

Abstract
Mitochondrial damage has been associated with early steps of cardiac dysfunction in heart subjected to ischemic stress, oxidative stress and hypertrophy. A common feature for the mitochondrial deterioration is the loss of the mitochondrial membrane potential (ΔΨ m) with the concomitant irreversible opening of the mitochondrial permeability transition pore (MPTP) which follows the mitochondrial Ca2+ overload, and the subsequent mitochondrial swelling. We have recently characterized the expression of the Na+/H+ exchanger 1 (mNHE1) in mitochondrial membranes. This surprising observation provided a unique target for the prevention of the Ca2+-induced MPTP opening, based on the inhibition of the NHE1 m. In this line, inhibition of NHE1 m activity and/or reduction of NHE1 m expression decreased the Ca2+-induced mitochondrial swelling and the release of reactive oxygen species (ROS) in isolated cardiac mitochondria and preserved the ΔΨ m in isolated cardiomyocytes. Mitochondrial NHE1 thus represents a novel target to prevent cardiac disease, opening new avenues for future research.
Keywords: ischemia, mitochondrial permeability transition pore, NHE1, mitochondrial swelling, siRNA
Sodium/proton exchanger (NHE)
Sodium/proton exchangers (NHE) are a family of integral membrane proteins present in most organisms. These transporters which catalyze the electroneutral exchange of one intracellular H+ for one extracellular Na+ across membrane along their concentrations gradient are crucial for control of intracellular pH (pHi) and cell volume, and, cell migration and proliferation.
The first NHE isoform to be identified was NHE1, which has a ubiquitous tissue distribution in mammals (Sardet et al., 1989). Since its discovery, nine other human isoforms have been identified (NHE2–NHE10) (Fliegel, 2008; Lee et al., 2008). While NHE1–NHE6 reside in the plasma membrane or recycling endosomes, NHE7–NHE9 are located inside the cell rather than the plasma membrane (Fliegel, 2008). The NHE10 is expressed in the surface of osteoclast (Lee et al., 2008).
NHE1 in the heart
NHE1 is the most studied isoform that accumulates preferentially in microdomains of cells membranes, concentrating along the basolateral membrane of epithelia (Biemesderfer et al., 1992) and the intercalated disks and t-tubules of cardiac myocytes (Petrecca et al., 1999). The sarcolemmal NHE1 is the major Na+ influx pathway found in the plasma membrane of cardiac cells.
NHE1 is an integral membrane glycoprotein with a predicted molecular mass of 85 kDa. NHE1 can be separated into an N-terminal, membrane-associated domain, and a long C-terminal tail, with both the N-and C-terminal domains being cytoplasmic (Wakabayashi et al., 1997), (Orlowski and Grinstein, 1997). NHE1 is expressed ubiquitously in mammalian cells, where it electroneutrally exchanges one intracellular H+ for one extracellular Na+, thus regulating pHi. The membrane domain composed of 12 transmembrane regions is associated with ionic transport (Wakabayashi et al., 1992), and contains the allosteric H+ sensor site that determines the exquisite sensitivity of the exchanger to intracellular H+. The cytoplasmic domain is involved in the regulation of the activity of the exchanger by several mechanisms. Removal of the distal region of the cytosolic tail causes a shift of the pHi sensitivity to the acidic side and an important inhibition of NHE1 activation by growth factors (Fliegel and Karmazyn, 2004). Cytoplasmic tail contains several phosphorylation sites and a high affinity and a low-affinity calmodulin binding sites (Bertrand et al., 1994). The high-affinity binding site functions as an “autoinhibitory domain” that binds Ca2+-bound calmodulin and allows activation of the exchanger. Deletion of this domain constitutively activates NHE1 and mimics elevated intracellular [Ca2+] (Wakabayashi et al., 1994). In addition, the cytoplasmic tail contains a binding site for the calcineurin B homolog protein CHP1, an essential cofactor for NHE1 (Pang et al., 2001). The exchanger is phosphorylated by different protein kinases in response to hormone and growth factor stimulation, as well as sustained acidosis (Sardet et al., 1990, 1991; Haworth et al., 2003).
Under basal conditions, NHE1 exchanger is relatively quiescent and its activity relies only on the extrusion of the H+ produced by the metabolic activity of cells as well as the H+ that enters the cell through acid-loading mechanisms. However, the exchanger has an exquisite sensitivity to the increase in intracellular H+ levels and enhances its activity once pHi drops below a threshold level by allosterically “sensing” pHi, and by being phosphorylated or by interacting with some associated proteins, thus promoting the rapid extrusion of acid (Leem et al., 1999). NHE1 is constitutively phosphorylated in resting cells, but further phosphorylation is induced by several stimuli acting through G-protein-coupled receptors such as α1-adrenergic receptors, angiotensin II (Ang II), and endothelin-1 (ET-1). Kinases such as the Ca2+-/calmodulin-dependent kinase II (Fliegel et al., 1992), the extracellular signal-regulated kinase (ERK) (Moor and Fliegel, 1999), 90 kDa ribosomal S6 kinase (p90 rsk) (Takahashi et al., 1999), p38 mitogen-activated kinase (p38 MAPK) (Khaled et al., 2001), p160 ROCK (Tominaga et al., 1998), and the Nck-interacting kinase (NIK) (Yan et al., 2001) putatively phosphorylate NHE1 to modulate NHE1 activity. In addition, both protein kinases C and D are thought to influence NHE1 activity in response to growth factor and hormone stimulation without a direct phosphorylation of the exchanger (Fliegel et al., 1992; Haworth et al., 1999). NHE1 is also susceptible to dephosphorylation by protein phosphatases such as PP1 (Misik et al., 2005) and PP2A (Snabaitis et al., 2006). Carbonic anhydrase II (CAII) binds to the regulatory cytosolic domain of NHE1 enhancing its activity. Phosphorylation of the C terminus of NHE1 greatly increased the binding of CAII. This binding was shown to involve a protein–protein interaction, suggesting that both proteins constitute a complex. The inhibition of CAII decreased NHE1 activity significantly (Li et al., 2002).
As previously described, the NHE1, relatively quiescent under basal conditions, however, becomes highly active during ischemia in response to intracellular acidosis, leading to NHE1-mediated Na+ entry into the cell (Karmazyn, 1999a; Karmazyn et al., 1999). Inhibition of [Na+]i accumulation by increasing NHE activity and prevention of Ca2+ overload via Na+/Ca2+ exchanger have been proposed as potential mechanisms of cardioprotection by NHE1 blockade (Avkiran, 1999). NHE exchanger inhibitors have proven to protect the heart against ischemia/reperfusion (I/R) injury (Karmazyn, 1999a,b). Moreover, the positive effect that NHE1 inhibition exerts on left ventricular systolic function during revascularization therapy after acute myocardial infarction has been documented (Rupprecht et al., 2000). The protection of the ischemic myocardium by NHE1 inhibition after the onset of reperfusion has been described (Rohmann et al., 1995; Gumina et al., 1998; Hurtado and Pierce, 2000). The decrease in myocardial infarct size, through interference with the action of reactive oxygen species (ROS) generation at the beginning of reperfusion, has also been reported (Koerner et al., 1991; Tanaka et al., 1994; McDonald et al., 1999; Sahna et al., 2002). Fantinelli et al. (2006) described that pharmacological interventions, ROS scavenging and NHE1 inhibition, when applied together and at their maximal effective concentration, did not induce protection further than that obtained separately by each pharmacologic procedure. These findings suggest that both interventions act through a common pathway. The authors proposed that in addition to the effect of preventing intracellular Na+ increase, NHE1 inhibition by cariporide decreases ROS-induced damage.
NHE1 activity is controlled by pHi and numerous other factors, such as hormones, catecholamines, enzymes, and mechanical stimuli, known to be associated with heart disease (Avkiran and Haworth, 2003; Cingolani et al., 2008; Villa-Abrille et al., 2010; De Giusti et al., 2011). Furthermore, cardiac expression of an activated form of NHE1 that lacks the calmodulin-binding inhibitory domain was sufficient by itself to initiate cardiac hypertrophy and heart failure (Nakamura et al., 2008). Constitutively, active NHE1 leads to pathological changes in activation of the Ca2+-dependent pro-hypertrophic signaling molecules, calcineurin and CaMKII (Nakamura et al., 2008). Similarly, in heart over-expression of activated NHE1 was recently found to elicit specific pathways of gene activation, inducing an increase in cross-sectional area of cardiomyocyte, interstitial fibrosis, and depressed cardiac function, in transgenic mice (Xue et al., 2010).
NHE1 and mitochondria
Emerging evidence supports the fact that mitochondrial dysfunction underlies the causes of numerous cardiac diseases (for review, see Baines, 2010). The mitochondrial death pathway features the sequential loss of mitochondrial membrane potential (ΔΨ m), which is accompanied by opening of the mitochondrial permeability transition pore (MPTP), release of ROS and diverse toxic proteins which promote the activation of proteolytic activity of caspases (Teshima et al., 2003). When the MPTP opens, the permeability barrier of the inner membrane becomes disrupted and causes the free movement of protons across it, inducing uncoupling of the oxidative phosphorylation and mitochondria swelling. The MPTP is a large conductance pore thought to be activated by ROS, by increased mitochondrial Ca2+ levels and by dissipation of the mitochondrial ΔΨ m (Akao et al., 2003; Javadov and Karmazyn, 2007). The MPTP opening can be further increased when Ca2+ overload is accompanied by oxidative stress, adenine nucleotide depletion, and elevated phosphate concentrations. Moreover, a decrease in mitochondrial anion superoxide production induced by two known enhancers of ROS production, Ang II and ET-1, upon NHE1 inhibition has been reported (Garciarena et al., 2008). Furthermore, the increase in ROS production induced by the opening of the mitochondrial ATP-dependent K+ channel was abolished by cariporide (Garciarena et al., 2008).
Studies on the MPTP and its role in reperfusion injury and cardiac hypertrophy have proven fruitful (Piot et al., 2008; Halestrap and Pasdois, 2009). Duchen et al. (1993) and Griffiths and Halestrap (1993) showed that delayed opening of MPTP by the MPTP inhibitor, cyclosporine A (CsA), protects ischemic/reperfusion (I/R) injury in perfused rat hearts. Recently, Piot et al. (2008) reported a decrease in infarct size in patients with acute myocardial infarction by CsA administered at the time of reperfusion. Interestingly, a role of mitochondria in determining cardiac hypertrophy after myocardial infarction has also been proposed (Karmazyn, 1988; Caldiz et al., 2007; Javadov et al., 2008, 2009; Cingolani et al., 2010). NHE1 inhibition was one of the most promising therapeutic strategies for I/R injury based on experimental animal studies. However controversial results were obtained using NHE1 inhibitors in clinical trials: cariporide (GUARDIAN) (Theroux et al., 2000) or EXPEDITION (Mentzer et al., 2008) and Eniporide (ESCAMI) (Zeymer et al., 2001) (for review, see Murphy and Allen, 2009). These trials were unable to demonstrate a significant reduction in mortality when these compounds were tested in patients after acute myocardial infarction (Theroux et al., 2000; Zeymer et al., 2001) or were suspended early due to undesired effects (Mentzer et al., 2008). In a subgroup of patients who underwent coronary artery bypass graft surgery and were treated with cariporide, a 25% improvement in cardiac performance was detected. Similar beneficial effects were seen in a small trial of 100 patients who received cariporide before percutaneous coronary angioplasty (Rupprecht et al., 2000).
The related myocardial protective effects induced by CsA and NHE1 inhibition prompted us to speculate about the possibility that NHE1 inhibition protects the heart by targeting the mitochondria (Robin et al., 2007). We recently demonstrated that reduction of mNHE1 protein expression (shRNA-NHE1) or specific inhibition of NHE1 (HOE642) at the rat heart mitochondria significantly reduced the Ca2+-induced mitochondrial swelling [Figures 1, 2, modified from Villa-Abrille et al. (2011)]. Interestingly, HOE642 did not provide additional effect on Ca2+-induced mitochondrial swelling, in mitochondria with diminished levels of mNHE1 protein expression, suggesting a direct involvement of mNHE1 on the susceptibility of the MPTP for opening following the Ca2+ overload of the mitochondria. Additionally, the immunosuppressive agent, CsA inhibited the MPTP opening and mitochondrial swelling to the same extent as observed in isolated rat heart mitochondria inhibited with HOE642 or isolated mitochondria with reduced NHE1 expression [Figures 1, 2, modified from Villa-Abrille et al. (2011)]. We speculate that a decrease in mitochondrial exchanger function (or reduced expression by siRNA) should increase H+ concentration on the external side of the MPTP. In this regard, a prevention of MPTP formation by acidosis after an ischemic episode has been reported. Thus, accumulation of H+ following mNHE1 inhibition or expression reduction should decrease MPTP opening in heart mitochondria subjected to stress conditions. Though numerous studies (Karmazyn, 1988; Kusumoto et al., 2001; Wang et al., 2003; Fantinelli et al., 2006) demonstrated a protective role of NHE1 inhibition I/R injury in the heart, no mitochondrial effect of NHE1 inhibition has been considered. However, a report by Ruiz-Meana et al. (2003) drew attention to this possibility. Furthermore, Aldakkak et al. (2008) demonstrated that the enhanced activation of NHE with alkaline pHi during ischemic stress leads to an additional increase in mitochondrial Ca2+ load, which contributes to greater deterioration of mitochondrial bioenergetics and ROS production on reperfusion. Authors proved that NHE1 blockade at an alkaline pH improved the functional recovery after reperfusion, minimized the increase in mitochondrial [Ca2+], preserved the mitochondrial redox state, and reduced ROS production. These data suggested that both sarcolemmal and mitochondrial NHE1 may be involved in promoting mitochondrial Ca2+ loading with I/R injury. In connection with this, Teshima and collaborators (2003) demonstrated that cariporide protects cardiomyocyte against cell death induced by oxidative stress, preserving intracellular Na+ and Ca2+ homeostasis and mitochondrial integrity. The mitochondrial Ca2+ overload and the mitochondrial ΔΨ m loss induced by oxidative stress were remarkably prevented by cariporide (Teshima et al., 2003). Additionally, Toda et al. (2007) demonstrated that the NHE1 inhibitor, cariporide, diminished the mitochondrial Ca2+ overload and MPTP opening induced by Na+/K+ ATPase inhibition in isolated cardiomyocytes. Although these effects were proposed to be secondary to prevention of the cytosolic increase in Ca2+, more recently a direct mitochondrial action of NHE1 inhibitors related to the suppression of the myocardial superoxide production has been reported (Garciarena et al., 2008). A “cyclosporine like effect” of NHE1 inhibition was proposed, but the site of action of those compounds was not identified (Garciarena et al., 2008). Garciarena et al. (2008) studied the “anti-ROS effect” of NHE1 inhibitors. The authors analyzed NADPH oxidase-dependent mitochondrial O2-generation induced by ANG II or ET-1. The NADPH oxidase-dependent mitochondrial release of ROS is the basis of the so-called “ROS-induced ROS release” phenomenon proposed by Zorov et al. (2000) and Kimura et al. (2005). However, there is no clear evidence that sarcolemmal NADPH oxidase-derived ROS interacts with the mitochondria. Accordingly, Zhang et al. (2001) using reconstituted mKATP channels of bovine heart demonstrated that O2-directly stimulates the opening of these channels. The three NHE1 inhibitors used by Garciarena et al. (2008) blunted the increased mitochondrial ROS production and the redox activation of the kinases, well-known downstream targets of ROS (Sabri et al., 1998; Rothstein et al., 2002). The authors proposed a direct mitochondrial effect rather than a scavenging action of NHE1 inhibitors. Additionally, they showed that cariporide blunted not only the increased O2-production induced by ANG II/ET-1 but also the production induced by opening the mKATP channel with diazoxide. A mitochondrial action of NHE1 inhibition secondary to changes in cytosol was previously suggested by Javadov et al. (2005). In two previous publications, these authors concluded that the mitochondrial effect of NHE1 inhibitors is indirect and possibly mediated by the prevention of cytosolic Ca2+ overload. Thus, inhibition of the sarcolemmal NHE1 alters intracellular Na+ concentration and promotes Ca2+ overload (Javadov et al., 2005, 2008). Accordingly, they did not detect any mitochondria direct effect of the NHE1 inhibitors, and they suggested an action on MPTP function through glycogen synthase kinase 3-β (Javadov et al., 2009). We were unable to determine whether or not under their experimental conditions the effect of NHE1 inhibitors on mitochondrial Ca2+ and/or H+ is prevented.
Figure 1.

Mitochondrial swelling induced by CaCl2. (A) Typical experiment showing the scattered light absorbance traces of Ca2+-induced swelling in response to addition of CaCl2 to heart mitochondria isolated from rats transduced with shRNA-SCR (SCR, scrambled, control) in the presence or absence of cariporide (HOE642, 10 μ M) and Cyclosporine A (CsA, 10 mM). Cariporide and Cyclosporine attenuated Ca2+-induced mitochondrial swelling and the decrease in light scattering in mitochondrial suspensions. Cariporide inhibited the decrease in light scattering in a similar magnitude to CsA. (B) Scheme of a mitochondrion showing the CaCl2-induced swelling in mitochondria that expresses scrambled sequence and functional NHE1, and possible mitochondrial site of action of the cariporide. This inhibitor could act on different mitochondrial mechanisms, including NHE. They could act through a decrease in mitochondrial Ca2+, H+, and ΔΨ m affecting the MPTP formation or altering the sensitivity to those factors to induce MPTP formation. Modified from Villa-Abrille et al. (2011).
Figure 2.
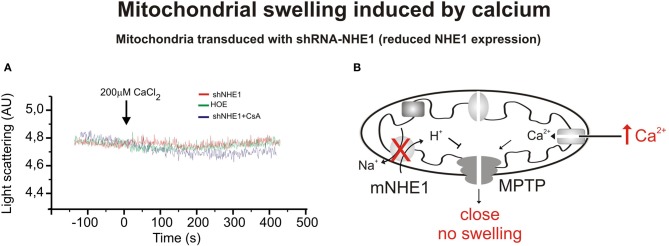
Mitochondrial swelling induced by CaCl2. (A) Typical experiment showing the scattered light absorbance traces of Ca2+-induced swelling in response to addition of CaCl2 for heart mitochondria isolated from rats transduced with shRNA-NHE1 in the presence and absence of cariporide. The shRNA-NHE1 transduction significantly inhibited Ca2+-induced mitochondrial swelling compared with SCR-transduced animals, (as previously shown in Figure 1). Cariporide and Cyclosporine attenuated the decrease in light scattering in SCR-transduced rats and did not affect the decrease in light scattering in shRNA-NHE1-transduced rats. These results indicate that the NHE1 participates in the mechanism by which the Ca2+ induces MPTP opening and swelling. (B) Scheme showing the absence of mitochondrial swelling and MPTP opening induced by Ca2+ when NHE1 is silencing. Modified from Villa-Abrille et al. (2011).
In 1969, Mitchell and Moyle (1969) proposed the existence of Na+/H+ exchange in mitochondria. Later, the existence of two systems capable of catalyze the H+ transport across the mitochondrial membrane, was described (Garlid et al., 1991). These systems—the unselective K+/H+ exchanger (KHE), which catalyzes the transport of virtually all alkali ions in the mitochondrion, and the NHE, which selectively transports Na+ or Li+ through mitochondrial membranes—were proposed as the main cation transporters of the mitochondria (Garlid et al., 1991). In addition, Garlid et al. (1991) demonstrated in beef heart mitochondria that solubilized NHE in the organelle can be purified in an active state.
In mitochondria, NHE mediates the exchange of matrix Na+ for intermembrane H+ generated by respiration (Garlid, 1988). Thus, H+ influx in the mitochondrial matrix through the mitochondrial NHE would constitute a form of H+ leakage not coupled to ATP synthesis, which in turn will dissipate the energy stored as transmitochondrial membrane H+ gradient.
NHE1 is a typical integral membrane protein with 10–12 predicted spanning segments, a long COOH terminus, and an NH2-terminal tail that possesses the mitochondrial localization signal. Previous studies using immunofluorescence and three-dimensional confocal microscopy techniques demonstrated the presence of NHE1 in the nuclear membranes isolated from the aortic vascular smooth muscle and liver of human, rabbit, and rat, suggesting a possible role of the nuclear NHE1 in the modulation of intranuclear pH (Bkaily et al., 2004). Our experiments, using isolated cardiomyocytes or mitochondrial lysates demonstrated the expression of the NHE1 protein in mitochondria isolated from rat ventricular myocardium and mitochondria isolated from HEK 293 cells transfected with NHE1-HA cDNA (Villa-Abrille et al., 2011). Besides the expression of NHE1 in the plasma membrane of cardiac cells, we have characterized the expression of NHE1 in mitochondria of cardiac muscle (Villa-Abrille et al., 2011) by several different experimental techniques (immunogold analysis combined with electron microscopy, immunohistochemistry combined with confocal microscopy or immunoblot analysis). Accordingly, Javadov et al. (2011) examined the expression of NHE1 and NHE6 in Percoll-purified rat heart mitochondria by immunoblot analysis and they only observed NHE1 in the mitochondria fraction (Javadov et al., 2011). Retention of NHE1 expression in isolated mitochondria subjected to digitonin allows them to conclude that NHE1 is expressed in the inner mitochondrial membrane (Javadov et al., 2011).
Dual distribution, at both plasma membrane and mitochondria, of others protein like the Na+/Ca+2 exchanger 1-3 (NCX1-3) (Gobbi et al., 2007), and Kv1.3 (Szabo et al., 2005), Kir6.2 (Garg and Hu, 2007), and Ca+2-activated BK potassium channels (Siemen et al., 1999), has been previously reported. In addition, Connexin 43 (Cx43), a constitutive protein that forms cardiac gap junctions contributing to cell-cell coupling, was also localized in mitochondria (Boengler et al., 2005, 2009). Cx43 contains four transmembrane domains as well as amino and carboxy termini located in the cytosol. Cx43 localized in subsarcolemmal mitochondria and its carboxy terminus directed toward the intermembrane space (Boengler et al., 2009).
With the assistance of the RNA interference technique, we evaluated the function and expression of NHE1 in cardiac tissues. With the objective of knocking down the expression of NHE1 in the heart, we injected a lentiviral vector expressing shRNA-NHE1 intramyocardically. The intramyocardial injection of lentivirus carrying a shRNA-NHE1 (silencing of NHE1) not only reduced the expression of NHE1 at the level of mitochondria but also prevented Ca2+-induced swelling of rat heart mitochondria [Figures 1, 2, modified from Villa-Abrille et al. (2011)]. The silencing of NHE also prevents the development of the slow force response (Perez et al., 2011). A decrease in mitochondrial NHE function (or reduced expression by siRNA) should increase H+ concentration in mitochondrial matrix. In connection with this, the prevention of MPTP formation by acidosis in reperfusion after ischemia has been reported (Cohen et al., 2007; Rodriguez-Sinovas et al., 2009). We reported a delay of MPTP formation in isolated mitochondria by NHE1 inhibition and posttranscriptional NHE1 gene silencing. These findings open a new avenue of research about the mechanism of protection achieved by NHE1 inhibition in reperfusion injury, cardiac hypertrophy, and heart failure. The mechanism by which the opening of the MPTP is prevented at low NHE1 expression is unknown. As previously described under physiological conditions, the mitochondrial NHE introduces cytosolic H+ into the mitochondrial matrix in exchange for mitochondrial Na+. Therefore, a decrease in exchanger activity or expression should reduce H+ and increase Na+ concentration in the mitochondrial matrix and perhaps increase H+ concentration on the cytosolic side of the MPTP. The decrease in H+ matrix concentration would favor and not prevent MPTP formation, but the increase of H+ on the cytosolic side of the MPTP may inhibit pore formation. In connection with this scenario, the prevention of MPTP formation by acidosis in reperfusion after ischemia has been reported (Cohen et al., 2007). An increase in mitochondrial Na+ would decrease the inwardly directed Na+ gradient, affecting the mitochondrial Na+/Ca2+ exchanger, other factors being constant (Murphy and Steenbergen, 2008). A decrease in Ca2+ efflux from the mitochondria would increase mitochondrial Ca2+ concentration and favor rather than reduce MPTP formation.
The main objective of this review was to emphasize the presence of NHE1 in the mitochondrial membrane and its role in MPTP opening. We are proposing the mitochondrial NHE1 as a novel target to prevent cardiac disease including I/R injury, cardiac hypertrophy, and heart failure. However, we could not rule out the concomitant participation of the sarcolemmal NHE1 in the protective effects achieved by NHE inhibition.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Akao M., O'Rourke B., Kusuoka H., Teshima Y., Jones S. P., Marban E. (2003). Differential actions of cardioprotective agents on the mitochondrial death pathway. Circ. Res. 92, 195–202 10.1161/01.RES.0000051862.16691.F9 [DOI] [PubMed] [Google Scholar]
- Aldakkak M., Stowe D. F., Heisner J. S., Spence M., Camara A. K. (2008). Enhanced Na+/H+ exchange during ischemia and reperfusion impairs mitochondrial bioenergetics and myocardial function. J. Cardiovasc. Pharmacol. 52, 236–244 10.1097/FJC.0b013e3181831337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avkiran M. (1999). Rational basis for use of sodium-hydrogen exchange inhibitors in myocardial ischemia. Am. J. Cardiol. 83, 10G–17G discussion: 17G–18G. 10.1016/S0002-9149(99)00215-5 [DOI] [PubMed] [Google Scholar]
- Avkiran M., Haworth R. S. (2003). Regulatory effects of G protein-coupled receptors on cardiac sarcolemmal Na+/H+ exchanger activity: signalling and significance. Cardiovasc. Res. 57, 942–952 10.1016/S0008-6363(02)00782-4 [DOI] [PubMed] [Google Scholar]
- Baines C. P. (2010). The cardiac mitochondrion: nexus of stress. Annu. Rev. Physiol. 72, 61–80 10.1146/annurev-physiol-021909-135929 [DOI] [PubMed] [Google Scholar]
- Bertrand B., Wakabayashi S., Ikeda T., Pouyssegur J., Shigekawa M. (1994). The Na+/H+ exchanger isoform 1 (NHE1) is a novel member of the calmodulin-binding proteins. Identification and characterization of calmodulin-binding sites. J. Biol. Chem. 269, 13703–13709 [PubMed] [Google Scholar]
- Biemesderfer D., Reilly R. F., Exner M., Igarashi P., Aronson P. S. (1992). Immunocytochemical characterization of Na(+)-H+ exchanger isoform NHE-1 in rabbit kidney. Am. J. Physiol. 263, F833–F840 [DOI] [PubMed] [Google Scholar]
- Bkaily G., Nader M., Avedanian L., Jacques D., Perrault C., Abdel-Samad D., et al. (2004). Immunofluorescence revealed the presence of NHE-1 in the nuclear membranes of rat cardiomyocytes and isolated nuclei of human, rabbit, and rat aortic and liver tissues. Can. J. Physiol. Pharmacol. 82, 805–811 10.1139/y04-119 [DOI] [PubMed] [Google Scholar]
- Boengler K., Dodoni G., Rodriguez-Sinovas A., Cabestrero A., Ruiz-Meana M., Gres P., et al. (2005). Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc. Res. 67, 234–244 10.1016/j.cardiores.2005.04.014 [DOI] [PubMed] [Google Scholar]
- Boengler K., Stahlhofen S., Van De Sand A., Gres P., Ruiz-Meana M., Garcia-Dorado D., et al. (2009). Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res. Cardiol. 104, 141–147 10.1007/s00395-009-0007-5 [DOI] [PubMed] [Google Scholar]
- Caldiz C. I., Garciarena C. D., Dulce R. A., Novaretto L. P., Yeves A. M., Ennis I. L., et al. (2007). Mitochondrial reactive oxygen species activate the slow force response to stretch in feline myocardium. J. Physiol. 584, 895–905 10.1113/jphysiol.2007.141689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani H. E., Perez N. G., Aiello E. A., Ennis I. L., Garciarena C. D., Villa-Abrille M. C., et al. (2008). Early signals after stretch leading to cardiac hypertrophy. Key role of NHE-1. Front. Biosci. 13, 7096–7114 10.2741/3213 [DOI] [PubMed] [Google Scholar]
- Cingolani H. E., Perez N. G., Caldiz C. I., Garciarena C. D., De Giusti V. C., Correa M. V., et al. (2010). Early hypertrophic signals after myocardial stretch. Role of reactive oxygen species and the sodium/hydrogen exchanger, in Mechanosensitivity in Cells and Tissues: Mechanosensitivy of the Heart, eds Kamkin A., Kiseleva I. (Moscow: Springer; ), 327–371 [Google Scholar]
- Cohen M. V., Yang X. M., Downey J. M. (2007). The pH hypothesis of postconditioning: staccato reperfusion reintroduces oxygen and perpetuates myocardial acidosis. Circulation 115, 1895–1903 10.1161/CIRCULATIONAHA.106.675710 [DOI] [PubMed] [Google Scholar]
- De Giusti V. C., Nolly M. B., Yeves A. M., Caldiz C. I., Villa-Abrille M. C., Chiappe De Cingolani G. E., et al. (2011). Aldosterone stimulates the cardiac Na(+)/H(+) exchanger via transactivation of the epidermal growth factor receptor. Hypertension 58, 912–919 10.1161/HYPERTENSIONAHA.111.176024 [DOI] [PubMed] [Google Scholar]
- Duchen M. R., McGuinness O., Brown L. A., Crompton M. (1993). On the involvement of a cyclosporin A sensitive mitochondrial pore in myocardial reperfusion injury. Cardiovasc. Res. 27, 1790–1794 10.1093/cvr/27.10.1790 [DOI] [PubMed] [Google Scholar]
- Fantinelli J. C., Cingolani H. E., Mosca S. M. (2006). Na+/H+ exchanger inhibition at the onset of reperfusion decreases myocardial infarct size: role of reactive oxygen species. Cardiovasc. Pathol. 15, 179–184 10.1016/j.carpath.2006.04.005 [DOI] [PubMed] [Google Scholar]
- Fliegel L. (2008). Molecular biology of the myocardial Na+/H+ exchanger. J. Mol. Cell. Cardiol. 44, 228–237 10.1016/j.yjmcc.2007.11.016 [DOI] [PubMed] [Google Scholar]
- Fliegel L., Karmazyn M. (2004). The cardiac Na-H exchanger: a key downstream mediator for the cellular hypertrophic effects of paracrine, autocrine and hormonal factors. Biochem. Cell Biol. 82, 626–635 10.1139/o04-129 [DOI] [PubMed] [Google Scholar]
- Fliegel L., Walsh M. P., Singh D., Wong C., Barr A. (1992). Phosphorylation of the C-terminal domain of the Na+/H+ exchanger by Ca2+/calmodulin-dependent protein kinase II. Biochem. J. 282(Pt 1), 139–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garciarena C. D., Caldiz C. I., Correa M. V., Schinella G. R., Mosca S. M., Chiappe De Cingolani G. E., et al. (2008). Na+/H+ exchanger-1 inhibitors decrease myocardial superoxide production via direct mitochondrial action. J. Appl. Physiol. 105, 1706–1713 10.1152/japplphysiol.90616.2008 [DOI] [PubMed] [Google Scholar]
- Garg V., Hu K. (2007). Protein kinase C isoform-dependent modulation of ATP-sensitive K+ channels in mitochondrial inner membrane. Am. J. Physiol. Heart Circ. Physiol. 293, H322–H332 10.1152/ajpheart.01035.2006 [DOI] [PubMed] [Google Scholar]
- Garlid K. D. (1988). Sodium/proton antiporters in the mitochondrial inner membrane. Adv. Exp. Med. Biol. 232, 37–46 10.1007/978-1-4757-0007-7_4 [DOI] [PubMed] [Google Scholar]
- Garlid K. D., Shariat-Madar Z., Nath S., Jezek P. (1991). Reconstitution and partial purification of the Na(+)-selective Na+/H+ antiporter of beef heart mitochondria. J. Biol. Chem. 266, 6518–6523 [PubMed] [Google Scholar]
- Gobbi P., Castaldo P., Minelli A., Salucci S., Magi S., Corcione E., Amoroso S. (2007). Mitochondrial localization of Na+/Ca2+ exchangers NCX1-3 in neurons and astrocytes of adult rat brain in situ. Pharmacol. Res. 56, 556–565 10.1016/j.phrs.2007.10.005 [DOI] [PubMed] [Google Scholar]
- Griffiths E. J., Halestrap A. P. (1993). Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J. Mol. Cell. Cardiol. 25, 1461–1469 10.1006/jmcc.1993.1162 [DOI] [PubMed] [Google Scholar]
- Gumina R. J., Mizumura T., Beier N., Schelling P., Schultz J. J., Gross G. J. (1998). A new sodium/hydrogen exchange inhibitor, EMD 85131, limits infarct size in dogs when administered before or after coronary artery occlusion. J. Pharmacol. Exp. Ther. 286, 175–183 [PubMed] [Google Scholar]
- Halestrap A. P., Pasdois P. (2009). The role of the mitochondrial permeability transition pore in heart disease. Biochim. Biophys. Acta 1787, 1402–1415 10.1016/j.bbabio.2008.12.017 [DOI] [PubMed] [Google Scholar]
- Haworth R. S., McCann C., Snabaitis A. K., Roberts N. A., Avkiran M. (2003). Stimulation of the plasma membrane Na+/H+ exchanger NHE1 by sustained intracellular acidosis. Evidence for a novel mechanism mediated by the ERK pathway. J. Biol. Chem. 278, 31676–31684 10.1074/jbc.M304400200 [DOI] [PubMed] [Google Scholar]
- Haworth R. S., Sinnett-Smith J., Rozengurt E., Avkiran M. (1999). Protein kinase D inhibits plasma membrane Na(+)/H(+) exchanger activity. Am. J. Physiol. 277, C1202–C1209 [DOI] [PubMed] [Google Scholar]
- Hurtado C., Pierce G. N. (2000). Inhibition of Na(+)/H(+) exchange at the beginning of reperfusion is cardioprotective in isolated, beating adult cardiomyocytes. J. Mol. Cell. Cardiol. 32, 1897–1907 10.1006/jmcc.2000.1222 [DOI] [PubMed] [Google Scholar]
- Javadov S., Choi A., Rajapurohitam V., Zeidan A., Basnakian A. G., Karmazyn M. (2008). NHE-1 inhibition-induced cardioprotection against ischaemia/reperfusion is associated with attenuation of the mitochondrial permeability transition. Cardiovasc. Res. 77, 416–424 10.1093/cvr/cvm039 [DOI] [PubMed] [Google Scholar]
- Javadov S., Huang C., Kirshenbaum L., Karmazyn M. (2005). NHE-1 inhibition improves impaired mitochondrial permeability transition and respiratory function during postinfarction remodelling in the rat. J. Mol. Cell. Cardiol. 38, 135–143 10.1016/j.yjmcc.2004.10.007 [DOI] [PubMed] [Google Scholar]
- Javadov S., Karmazyn M. (2007). Mitochondrial permeability transition pore opening as an endpoint to initiate cell death and as a putative target for cardioprotection. Cell. Physiol. Biochem. 20, 1–22 10.1159/000103747 [DOI] [PubMed] [Google Scholar]
- Javadov S., Rajapurohitam V., Kilic A., Hunter J. C., Zeidan A., Said Faruq N., et al. (2011). Expression of mitochondrial fusion-fission proteins during post-infarction remodeling: the effect of NHE-1 inhibition. Basic Res. Cardiol. 106, 99–109 10.1007/s00395-010-0122-3 [DOI] [PubMed] [Google Scholar]
- Javadov S., Rajapurohitam V., Kilic A., Zeidan A., Choi A., Karmazyn M. (2009). Anti-hypertrophic effect of NHE-1 inhibition involves GSK-3beta-dependent attenuation of mitochondrial dysfunction. J. Mol. Cell. Cardiol. 46, 998–1007 10.1016/j.yjmcc.2008.12.023 [DOI] [PubMed] [Google Scholar]
- Karmazyn M. (1988). Amiloride enhances postischemic ventricular recovery: possible role of Na+-H+ exchange. Am. J. Physiol. 255, H608–H615 [DOI] [PubMed] [Google Scholar]
- Karmazyn M. (1999a). Mechanisms of protection of the ischemic and reperfused myocardium by sodium-hydrogen exchange inhibition. J. Thromb. Thrombolysis 8, 33–38 [DOI] [PubMed] [Google Scholar]
- Karmazyn M. (1999b). The role of the myocardial sodium-hydrogen exchanger in mediating ischemic and reperfusion injury. Ann. N.Y. Acad. Sci. 874, 326–334 [DOI] [PubMed] [Google Scholar]
- Karmazyn M., Gan X. T., Humphreys R. A., Yoshida H., Kusumoto K. (1999). The myocardial Na(+)-H(+) exchange: structure, regulation, and its role in heart disease. Circ. Res. 85, 777–786 10.1161/01.RES.85.9.777 [DOI] [PubMed] [Google Scholar]
- Khaled A. R., Moor A. N., Li A., Kim K., Ferris D. K., Muegge K., et al. (2001). Trophic factor withdrawal: p38 mitogen-activated protein kinase activates NHE1, which induces intracellular alkalinization. Mol. Cell. Biol. 21, 7545–7557 10.1128/MCB.21.22.7545-7557.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S., Zhang G. X., Nishiyama A., Shokoji T., Yao L., Fan Y. Y., et al. (2005). Mitochondria-derived reactive oxygen species and vascular MAP kinases: comparison of angiotensin II and diazoxide. Hypertension 45, 438–444 [DOI] [PubMed] [Google Scholar]
- Koerner J. E., Anderson B. A., Dage R. C. (1991). Protection against postischemic myocardial dysfunction in anesthetized rabbits with scavengers of oxygen-derived free radicals: superoxide dismutase plus catalase, N-2-mercaptopropionyl glycine and captopril. J. Cardiovasc. Pharmacol. 17, 185–191 10.1097/00005344-199102000-00002 [DOI] [PubMed] [Google Scholar]
- Kusumoto K., Haist J. V., Karmazyn M. (2001). Na(+)/H(+) exchange inhibition reduces hypertrophy and heart failure after myocardial infarction in rats. Am. J. Physiol. Heart Circ. Physiol. 280, H738–H745 [DOI] [PubMed] [Google Scholar]
- Lee S. H., Kim T., Park E. S., Yang S., Jeong D., Choi Y., et al. (2008). NHE10, an osteoclast-specific member of the Na+/H+ exchanger family, regulates osteoclast differentiation and survival [corrected]. Biochem. Biophys. Res. Commun. 369, 320–326 10.1016/j.bbrc.2008.01.168 [DOI] [PubMed] [Google Scholar]
- Leem C. H., Lagadic-Gossmann D., Vaughan-Jones R. D. (1999). Characterization of intracellular pH regulation in the guinea-pig ventricular myocyte. J. Physiol. 517(Pt 1), 159–180 10.1111/j.1469-7793.1999.0159z.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Alvarez B., Casey J. R., Reithmeier R. A., Fliegel L. (2002). Carbonic anhydrase II binds to and enhances activity of the Na+/H+ exchanger. J. Biol. Chem. 277, 36085–36091 10.1074/jbc.M111952200 [DOI] [PubMed] [Google Scholar]
- McDonald M. C., Zacharowski K., Bowes J., Cuzzocrea S., Thiemermann C. (1999). Tempol reduces infarct size in rodent models of regional myocardial ischemia and reperfusion. Free Radic. Biol. Med. 27, 493–503 10.1016/S0891-5849(99)00100-8 [DOI] [PubMed] [Google Scholar]
- Mentzer R. M., Jr., Bartels C., Bolli R., Boyce S., Buckberg G. D. (2008). Sodium-hydrogen exchange inhibition by cariporide to reduce the risk of ischemic cardiac events in patients undergoing coronary artery bypass grafting: results of the EXPEDITION study. Ann. Thorac. Surg. 85, 1261–1270 10.1016/j.athoracsur.2007.10.054 [DOI] [PubMed] [Google Scholar]
- Misik A. J., Perreault K., Holmes C. F., Fliegel L. (2005). Protein phosphatase regulation of Na+/H+ exchanger isoform I. Biochemistry 44, 5842–5852 10.1021/bi047659s [DOI] [PubMed] [Google Scholar]
- Mitchell P., Moyle J. (1969). Translocation of some anions cations and acids in rat liver mitochondria. Eur. J. Biochem. 9, 149–155 10.1111/j.1432-1033.1969.tb00588.x [DOI] [PubMed] [Google Scholar]
- Moor A. N., Fliegel L. (1999). Protein kinase-mediated regulation of the Na(+)/H(+) exchanger in the rat myocardium by mitogen-activated protein kinase-dependent pathways. J. Biol. Chem. 274, 22985–22992 10.1074/jbc.274.33.22985 [DOI] [PubMed] [Google Scholar]
- Murphy E., Allen D. G. (2009). Why did the NHE inhibitor clinical trials fail. J. Mol. Cell. Cardiol. 46, 137–141 10.1016/j.yjmcc.2008.09.715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E., Steenbergen C. (2008). Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 88, 581–609 10.1152/physrev.00024.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T. Y., Iwata Y., Arai Y., Komamura K., Wakabayashi S. (2008). Activation of Na+/H+ exchanger 1 is sufficient to generate Ca2+ signals that induce cardiac hypertrophy and heart failure. Circ. Res. 103, 891–899 10.1161/CIRCRESAHA.108.175141 [DOI] [PubMed] [Google Scholar]
- Orlowski J., Grinstein S. (1997). Na+/H+ exchangers of mammalian cells. J. Biol. Chem. 272, 22373–22376 10.1074/jbc.272.36.22373 [DOI] [PubMed] [Google Scholar]
- Pang T., Su X., Wakabayashi S., Shigekawa M. (2001). Calcineurin homologous protein as an essential cofactor for Na+/H+ exchangers. J. Biol. Chem. 276, 17367–17372 10.1074/jbc.M100296200 [DOI] [PubMed] [Google Scholar]
- Perez N. G., Nolly M. B., Roldan M. C., Villa-Abrille M. C., Cingolani E., Portiansky E. L., et al. (2011). Silencing of NHE-1 blunts the slow force response to myocardial stretch. J. Appl. Physiol. 111, 874–880 10.1152/japplphysiol.01344.2010 [DOI] [PubMed] [Google Scholar]
- Petrecca K., Atanasiu R., Grinstein S., Orlowski J., Shrier A. (1999). Subcellular localization of the Na+/H+ exchanger NHE1 in rat myocardium. Am. J. Physiol. 276, H709–H717 [DOI] [PubMed] [Google Scholar]
- Piot C., Croisille P., Staat P., Thibault H., Rioufol G., Mewton N., et al. (2008). Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 359, 473–481 10.1056/NEJMoa071142 [DOI] [PubMed] [Google Scholar]
- Robin E., Guzy R. D., Loor G., Iwase H., Waypa G. B., Marks J. D., et al. (2007). Oxidant stress during simulated ischemia primes cardiomyocytes for cell death during reperfusion. J. Biol. Chem. 282, 19133–19143 10.1074/jbc.M701917200 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Sinovas A., Cabestrero A., Garcia Del Blanco B., Inserte J., Garcia A., Garcia-Dorado D. (2009). Intracoronary acid infusion as an alternative to ischemic postconditioning in pigs. Basic Res. Cardiol. 104, 761–771 10.1007/s00395-009-0032-4 [DOI] [PubMed] [Google Scholar]
- Rohmann S., Weygandt H., Minck K. O. (1995). Preischaemic as well as postischaemic application of a Na+/H+ exchange inhibitor reduces infarct size in pigs. Cardiovasc. Res. 30, 945–951 [PubMed] [Google Scholar]
- Rothstein E. C., Byron K. L., Reed R. E., Fliegel L., Lucchesi P. A. (2002). H(2)O(2)-induced Ca(2+) overload in NRVM involves ERK1/2 MAP kinases: role for an NHE-1-dependent pathway. Am. J. Physiol. Heart Circ. Physiol. 283, H598–H605 [DOI] [PubMed] [Google Scholar]
- Ruiz-Meana M., Garcia-Dorado D., Pina P., Inserte J., Agullo L., Soler-Soler J. (2003). Cariporide preserves mitochondrial proton gradient and delays ATP depletion in cardiomyocytes during ischemic conditions. Am. J. Physiol. Heart Circ. Physiol. 285, H999–H1006 [DOI] [PubMed] [Google Scholar]
- Rupprecht H. J., Vom Dahl J., Terres W., Seyfarth K. M., Richardt G., Schultheibeta H. P., et al. (2000). Cardioprotective effects of the Na(+)/H(+) exchange inhibitor cariporide in patients with acute anterior myocardial infarction undergoing direct PTCA. Circulation 101, 2902–2908 10.1161/01.CIR.101.25.2902 [DOI] [PubMed] [Google Scholar]
- Sabri A., Byron K. L., Samarel A. M., Bell J., Lucchesi P. A. (1998). Hydrogen peroxide activates mitogen-activated protein kinases and Na+-H+ exchange in neonatal rat cardiac myocytes. Circ. Res. 82, 1053–1062 10.1161/01.RES.82.10.1053 [DOI] [PubMed] [Google Scholar]
- Sahna E., Acet A., Ozer M. K., Olmez E. (2002). Myocardial ischemia-reperfusion in rats: reduction of infarct size by either supplemental physiological or pharmacological doses of melatonin. J. Pineal Res. 33, 234–238 10.1034/j.1600-079X.2002.02924.x [DOI] [PubMed] [Google Scholar]
- Sardet C., Counillon L., Franchi A., Pouyssegur J. (1990). Growth factors induce phosphorylation of the Na+/H+ antiporter, glycoprotein of 110 kD. Science 247, 723–726 10.1126/science.2154036 [DOI] [PubMed] [Google Scholar]
- Sardet C., Fafournoux P., Pouyssegur J. (1991). Alpha-thrombin, epidermal growth factor, and okadaic acid activate the Na+/H+ exchanger, NHE-1, by phosphorylating a set of common sites. J. Biol. Chem. 266, 19166–19171 [PubMed] [Google Scholar]
- Sardet C., Franchi A., Pouyssegur J. (1989). Molecular cloning, primary structure, and expression of the human growth factor-activatable Na+/H+ antiporter. Cell 56, 271–280 10.1016/0092-8674(89)90901-X [DOI] [PubMed] [Google Scholar]
- Siemen D., Loupatatzis C., Borecky J., Gulbins E., Lang F. (1999). Ca2+-activated K channel of the BK-type in the inner mitochondrial membrane of a human glioma cell line. Biochem. Biophys. Res. Commun. 257, 549–554 10.1006/bbrc.1999.0496 [DOI] [PubMed] [Google Scholar]
- Snabaitis A. K., D'Mello R., Dashnyam S., Avkiran M. (2006). A novel role for protein phosphatase 2A in receptor-mediated regulation of the cardiac sarcolemmal Na+/H+ exchanger NHE1. J. Biol. Chem. 281, 20252–20262 10.1074/jbc.M600268200 [DOI] [PubMed] [Google Scholar]
- Szabo I., Bock J., Jekle A., Soddemann M., Adams C., Lang F., et al. (2005). A novel potassium channel in lymphocyte mitochondria. J. Biol. Chem. 280, 12790–12798 10.1074/jbc.M413548200 [DOI] [PubMed] [Google Scholar]
- Takahashi E., Abe J., Gallis B., Aebersold R., Spring D. J., Krebs E. G., Berk B. C. (1999). p90(RSK) is a serum-stimulated Na+/H+ exchanger isoform-1 kinase. Regulatory phosphorylation of serine 703 of Na+/H+ exchanger isoform-1. J. Biol. Chem. 274, 20206–20214 10.1074/jbc.274.29.20206J [DOI] [PubMed] [Google Scholar]
- Tanaka M., Fujiwara H., Yamasaki K., Sasayama S. (1994). Superoxide dismutase and N-2-mercaptopropionyl glycine attenuate infarct size limitation effect of ischaemic preconditioning in the rabbit. Cardiovasc. Res. 28, 980–986 10.1093/cvr/28.7.980 [DOI] [PubMed] [Google Scholar]
- Teshima Y., Akao M., Jones S. P., Marban E. (2003). Cariporide (HOE642), a selective Na+-H+ exchange inhibitor, inhibits the mitochondrial death pathway. Circulation 108, 2275–2281 10.1161/01.CIR.0000093277.20968.C7 [DOI] [PubMed] [Google Scholar]
- Theroux P., Chaitman B. R., Danchin N., Erhardt L., Meinertz T., Schroeder J. S., et al. (2000). Inhibition of the sodium-hydrogen exchanger with cariporide to prevent myocardial infarction in high-risk ischemic situations. Main results of the GUARDIAN trial. Guard during ischemia against necrosis (GUARDIAN) Investigators. Circulation 102, 3032–3038 10.1161/01.CIR.102.25.3032 [DOI] [PubMed] [Google Scholar]
- Toda T., Kadono T., Hoshiai M., Eguchi Y., Nakazawa S., Nakazawa H., et al. (2007). Na+/H+ exchanger inhibitor cariporide attenuates the mitochondrial Ca2+ overload and PTP opening. Am. J. Physiol. Heart Circ. Physiol. 293, H3517–H3523 10.1152/ajpheart.00483.2006 [DOI] [PubMed] [Google Scholar]
- Tominaga T., Ishizaki T., Narumiya S., Barber D. L. (1998). p160ROCK mediates RhoA activation of Na-H exchange. EMBO J. 17, 4712–4722 10.1093/emboj/17.16.4712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa-Abrille M. C., Caldiz C. I., Ennis I. L., Nolly M. B., Casarini M. J., Chiappe De Cingolani G. E., et al. (2010). The Anrep effect requires transactivation of the epidermal growth factor receptor. J. Physiol. 588, 1579–1590 10.1113/jphysiol.2009.186619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa-Abrille M. C., Cingolani E., Cingolani H. E., Alvarez B. V. (2011). Silencing of cardiac mitochondrial NHE1 prevents mitochondrial permeability transition pore opening. Am. J. Physiol. Heart Circ. Physiol. 300, H1237–H1251 10.1152/ajpheart.00840.2010 [DOI] [PubMed] [Google Scholar]
- Wakabayashi S., Bertrand B., Ikeda T., Pouyssegur J., Shigekawa M. (1994). Mutation of calmodulin-binding site renders the Na+/H+ exchanger (NHE1) highly H(+)-sensitive and Ca2+ regulation-defective. J. Biol. Chem. 269, 13710–13715 [PubMed] [Google Scholar]
- Wakabayashi S., Fafournoux P., Sardet C., Pouyssegur J. (1992). The Na+/H+ antiporter cytoplasmic domain mediates growth factor signals and controls “H(+)-sensing.” Proc. Natl. Acad. Sci. U.S.A. 89, 2424–2428 10.1073/pnas.89.6.2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi S., Shigekawa M., Pouyssegur J. (1997). Molecular physiology of vertebrate Na+/H+ exchangers. Physiol. Rev. 77, 51–74 [DOI] [PubMed] [Google Scholar]
- Wang Y., Meyer J. W., Ashraf M., Shull G. E. (2003). Mice with a null mutation in the NHE1 Na+-H+ exchanger are resistant to cardiac ischemia-reperfusion injury. Circ. Res. 93, 776–782 10.1161/01.RES.0000094746.24774.DC [DOI] [PubMed] [Google Scholar]
- Xue J., Mraiche F., Zhou D., Karmazyn M., Oka T., Fliegel L., et al. (2010). Elevated myocardial Na+/H+ exchanger isoform 1 activity elicits gene expression that leads to cardiac hypertrophy. Physiol. Genomics 42, 374–383 10.1152/physiolgenomics.00064.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W., Nehrke K., Choi J., Barber D. L. (2001). The Nck-interacting kinase (NIK) phosphorylates the Na+-H+ exchanger NHE1 and regulates NHE1 activation by platelet-derived growth factor. J. Biol. Chem. 276, 31349–31356 10.1074/jbc.M102679200 [DOI] [PubMed] [Google Scholar]
- Zeymer U., Suryapranata H., Monassier J. P., Opolski G., Davies J., Rasmanis G., et al. (2001). The Na(+)/H(+) exchange inhibitor eniporide as an adjunct to early reperfusion therapy for acute myocardial infarction. Results of the evaluation of the safety and cardioprotective effects of eniporide in acute myocardial infarction (ESCAMI) trial. J. Am. Coll. Cardiol. 38, 1644–1650 10.1016/S0735-1097(01)01608-4 [DOI] [PubMed] [Google Scholar]
- Zhang D. X., Chen Y. F., Campbell W. B., Zou A. P., Gross G. J., Li P. L. (2001). Characteristics and superoxide-induced activation of reconstituted myocardial mitochondrial ATP-sensitive potassium channels. Circ. Res. 89, 1177–1183 10.1161/hh2401.101752 [DOI] [PubMed] [Google Scholar]
- Zorov D. B., Filburn C. R., Klotz L. O., Zweier J. L., Sollott S. J. (2000). Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 192, 1001–1014 10.1084/jem.192.7.1001 [DOI] [PMC free article] [PubMed] [Google Scholar]