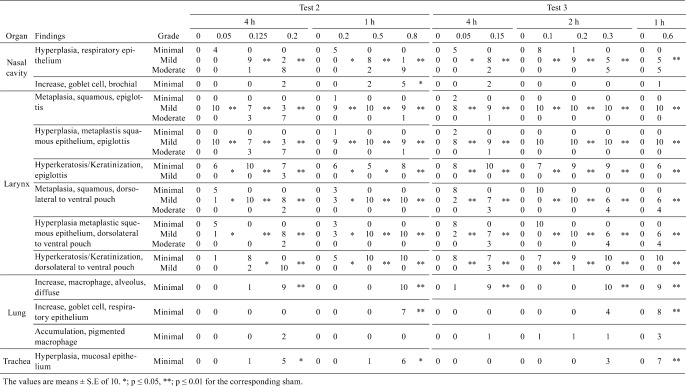
Fig. 3.
Histological changes of the nose. Hyperplasia and squamous metaplasia of the epithelial cells in the nasal septum were observed both after 2wks (B, C) and 13 wks (E, F) compared with the respective air groups (A for 2 weeks, D for 13 weeks), while the severity for both exposure conditions was similar.
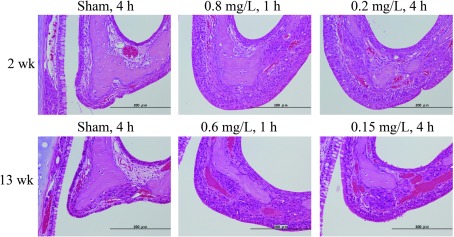
Fig. 4.
Representative histological changes in the epiglottis. Hyperplasia, squamous metaplasia and keratosis of the epithelial cells in the epiglottis were observed both after 2 weeks (B, C) and 13 weeks (E, F) compared with the respective air groups (A for 2 weeks, D for 13 weeks), while the severity for both exposure conditions was similar.
Fig. 5.

Representative histological changes in the lungs from F344 rats exposed to air (A) or cigarette smoke (B) after 2 weeks of exposure. Hyperplasia of bronchial epithelial cells and an increase of alveolar macrophages were induced after cigarette smoke exposure (B) compared with air exposure (A).
Nasal cavity/turbinate: In Test 2, hyperplasia of the respiratory epithelium was statistically increased in a dose-dependent manner in both exposure regimens. Squamous metaplasia of the respiratory epithelium was significantly increased in the 1-h 0.8 mg/L groups (10/10), but only slightly increased in the 4-h 0.2 mg/L group (4/10). Regeneration of the olfactory epithelium was significantly increased only in the 4-h 0.2 mg/L group. AB/PAS staining showed a significant increase of goblet cells in the 1-h 0.8 mg/L group (5/10), while a few animals (2/10) showing significant increases were observed in the 4-h 0.2 mg/L and 1-h 0.8 mg/L groups.
In Test 3, hyperplasia of the respiratory epithelium was statistically increased dose dependently in all exposed groups. The incidence and severity of the lesion was similar overall between 2-week (Test 2) and 13-week (Test 3) exposure durations.
Larynx: In Test 2, the incidence and severity of hyperkeratosis/keratinization and hyperplasia/squamous metaplasia of the epithelium at the epiglottis was significantly increased in all smoke-exposed groups generally in a dose-dependent manner in both regimens. Squamous metaplasia at the dorsolateral to ventral pouch and epiglottis was also significantly increased in all exposed groups from both regimens. Hyperkeratosis/keratinization, hyperplasia/squamous metaplasia of the epithelium, and squamous metaplasia at the caudal larynx were significantly observed only in the 1-h 0.8 mg/L group (data not shown). Hyperkeratosis/keratinization at dorsolateral to ventral pouch was significantly observed in all exposed groups except for the 4-h 0.05 mg/L group. Hyperplasia of the respiratory epithelium at the caudal larynx was increased significantly only for the high dose groups in both regimens. Chronic inflammation at the epiglottis was observed sporadically in a few animals in the 4-h groups. Hyperkeratosis/ keratinization, hyperplasia of the metaplastic squamous epithelium, and squamous metaplasia at the caudal larynx were observed in the 4-h 0.2 mg/L group in Test 2.
In Test 3, the following lesions were significantly increased in all exposed groups regardless of exposure regimens: hyperplasia/squamous metaplasia, squamous metaplasia, hyperkeratosis/keratinization at the dorsolateral to ventral pouch and epiglottis. These lesions at the caudal larynx were significantly increased in all exposed groups, except for the low dose 2- and 4-h groups (data not shown). Hyperplasia of the respiratory epithelium and hyperplasia of the metaplastic squamous epithelium at the caudal larynx were observed in all top-dose groups of each regimen in Test 2 and in Test 3. Squamous metaplasia at the caudal larynx was statistically increased in 4-h 0.2 mg/L group in Test 2, and in all top-dose groups in Test 3. The trend of these laryngeal lesions was overall similar to Test 2 in their incidences and severities, and the lesions were mainly affected at higher WTPM concentrations.
Trachea: In Test 2, the incidence of hyperplasia of the mucosal epithelium was significantly increased in the high-dose groups in both regimens. Focal lymphocyte infiltration was observed in a few animals in most groups including the sham, but without dose dependency or statistical significance. Similar findings were observed in Test 3, and hyperplasia of the mucosal epithelium was significantly increased only in the 1-h high-dose group.
Lung: In Test 2, diffused macrophages in alveoli were significantly increased in the high-dose groups in both regimens, while the increase of goblet cells was significant only in the 1-h high-dose group. Other findings were sporadically observed in a few animals, without dose dependency and statistical significance. Similar findings were observed in Test 3 in that diffused macrophages in alveoli were significantly increased in all the high-dose groups regardless of regimen. The goblet cells at the bronchial epithelium was increased only in the 1-h high-dose group.
In considering both Test 2 and 3, smoke-induced histopathological findings were mainly observed at the epithelial cells in the nasal cavity and larynx (e.g., hyperplasia, squamous metaplasia, hyperkeratosis of the epithelial cells). In the lung, inflammatory cell infiltration (e.g., macrophages and neutrophils) was increased in smoke-exposed groups. As these lesions observed in 13-week exposure at 0.6 mg/L in Test 3 were comparable with those observed in 2-week exposure at 0.8 mg/L, the possibly exacerbating effects of a longer smoke exposure (2 versus 13 weeks) were not observed in the histopathological examination.
Discussion
In Test 1, a single instillation of LPS induced severe pulmonary inflammation based on changes in BALF parameters. LPS is a strong inflammatory endotoxin that binds TLR4, and chronic inhalation to LPS has shown emphysema-like changes in mice30. When BALF parameters were qualitatively compared between the LPS and smoke-exposed groups, LPS strongly induced almost all of cytokines in all treated groups, while cigarette smoke induced only CINC-3 and Fractalkine mainly in females. Based on the acute responses with LPS and cigarette smoke, F344 rats tended to show greater pulmonary inflammation than SD rats, and males in both strains appeared to display slightly greater responses than females. Based on these results, male F344 rats were used in the subsequent 2-week (Test 2) and 13-week (Test 3) studies.
Blood COHb was increased with increasing WTPM concentration in both Test 2 and 3. In Test 2, the blood COHb concentration in the 800 µg/L group reached 46.1%, which seemed to be too high for longer-term studies and considering the reported heavy smokers’ COHb levels (10–15%)31. The mean COHb % of smokers increased approximately linearly dependent on consumed cigarette numbers up to 20 cigarettes per day, after which the increase in COHb was less than proportional to consumed cigarette numbers32. In both Tests, plasma nicotine plus cotinine contents (Fig. 1-b and c) were increased dose dependently according to the daily total dose (WTPM concentration x daily exposure duration), while urinary nicotine plus cotinine tended to be higher in the 4-h groups compared with 1-h groups in both tests. The apparent difference between plasma and urinary data was likely attributable to the difference in their elimination half-lives and the clearance observed in the previous report33.
In Test 2 and 3, smoke-exposed animals showed reduced weight gains with increasing exposure concentration. This is consistent with previous observations from cigarette smoke inhalation studies34,35 and speculated to be the consequence of a biological effect of nicotine and acrolein in cigarette smoke. In Test 2, the 4-h sham showed a lower body weight compared to the 1-h sham group, suggesting that the restriction stress during the acute exposure may have a negative impact on weight gain. Between regimen groups in Test 3, there was no difference in weight changes, and animals were otherwise unremarkable during up to 13 weeks of the study period.
BALF total cell and neutrophil counts from Test 2 and 3 did not increase markedly compared with the smoke group data in Test 1. The highest WTPM concentration under the 1 h regimen was set at 0.6 mg/L in Test 3, while it was set at 0.8 mg/L in Test 2. Comparison between Test 2 and 3, with Test 3 having about a 25% reduction in the WTPM concentration, showed that an extended exposre duration (2 to 13 weeks) didn’t exacerbte the pulmonary inflammation. In chronic pulmonary inflammation, activated alveolar macrophages36 and extravasated/activated neutrophils19, 37 could play a role in destruction of the alveolar structure by releasing various kinds of proteinases.
Among BALF parameters in our study, γGT was significantly increased dose dependently and could be an early marker for pulmonary injury following cigarette smoke exposure. In contrast, there were no consistent and notable changes in BALF cytokines when measured the day after exposure. It is possible that their levels could be higher at different time points, as the release of various cytokines differs depending on the time after exposure38. However, the increase in BALF neutrophil count was also marginal, further supporting that the degree of pulmonary inflammation was minimal under the employed exposure regimens.
In organ weight data from Test 2 and 3, relative lung weights were significantly increased in all smoke-exposed groups, and this has been observed commonly by others6 and thought to be due to the pulmonary inflammation. Relative heart weights were also significantly increased dose dependently regardless of exposure regimens, and this was associated with CO in the cigarette smoke39.
Upon microscopic examination of the nasal cavity, hyperplasia of the respiratory epithelium was commonly and significantly increased in all smoke-exposed groups in Test 2 and 3. Squamous metaplasia of the respiratory epithelium and regeneration of the olfactory epithelium were only increased in 4-h 0.2 mg/L group in Test 2 and were not increased in the smoke-exposed groups in Tests 3. In the Larynx, the lesions were observed at the epiglottis and dorsolateral to the ventral pouch for all smoke-exposed groups in Test 2 and 3 with statistical differences in; hyperkeratosis/keratinization, hyperplasia, and squamous metaplasia. These lesions were commonly observed at the larynx after smoke exposure regardless of the smoke concentration and exposure duration, similar to our previous study13. In the trachea, the incidence of hypertrophy of the mucosal epithelium was significantly increased in the top-dose groups for both regimens but only in the 1-h regimen group in Test 3. This suggested that under the 13-week exposure, the lesion is mainly affected by the WTPM concentration, not by the total daily exposure dose.
In the lung, the increase of diffused alveolar macrophages was significant in the high-dose groups for all regimens in both tests. Goblet cells were significantly increased only in the top-dose groups and under the 1-h regimen in both tests. This was similarly observed in our previous study13 in that the incidence and severity of goblet cell hyperplasia in the 1-h 0.6 mg/L group was higher in the 6-h 0.1 mg/L group. An increase of neutrophils in the alveolus was not observed in the histopathological examination, while the neutrophil content in BALF was increased in all smoke-exposed groups compared with the sham group.
Considering the histopathological changes in Test 2 and 3, the lesions were observed mainly in the epithelium of the nose and larynx, and not in the lower respiratory tract including the lungs. Comparison between Test 2 and 3 showed that the lesions were not exacerbated between 2 and 13 weeks; in fact, in some cases, a lesion observed at 2 weeks was more severe than that at 13 weeks. This suggested that the daily exposure WTPM concentrations of 0.6 mg/L or 0.8 mg/L, whether the total daily dose was delivered over 1 or 4 h, was insufficient to induce the magnitude of pulmonary inflammation associated with tobacco-related lung diseases.
Between the regimens, parameters indicative of pulmonary responses (e.g., BALF parameters, organ weight ratio, and the incidence and severity of the histopathological lesions) were overall comparable between groups at equivalent daily exposure amounts in Test 2 and 3. This suggests that Haber’s rule may be applicable for exposure studies of mixtures like cigarette mainstream smoke under these study design in F344 rats. F344 rat was commonly used in chronic bioassays such as for the National Toxicology Program (NTP), while the SD rat has been used in various standard toxicological studies, including tobacco ingredient testing. Our previous study13 with SD rats reported similar findings in that the equivalent exposure amount of cigarette smoke (based on Haber’s rule) from different 90-day exposure regimens resulted in comparable biological impact.
In the literature, March et al. employed a much more intense exposure regimen under a longer exposure duration: they exposed F344 rats and B6C3F1 mice at 0.75 mg/L for 6 h/day for 2 years under whole-body inhalation exposure40. The accumulated daily exposure amount was up to 4.5 mg WTPM/L/day, which was more than 5- or 7-fold higher than in the current study (0.6 or 0.8 mg WTPM/L/day), and the duration was much longer (up to 2 years vs. 13 weeks). Their stereological examination showed significant enlargement of alveoli, a signature phenotype for emphysema, by 13 months exposure, with the greater severity being observed in mice compared with rats. Similarly, Zheng et al.22 exposed male Wistar rats to sidestream cigarette smoke under whole-body exposure and reported a significant increase in mean linear intercept (Lm) after 24 wks of exposure. Stevenson et al. (2007) exposed cigarette smoke to male SD rats for 1 day to 34 weeks, and reported a significant increase of Lm after 34 weeks. Although the exact daily exposure dose was not reported in these whole-body studies, these results suggested that the pathogenesis of COPD (emphysema) in rats required a longer exposure duration than the typical 13-week duration used for standard toxicity testing. Our results and previous reports again stress that defining biologically relevant exposure regimens (the smoke concentration, daily exposure duration, and the days of exposures) is critical to inducing pathophysiological responses in animal disease models.
Acknowledgments
This study was conducted in an AAALAC-accredited laboratory sponsored by JT. We are grateful to Dr. Yoshimi Yoshida, and Dr. Satoshi Numazawa of the Department of Biochemical Toxicology, School of Pharmaceutical Sciences, Showa University, for his helpful discussion and suggestions. We thank Hiroshi Yoinara, Satoshi Kitao, and Kazushige Inada for their thoughtful discussion.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-Commercial No Derivatives (by-nc-nd) License <http://creativecommons.org/licenses/by-nc-nd/3.0/>.
References
- 1.Taraseviciene-Stewart L,Voelkel NF. Molecular pathogenesis of emphysema. J Clin Invest. 118: 394–402 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stinn W, Arts JHE, Buettner A, Duistermaat E, Janens K, Kuper CF, and Haussmann HJ. Murine lung tumor response after exposure to cigarette mainstream smoke or its particulate and gas/vapor phase fraction. Toxicol. 275: 10–20 2010. [DOI] [PubMed] [Google Scholar]
- 3.Schroeter MR, Sawalich M, Humboldt T, Leifheit M, Meurrens K, Berges A, Xu H, Lebrun S, Wallerach T, Konstantinides S, Schleef R, and Schaefer K. Cigarette smoke exposure promotes arterial thrombosis and vessel remodeling after vascular injury in apolipoprotein E-deficient mice. J Vasc Res. 45: 480–492 2008. [DOI] [PubMed] [Google Scholar]
- 4.Chen BT, Bechtold WE, Barr EB, Chang Y-S, Mauderly JL, and Cuddihy RG. Comparison of cigarette smoke exposure atmospheres in different exposure and puffing models. Inhal Toxicol. 1: 331–347 1989. [Google Scholar]
- 5.De Flora S, Balansky RM, Agostini FD, Lzzotti A, Camoirano A, Bennicelli C, Zhang Z, Wang Y, Lubet R, and You M. Molecuar alterations and lung tumors in p53 mutant mice exposed to cigarette smoke. Cancer Res. 63: 793–800 2003. [PubMed] [Google Scholar]
- 6.Mauderly JL, Gigliotti AP, Barr EB, Bechtld WE, Blinsky SA, Hahn FF, Hobbs CA, March TH, Seilkop SK, and Finch GL. Chronic inhalation exposure to mainstream cigarette smoke increases lung and nasal tumor incidence in rats. Toxicol Sci. 81: 280–292 2004. [DOI] [PubMed] [Google Scholar]
- 7.Curtin GM, Higuchi MA, Ayres PH, Swauger JE, and Mosberg AT. Lung tumorigenicity in A/J and ras H2 transgenic mice following mainstream tobacco smoke inhalation. Toxicol Sci. 81: 26–34 2004. [DOI] [PubMed] [Google Scholar]
- 8.Baker RR, Massey EH, and Smith G. An overview of the effects of tobacco ingredients on smoke chemistry and toxicity. Food Chem Toxicol. 42: S53–S83 2004. [DOI] [PubMed] [Google Scholar]
- 9.Gebel S, Gerstmayer B, Kuhl P, Boriak J, Meurrens K, and Muller T. The kinetics of transcriptomic changes induced by cigarette smoke in rat lungs reveals a specific program of defence, inflammation, and circadian clock gene expression. Toxicol Sci. 93: 422–431 2006. [DOI] [PubMed] [Google Scholar]
- 10.Coggins CRE. An updated review of inhalation studies with cigarette smoke in laboratory animals. Int J Toxicol. 26: 331–338 2007. [DOI] [PubMed] [Google Scholar]
- 11.Hamm JT, Yee S, Rajendran N, Morrisey RL, Richter SJ, and Misra M. Histological alterations in male A/J mice following nose-only exposure to tobacco smoke. Inhal Toxicol. 19: 405–418 2007. [DOI] [PubMed] [Google Scholar]
- 12.Bolton SJ, Pinnion K, Oreffo V, Foster M, and Pinkerton KE. Characterization of the proximal airway squamous metaplasia induced by chronic tobacco smoke exposure in spontaneously hypertensive rats. Respir Res. 10: 118–133 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsuji H, Lee KM, Yoshino K, Nakamura H, Lulham G, Renne R, and Yoshimura H. Comparison of the physiological and morphological effects of cigarette smoke exposure at comparable weekly doses on Spraque-Dawley rats. Inhal Toxicol. 23: 17–32 2011. [DOI] [PubMed] [Google Scholar]
- 14.Gaworski CL, Shramke H, Diekmann J, Meisgen TJ, Tewes FJ, Veltel DJ, Vansheeuwijck PM, Rajendran N, Muzzio M, and Haussmann HJ. Effect of filtration by activated charcoal on the toxicological activity of cigarette mainstream smoke from experimental cigarette. Inhal Toxicol. 21: 688–704 2009. [DOI] [PubMed] [Google Scholar]
- 15.Carmines EL. Evaluation of potential effects of ingredients added to cigarettes. Part 1: Cigarette design, testing approach, and review of results. Food Chem Toxicol. 40: 77–91 2002. [DOI] [PubMed] [Google Scholar]
- 16.Renne RA, Yoshimura H, Yoshino K, Lulham G, Minamisawa S, Tribukait A, Dennis DD, Lee KM, and Westerberg RB. Effects of flavoring and casing ingredients on the toxicity of mainstream cigarette smoke in rats. Inhal Toxicol. 18: 685–706 2006. [DOI] [PubMed] [Google Scholar]
- 17.OECD. OECD guideline for the testing of chemicals; Subchronic inhalation Toxicity: 90-day study. Environmental Health and Safety Monograph Series on Testing and Assessment No.413. 2009. http://iccvam.niehs.nih.gov/SuppDocs/FedDocs/OECD/OECD-TG413.pdf
- 18.NTP Specifications for the Conduct of Studies to Evaluate the Reproductive and Developmental Toxicity of Chemical, Biological and Physical Agents in Laboratory Animals for the National Toxicology Program (NTP). 2011. http://ntp.niehs.nih.gov/ntp/Test_Info/FinalNTP_ToxCarSpecsJan2011.pdf
- 19.Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, and Belaaouaj A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Pathol. 163: 2329–2335 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guerassimov A, Hoshino Y, Takubo Y, Turcotte A, Yamamoto M, Ghezzo H, Triantafillopoulos A, Whittaker K, Hoidal JR, and Cosio MG. The development of emphysema in cigarette smoke-exposed mice is strain dependent. Am J Respir Crit Care Med. 170: 974–980 2004. [DOI] [PubMed] [Google Scholar]
- 21.Stevenson CS, Docx C, Webster R, Battram C, Hynx D, Giddings J, Cooper PR, Chakravarty P, Rahman I, Marwick JA, Kirkham PA, Charman C, Richardson DL, Nirmala NR, Whittaker P, and Butler K. Comprehensive gene expression profiling of rat lung reveals distinct acute and chronic response to cigarette smoke inhalation. Am J Physiol Lung Cell Mol Physiol. 293: L1183–L1193 2007 [DOI] [PubMed] [Google Scholar]
- 22.Zheng H, Liu Y, Huang T, Fang Z, Li G, and He S. Development and characterization of a rat model of chronic obstructive pulmonary disease (COPD) induced by sidestream cigarette smoke. Toxicol Lett. 189: 225–234 2009. [DOI] [PubMed] [Google Scholar]
- 23.Gaylor DW. The use of Haber’s Law in standard setting and risk assessment. Toxicology. 149: 17–19 2000. [DOI] [PubMed] [Google Scholar]
- 24.Mioduszewski R, Manthei J, Way R, Burnett D, Gaviola B, Muse W, Thomson S, Sommerville D, and Crosier R. Interaction of exposure concentration and duration in determining acute toxic effects of sarin vapor in rats. Toxicol Sci. 66: 176–184 2002. [DOI] [PubMed] [Google Scholar]
- 25.Hoyle GW, Chang W, Chen J, Schlueter CF, and Rando RJ. Deviations from Haber’s Law for multiple measures of acute lung injury in chlorine-exposed mice. Toxicol Sci. 118: 696–703 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coggins CR, Duchosal F, Musy C, and Ventrone R. Measurement of respiratory patterns in rodents using whole-body plethysmography and a pneumotachograph. Lab Anim. 15: 137–140 1981. [DOI] [PubMed] [Google Scholar]
- 27.Young JT. Histopathological examination of the rat nasal cavity. Fundam Appl Toxicol. 1: 309–312 1981. [DOI] [PubMed] [Google Scholar]
- 28.Renne RA, Gideon KM, Miller RA, Mellick PW, and Grumbein SL. Histologic methods and interspecies variations in the laryngeal histology of F344/N rats and B6C3F1 mice. Toxicol Pathol. 20: 44–51 1992. [DOI] [PubMed] [Google Scholar]
- 29.Terpstra PM, Teredesal A, Vansheeuwijck PM, Verbeeck J, Schepers G, Radtke F, Kuhl P, Gomm W, Anskeit E, and Patskan G. Toxicological evaluation of an electrically heated cigarette. Part 4. Subchronic inhalation toxicology. J Appl Toxicol. 23: 349–362 2003. [DOI] [PubMed] [Google Scholar]
- 30.Brass DM, Hollingsworth JW, Cinque M, Li Z, Potts E, Toloza E, Foster WM, and Schwartz DA. Chronic LPS inhalation causes emphysema-like changes in mouse lung that are associated with apoptosis. Am J Respir Cell Mol Biol. 39: 584–590 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ernst A, and Zibrak JD. Carbon monoxide poisoning. N Engl J Med. 339: 1603–1608 1998. [DOI] [PubMed] [Google Scholar]
- 32.Law MR, Morris JK, Watt HC, and Wald NJ. The dose-response relationship between cigarette consumption, biochemical markers and risk of lung cancer. Br J Cancer. 75: 1690–1693 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamazaki H, Horiuchi K, Takano R, Nagano T, Shimizu M, Kitajima M, Murayama N, and Shono F. Human blood concentrations of cotinine, a biomonitoring marker for tobacco smoke, extrapolated from nicotine metabolism in rats and humans and physiologically based pharmacokinetic modeling. Int J Environ Res Public Health. 7: 3406–3421 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bouley G, Dubreuil A, Godin J, and Boudene C. Effects in the rat of a week dose of acrolein inhaled continuously. Eur J Toxicol Environ Hyg. 8: 291–297 1975. [PubMed] [Google Scholar]
- 35.Chowdhury P. Endocrine and metabolic regulation of body mass by nicotine: role of growth hormone. Ann Clin Lab Sci. 20: 415–419 1990. [PubMed] [Google Scholar]
- 36.Russell RE, Thorley A, Culpitt SV, Dodd S, Donnelly LE, Demattos C, Fitzgerald M, and Barnes PJ. Alveolar macrophage-mediated elastolysis: roles of matrix metalloproteinases, syteine, and serine proteases. Am J Physiol Lung Cell Mol Physiol. 283: L867–L873 2002. [DOI] [PubMed] [Google Scholar]
- 37.Churg A, Wang RD, Tai H, Wang X, Xie C, and Wright JL. Tumor necrosis factor-α drives 70% of cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med. 170: 492–498 2004. [DOI] [PubMed] [Google Scholar]
- 38.Moriyama C, Betsuyaku T, Ito Y, Hamamura I, Hata J, Takahashi H, Nasuhara Y, and Nishimura M. Aging enhances susceptibility to cigarette smoke-induced inflammation through bronchiolar chemokines. Am J Respir Cell Mol Biol. 42: 304–311 2010. [DOI] [PubMed] [Google Scholar]
- 39.Penney D, Benjamin M, and Dunham E. Effect of carbon monoxide on cardiac weight as compared with altitude effects. J Appl Physiol. 37: 80–84 1974. [DOI] [PubMed] [Google Scholar]
- 40.March TH, Barr EB, Finch GL, Hahn FF, Hobbs CH, Menache MG, and Nikula KJ. Cigarette smoke exposure produces more evidence of emphysema in B6C3F1 mice than F344 rats. Toxicol Sci. 51: 289–299 1999. [DOI] [PubMed] [Google Scholar]