

Abstract
The role that water plays in the salt-based stabilization of proteins is central to our understanding of protein biophysics. Ion hydration and the ability of ions to alter water surface tension are typically invoked, along with direct ion-protein binding, to describe Hofmeister stabilization phenomena observed for proteins experimentally, but the relative influence of these forces has been extraordinarily difficult to measure directly. Recently, we have used gas-phase measurements of proteins and large multiprotein complexes, using a combination of innovative ion mobility (IM) and mass spectrometry (MS) techniques, to assess the ability of bound cations and anions to stabilize protein ions in the absence of the solvation forces described above. Our previous work has studied a broad set of 12 anions bound to a range of proteins and protein complexes, and while primarily motivated by the analytical challenges surrounding the gas-phase measurement of solution-phase relevant protein structures, our work has also lead to a detailed physical mechanism of anion-protein complex stabilization in the absence of bulk solvent. Our more-recent work has screened a similarly-broad set of cations for their ability to stabilize gas-phase protein structure, and we have discovered surprising differences between the operative mechanisms for cations and anions in gas-phase protein stabilization. In both cases, cations and anions affect protein stabilization in the absence of solvent in a manner that is generally reversed relative to their ability to stabilize the same proteins in solution. In addition, our evidence suggests that the relative solution-phase binding affinity of the anions and cations studied here is preserved in our gas-phase measurements, allowing us to study the influence of such interactions in detail. In this report, we collect and summarize such gas-phase measurements to distill a generalized picture of salt-based protein stabilization in the absence of bulk water. Further, we communicate our most recent efforts to study the combined effects of stabilizing cations and anions on gas-phase proteins, and identify those salts that bear anion/cation pairs having the strongest stabilizing influence on protein structures in vacuo.
1. Introduction
Proteins are central molecular machines in the critical biological processes necessary for life. In many cases, these essential functions are regulated by protein structure, dynamics, and stability.1 As such, a deep understanding of these properties has been sought by biochemists for well over a century. In that time, we have learned that many of the important biophysical properties of proteins can be influenced dramatically by the presence or absence of salts in vivo.2–5 Indeed, such disparate biochemical properties as cell growth and protein crystallization have been directly linked to the influences of critical anions and cations.5, 6 In pioneering work, Hofmeister discovered that such salts can either stabilize or destabilize proteins differentially, and that empirical observations can be used to construct a generally predictive rank order.7 Understanding the basic physical mechanism(s) that underlie the series that now bears his name has become an active area of research, due to its central importance in our understanding of protein biophysics.
While our knowledge of Hofmeister-type protein-salt interactions is still evolving, several experimental results, some of them fairly recent, have provided tremendous insight into the important aspects of their stabilization mechanism. Originally, the structure of bulk water, and its alteration through specific long range forces generated by anions and cations in solution, was thought to be critical for understanding Hofmeister-type protein stabilization.8, 9 Anions and cations were classified as either water structure makers (kosmotropes) or breakers (chaotropes), but recent experiments have indicated strongly that such structural effects are minimal at relevant solute concentrations, and have no direct causal relationship to Hofmeister-type protein stabilization.10–14 Revised theories center on direct anion/cation interactions with proteins in three main ways.15 First, anions and cations may directly interact with the protein backbone and side chains through ion pairing interactions.16, 17 Anions are known to have high affinity for amino functional groups within proteins,18–21 and cations are likely to interact with an array of sites,22, 23 in many cases involving carboxylate groups.20, 24, 25 Arguably more important, in light of current data, are the more-indirect interactions between Hofmeister ions and the layer of water closely associated with proteins. Anions and cations can alter both the surface tension and hydrogen-bonding network surrounding proteins significantly, such that hydration entropy and protein stability are dramatically affected.26–29 Although canonical Hofmeister series are operative in many cellular processes, reversed Hofmeister series have also been observed, and rationalized through alterations to local water structures and direct protein-ion interactions, as above.15, 30–32 Thus, while local protein-water interactions have been deemed important in Hofmeister stabilization, the magnitude of their importance is a somewhat malleable concept and subject to change based on the specific process or proteins being studied.
Based on the potential importance of solvent in Hofmeister-type protein stabilization, a number of groups have undertaken experiments carried out in environments of rarefied solvation (e.g., the gas-phase) to study both the local water structure surrounding small ions, and their interactions with proteins. Many of these experiments have been carried out using mass spectrometry (MS), where shifts in ion molecular mass can be interpreted relative to direct protein-counterion binding in solution.20, 21, 33–36 For example, precise measurements of molecular mass allowed Kebarle and co-workers to define the binding mode of many anions and cations to specific proteins.20 More recently, MS conditions have been identified whereby anion binding observed in the gas-phase can correlate precisely to the number of solvent accessible basic sites on a protein, thus mirroring bound populations in solution.21 Similarly, wavelength-resolved action spectroscopy, using MS detection, has been utilized to deduce the relative population of charge-solvated and zwitterionic structures in a range of amino acid-cation complexes.37–43 Similar measurements of anions and cations clustered with varying amounts of water have also been used to deduce ion specific effects on both local and bulk water structure.44–51 While these data have demonstrated significant local water structure effects for specific ions, in some cases extending out to long ranges, they have also shown that bulk water structure is largely unaffected by cations and anions in solution at relevant concentrations.
The desolvated structures of proteins can also be measured using ion mobility (IM) spectrometry,52,53 where ions are separated according to their orientiationally-averaged collision cross sections (CCSs) by measuring protein transit times through a gas-filled chamber under the influence of a relatively-weak electric field. IM data has been used to study the general dependence of protein structure as a function of bound solvent, revealing both the complexity of protein structural states that exist in the gas-phase and the level of solvation necessary to compact Coulombically unfolded proteins.54–56 In addition, IM data have recently been used to deduce the influence of a range of anions and cations on protein stability and structure in the absence of bulk solvent. For example, anion binding can result in the recovery of compact protein structures when the unbound gas-phase protein ions occupy unfolded states in the gas-phase.35 Similarly, our group has demonstrated the differential effects on desolvated protein ion stability for various cations and anions, showing that the identity of condensed salt can stabilize gas-phase protein structure through mechanisms vastly different from those found in solution.34, 36 In all of these cases, gas-phase data have served both to inform on the importance of solvent in protein structure and also highlighted examples where such structures can be conserved in the absence of bulk solvent.
The correlation between solution and gas-phase protein structure has, in part, driven the development and application of IM-MS in structural biology. For example, MS measurements have been used to study bioactive peptide aggregation,57 membrane protein structure,58, 59 and protein stability changes upon ligand binding.60, 61 Applying IM and MS to protein quaternary structure has rapidly developed in recent years, enabling the determination of multiprotein stoichiometry, dynamics, and 3D topology.62–67 In IM-MS experiments aimed at the determination of protein quaternary structure, detailed MS measurements of protein connectivity are combined with equally detailed IM measurements of gas-phase protein size in order to generate topology models.67 As such, the above-mentioned correlation between solution and gas-phase protein size and structure has become a critical component in protocols aimed at deducing multiprotein topology. Our group aims to use Hofmeister salts to stabilize protein complex structure in the absence of solvent, and our efforts to deduce the mechanisms driving cation and anion-mediated protein stability in the gas phase have primarily been motivated by our work to develop general IM-MS methods for determining protein assembly structure.
Here, we describe and summarize our previous results where we have used Hofmeister anions and cations as gas-phase stabilizers for protein structure. After providing a detailed account of the IM-MS methods we have developed to measure multiprotein complex stability, we then discuss the evidence to date illuminating the operative mechanisms of cation and anion protein stabilization in the gas phase. Anions primarily stabilize gas-phase proteins through a ‘dissociative-cooling’ mechanism that requires bound anions to dissociate from complexes to carry away excess energy from the system,34 and stabilizing cations often bind tightly to tether regions of the protein together and limit charge mobility.36 While our previous results focus primarily on measuring the influence of isolated anions and cations or protein structure, we also describe new experiments in which we seek to combine the influence of both bound anions and cations on gas-phase protein structure simultaneously. These results demonstrate that both anions and cations can be used in concert to stabilize protein structures in the absence of bulk solvent to an extent not previously accessible using either alone. We conclude by discussing the general relevance of gas-phase protein stability measurements for Hofmeister stabilization in solution, and emphasize the role that such measurements can play in elucidating the role of solvent in protein-salt interactions.
2. The IM-MS Approach for Measuring the Stability of Desolvated Protein Structure
2.1 A Multidimensional Measurement of Protein Stability
Our experimental approach for measuring the stability of gas-phase multiprotein complexes employs IM-MS to make rapid serial measurements of protein size and mass as a function of experimental variables. The measurements described in this report were carried out on a Synapt G2 HDMS (Waters, Milford, MA) platform.68–70 The instrument is equipped with a quadrupole mass analyzer, a travelling-wave ion mobility separator, and a time-of-flight (ToF) mass analyzer arranged in tandem.69 In order to generate useful information regarding the structure of proteins, the non-covalent contacts and course-grained structures of protein ions must to be maintained during their transfer to the gas phase. Nano-electrospray ionization (nESI, typically using ~ 5μL of sample, operating in the positive ion mode) is used in our experiments for both its broad tolerance to aqueous salt concentrations and its comparatively direct route in generating gas-phase protein ions from such solutions. Emitters were prepared as previously described,71 and ions were generated using capillary voltages ranging from 1.4–1.7 kV. The sampling cone voltage and source pressures were also optimized in order to transmit intact protein complexes and to preserve non-covalent interactions, as described previously.71
Ionized proteins and complexes are introduced into the vacuum of the IM-MS instrument via a series of differentially pumped regions, each having tightly controlled pressures and temperatures such that volatile solution additives are gradually removed to limit unwanted ion heating. Upon arrival at the quadrupole mass filter, ions of interest can be selected prior to IM-MS analysis based on their m/z. In this tandem-MS mode, ions in a narrow m/z range are selected and accumulated in an ion trap region situated in front of the IM separator (containing 3.3 × 10−2 mbar of argon gas). Each mass-selected ion can be activated by increasing the acceleration voltage used to introduce ions into the ion trap just prior to the mobility separator. Increases in this acceleration cause controlled ion heating, which in turn causes multiprotein complex ions to first unfold and then dissociate into both highly-charged protein monomers and complexes stripped of individual subunits. The monomers produced by this collision induced dissociation (CID) process are highly-charged primarily because of the relatively unfolded transition states occupied by the heated ions during the preceding collision induced unfolding (CIU) process.72, 73 For the CID and CIU data shown in this report, trap collision voltage (the accelerating potential described above) was varied in a step wise fashion while keeping other settings constant in order to construct collision energy-dependant datasets.
The IM separator within the instrument utilizes a series of low-voltage ‘waves’ that ‘travel’ at relatively low speeds by altering the potentials on a series of individually addressable electrode rings within the device (typical values are 40 V wave amplitudes, traveling at 800–1000 m/s).68 Under high-vacuum conditions, all ions are transported through the device at the velocity of these traveling waves. To undergo IM separation, the device is pressurized with ~3.5 mBar N2 gas such that the drift times of large ions, which collide frequently with the neutral gas, are slowed relative to ions having a smaller size, or CCS. A single IM separation typically takes ~30 milliseconds during which 200 ToF datasets can be recorded over a broad m/z range. Drift time measurements using a traveling-wave type IM separator are typically calibrated externally using a series of proteins having known gas-phase sizes to generate CCS values for unknowns. It should be noted that the general approach described above is not exclusive to the Synapt instrument geometry. In fact, many previous reports of monomeric protein unfolding have been described in the literature using both drift-tube ion mobility spectrometry (DT-IMS) 74–76 and field asymmetric IM (FAIMS)77 coupled to MS detectors.
The IM-MS approach that we have used to measure the stability of desolvated proteins and their complexes is illustrated in Figure 1, which shows a basic schematic diagram of the inner-workings of the IM-MS instrumentation. Our illustration depicts two simplified ion classes: those ions that lack stabilizing salts bound to their exterior (blue), and those proteins bound to a large numbers of such stabilizers (red). After creation through nESI, IM analysis of stabilized protein complexes exhibit narrow, monomodal IM drift time distributions (Figure 1B, red). In contrast, less stable protein complex ions unfold prior to IM analysis and exhibit broad, multimodal drift time spectra (Figure 1B, blue). The clear differences in the IM arrival time distributions recorded for these two ion classes allows us to infer that protein ions better maintain their compact states when bound to stabilizing salts (Figure 1A and Figure 1B). MS spectra for these same two protein ion classes, recorded on a ToF mass analyzer (m/z range of 800–15000 operated at a pressure of 1.6 ×10−6 mbar), can be used to determine the composition of the complex and verify protein binding stoichiometry. When observed at relatively low activation energies, the measured masses of protein ions bound to salt adducts are significantly larger than the known sequence mass of the assembly, as illustrated by the higher m/z values observed for those ions in Figures 1A and 1C. While this excess can be quantified using precise mass measurements and comparison with control samples, ion heating can also be used to drive the evaporation of salt adducts from protein ions, reducing their masses toward that predicted by sequence alone.54
Figure 1.
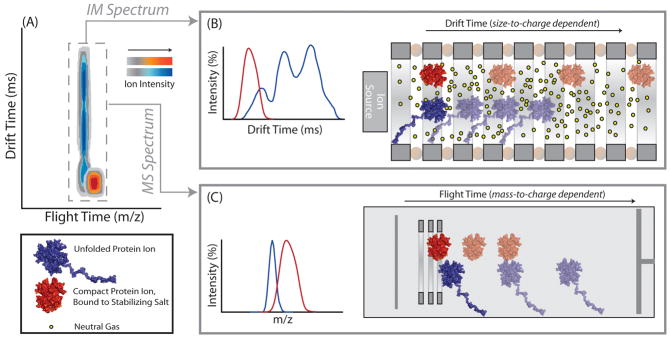
A schematic diagram of the IM-MS experiment, in which collisional heating of two protein complexes has generated different CIU responses. A) IM-MS data is multi- dimensional, consisting of IM drift time (in ms), m/z (measured in a time-of-flight mass analyzer), and ion intensity (shown on a color axis, different for the two ion populations). Signal corresponding to stabilizing salt-bound protein is illustrated on a red color axis, and a blue color axis corresponds to signal from protein ions in the absence of stabilizing salt adducts. B) Equally-charged protein ions are separated in a travelling-wave type IM separator based on their conformational differences following CIU. Protein ions bound to stabilizing salts travel more quickly in the IM separator when compared to non-stabilized protein ions due to their more-compact, folded geometry. IM data for unfolded proteins typically reveal a multimodal drift time profile, indicative of multiple unfolded intermediate states, stable on the time scale of the experiment, created during the CIU process C) Subsequent mass analysis of ions using a time-of-flight mass analyzer reveals that salt molecules remain bound to protein ions during the analysis, resulting in an increased molecular mass for stabilized complexes.
2.2 Preparing Multiprotein-salt Complexes for IM-MS Measurements
Our approach involves the protection of protein complex structure through the addition of salts in solution prior to nESI. We screened a series of Hofmeister-type cations and anions for their ability to stabilize multiprotein complexes in the absence of bulk solvent. The proteins studied herein include the protein tetramers avidin (egg white), transthyretin (TTR, human), concanavalin A (ConA, jack bean), and alcohol dehydrogenase (ADH, yeast) as well as the β-lactoglobulin A dimer (BLA, bovine) purchased from Sigma (St. Louis, MO, USA). All protein samples were buffer exchanged into 100 mM ammonium acetate at pH 7 using Micro Bio-Spin 6 columns (Bio-Rad, Hercules, CA) and prepared to a final concentration of 5 μM (avidin, TTR, ConA, ADH tetramers) or 10 μM (BLA dimer).
We began our experiments by assessing the stabilizing effect of isolated cations and anions on gas-phase protein structure by adding salts where cations (acetate anion with ammonium, tetramethylammonium (TMA), sodium, potassium, rubidium, lithium, Tris (2-Amino-2-hydroxymethyl-propane-1,3-diol), calcium, barium, and magnesium counterions) or anions (ammonium cation with acetate, fluoride, chloride, nitrate, citrate, thiocyanate, bicarbonate, tartrate, iodide, hydrogen phosphate, sulfate and perchlorate counter-ions) were altered specifically so that their stabilizing influence could be evaluated individually. In later experiments, salts of varying anion and cation content are added to solution to observe the combined effects of simultaneous anion and cation adduction on protein ion stability. All salts were purchased from Sigma (St. Louis, MO, USA) and prepared in aqueous solution prior to nESI. In order to study the influence of different salts on protein stability without significantly altering buffer capacity or solution pH, the salts were prepared as stock solutions in 100 mM ammonium acetate at a concentration of 20 mM, each of which was then added to protein solutions. Final solutions contained added salt concentrations of less than or equal to 5 mM (2 mM for avidin, TTR, ConA, ADH and 0.5 mM for BLA samples for cation-based studies, 5 mM for avidin, TTR, ConA, ADH and 0.2 mM for BLA samples for anion-based studies). While these concentrations are much lower than that used for deriving Hofmeister-ion effects on proteins in solution (> 300 mM), and selected for our experiments primarily to avoid ion suppression effects, the mechanics of nESI desolvation allows all the samples tested to achieve very high relative salt concentrations prior to ionization.
2.3 IM-MS Data Analysis Techniques for the Measurement of Desolvated Protein Stability
Our IM-MS measurements of protein complex stability are all performed in a tandem-MS (quadrupole selection) mode. Ions were selected for stability measurements in the quadrupole mass filter at an m/z corresponding to the 16+ charge state of Avidin, the ConA 19+ state, the TTR 14+ state, the ADH 24+ state for the tetramers and the 11+ state for BLA dimers. Charge states were chosen based on their intensity across each solution state studied, and control IM data were screened for evidence of overlapping non-tetrameric or non-dimeric ions at the same m/z values respectively. Each of these mass-selected ions was activated by increasing the trap collision voltage. For all the protein-salt systems investigated here, trap collision voltages were incremented in 5 V steps. Upper voltage limits were considered to be where no further CID was observed.
All mass spectra were calibrated externally using a solution of cesium iodide (100 mg/ml) and were processed with Masslynx 4.1 software (Waters, UK). Spectra are shown with minimal smoothing and without background subtraction. The relative abundances of mass-selected tetrameric ions (or dimeric ions for BLA) (Itet) were calculated as a percentage of the total intensity for all the signals observed in the MS data corresponding to either intact protein complexes or identified product ions (i.e., monomer or stripped protein complexes). The relative abundances of the compact form observed for tetrameric (or dimeric for BLA) ions separated in IM experiments (If) were calculated as a percentage of the total intensity of the peaks observed in the arrival time distribution at a selected m/z value corresponding to intact tetramer (or dimer for BLA):
| (1) |
| (2) |
The average relative standard deviation for the determination of either Itet (%) or If (%) is 2–4%. Some figures contain axes labeled in collision energy (units of eV*). This axis is a normalized version of ion kinetic energy, which takes into account both the ion charge and the reduced mass of the ion-neutral collision complex, for making comparisons across large mass ranges.34
Typical protein stability data is shown in Figure 2, using the 64 kDa avidin tetramer as an example. The IM-MS dataset at low activation energies shows well-resolved peaks primarily corresponding to intact and folded tetrameric protein complex ions having 14+ to 17+ charges (Figure 2A, red). When the most intense charge state, the 16+ tetramer, is selected in the quadruopole mass filter, the ions are resolved in the IM separator as a single narrow peak requiring ~20 ms of drift time to traverse the IM separator (Figure 2C, red). When the ions are activated by increasing the trap collision energy to 65 V, the IM-MS dataset exhibits substantially different features. For example, the IM data for the same tetramer ions are now resolved as multiple conformational species populating significantly longer drift times (Figure 2C, blue). The appearance of new features is also prominent at both lower and higher m/z, corresponding to the 7+ to 9+ charge state avidin monomer ions and 8+ to 10+ charge state avidin trimer ions respectively (Figure 2B, blue). Thus, CIU and CID information can be obtained in a single dataset, and carefully monitoring this information over a wide range of collision energies enables a quantitative measure of protein quaternary structure stability (through CID) and subunit tertiary structure stability (through CIU).
Figure 2.

Example CIU and CID data for the avidin tetramer. A) Mass spectra of avidin acquired using instrument conditions that either preserve (trap collision voltage: 5 V, red), or activate protein complex ions (65 V, blue). Peaks corresponding to the 14 – 17+ charge states of the avidin tetramer and the 7 – 10+ charge states of the monomer are shown. B) Contour plots of IM drift time versus m/z acquired at a trap collision voltage of 5 V (red) and 65 V (blue). Major peaks from the charge state series corresponding to monomeric and tetrameric avidin are labeled. C) IM drift time data for only the selected 16+ tetramer ions reveals compact ions at low levels of ion activation (red), and extended tetrameric ions generated under activating conditions (blue).
To ease the interpretation of desolvated protein stability, we typically reduce IM-MS measurements to histograms that plot the relative structural integrity of the complexes observed against additives used for protein stabilization. This procedure is illustrated with data acquired from measurements of the avidin tetramer incubated in three different buffers (control/100% 100 mM ammonium acetate, 100 mM ammonium acetate with 5 mM added ammonium fluoride, and 100 mM ammonium acetate with 5 mM added ammonium nitrate). MS for avidin bound to nitrate anions reveal a broadened mass profile consistent with a statistical distribution of bound adducts (Figure 3A). In contrast, the MS data for avidin incubated in non-stabilizing fluoride-based salts is indistinguishable from control data acquired from sample buffers comprised entirely from salts containing highly volatile acetate anions. This result is consistent with previous observations indicating that nitrate binds to protein in large numbers.34 The tandem MS data for 16+ avidin tetramer, acquired under elevated trap collision energy, shows that avidin incubated with stabilizing nitrate anions yields significantly less monomers upon activation, compared to both fluoride-added and control datasets. (Figure 3B) Likewise, the arrival time distribution of the same charge state reveals that protein incubated with nitrate consists primarily of a relatively narrow peak at 20ms. By contrast, avidin incubated with fluoride anions populates longer drift times (~ 25 ms), indicating that these ions adopt significantly extended conformations by comparison (Figure 3C). Thus, this data demonstrates how anion binding can dramatically influence the stability of protein complexes in gas phase.
Figure 3.

Addition of anions in solution alters the dissociation and unfolding profiles of protein assemblies. A) Example MS data for the avidin tetramer incubated with anions (nitrate, fluoride, acetate) acquired at instrument conditions that preserve the tetrameric assembly. The peak corresponding to 16+ avidin is marked with a dashed box, and corresponds to those ions selected from this primary MS dataset for CID (B) and CIU (C) stability analysis, which use energy resolved datasets from MS and IM respectively to deduce the influence of selected anions on protein complex stability in the gas-phase. D) Plots of the relative intensities for intact (Itet, solid lines) and compact (If, dashed lines) avidin tetramer 16+ ions are shown as a function of trap collision voltage. Avidin ions were generated using solutions containing either added nitrate (circle), fluoride (triangle), or acetate anions (control, square). The energy at which the relative intensity of Itet or If reduces to 50% of its original value is marked with a horizontal dashed line. E) A histogram showing the energy at which 50% dissociation (filled) and unfolding (open) occurs for avidin tetramers generated from solutions having the indicated additives.
For quantitative measurements of protein stability, the trap collision voltage at which ions undergo CIU and CID is monitored, and plots of collision voltage versus the intensity observed for compact (If) and intact (Itet) tetramer ions are recorded (Figure 3D) while keeping the other parameters in the experiment constant. Both If and Itet decrease as the collision voltage is increased, and If is observed to decrease at lower voltages than Itet for ions generated from all three buffer compositions. We also observe that the addition of ammonium nitrate to sample solutions (circles) increases the voltage values at which If and Itet decrease, and that this change is significantly greater than that observed for the addition of ammonium fluoride (triangles), relative to control samples (squares). This result is more-clearly illustrated through a comparison of the normalized collision energy at which If and Itet decrease to 50% of their original intensity (Figure 3E). This general workflow is used throughout our experiments in order to quantitatively compare the influence of Hofmeister anions and cations on the stability of desolvated protein ions in the gas phase.
3. Gas-phase proteins and their complexes are stabilized by cationic and anionic additives through different mechanisms and to different extents
Using the protocol illustrated in Figure 3, we have developed a classification system that allows us to generally rank cations and anions in terms of the stability they afford to gas-phase protein ions (Figure 4A). Our data suggests that cations (red) influence the unfolding and dissociation processes of protein complexes to similar degrees. Bound cations increase the threshold dissociation/unfolding energy in the order: NH4+ ≈ TMA+ < Rb+ < K+ < Na+ < TrisH+ < Ba2+ ≈ Li+ < Ca2+ < Mg2+, progressively stabilizing gas-phase proteins to greater degrees. In contrast to cations, bound anions (blue) tend to stabilize protein complex CID and CIU to different extents. Therefore rather than a clearly-defined rank order, our IM-MS data reveals that anions can be categorized into three distinct groups. Tartrate2−, Cl−, citrate2− and NO3− are among the most efficient stabilizers of protein unfolding and dissociation, populating a ‘highly-stabilizing’ cluster. In contrast, HCO3−, I− and ClO4− provide little or no additional stability to protein complex ions, and are thus regarded as ‘weakly-stabilizing” salts in the gas-phase. The remaining anions including SO42−, HPO42−, SCN−, and F− form a final ‘medium-stabilizing’ cluster.
Figure 4.

Comparison of anion and cation-mediated stabilization afforded to gas-phase protein ions. A) A plot of the average CID versus average CIU energies (eV*) for the 5 protein complexes studied herein for cation (red) and anion (blue) additives. The plot reveals that the anions can be categorized into three distinct groups (blue background color), according to their ability to stabilize protein complexes relative to the control data set (OAc−). Both B) and C) show residual plots for stability data from best-fit linear relationships between CIU and CID energies, for cation and anion datasets respectively. The correlation between unfolding and dissociation stabilization is much higher for our cation data (R2 = 0.97) relative to equivalent data recorded for anion additives (R2 = 0.55). D) A diagram depicting our current mechanistic understanding of gas-phase protein structure stabilization through bound cations and anions. Anions with mid-range acidities bind the protein in high affinity and are released upon dissociation leading to high protein structural stability in the gas phase through dissociative cooling (green track). Anion additives that do not bind, nor dissociate from gas-phase protein ions do not provide significant structural stabilization (blue and yellow track). In contrast, high charge-per-unit-area cations, having much greater surface charge densities than any of the anions tested to date, bind in large numbers to protein complexes and remain bound to become less-mobile as charge carriers, thus affording increased stability enhancement to gas-phase protein structure (red track). E) A plot of the charge-per-unit-area of cations against Lewis acid strength reveals a high level of correlation to the best fit line (shown). F) A plot of the gas-phase acidity of anions against pKa reveals a positive correlation between the two parameters. The data are color-coded to indicate the membership of each anion in the three relevant mechanistic tracks depicted in Figure 4D, and a trend line is added to guide the eye.
For a more quantitative comparison of the correlation between gas-phase unfolding and dissociation found in our IM-MS data, linear regression analysis was used to derive residual plots and correlation coefficients for linear fits to the anion and cation data shown in Figure 4A. Figures 4B and 4C show these data, and support our earlier assertion regarding the superior linearity of cation-based IM-MS stability data when compared to anion data, resulting in average residual sum of squares (RSS) values of 0.004 and 0.057 respectively. Furthermore, this analysis is also consistent with the correlation coefficients calculated for the same data (Figure 4B, caption), therefore suggesting a link between local and global protein stabilization in the case of cation additives that is absent for bound anions. In addition, cationic additives seem to stabilize protein complex ions against CID to a greater extent, on average, than equivalent anions while anionic adducts are, in general, better stabilizers of gas-phase protein unfolding.
The above differences between anionic and cationic stabilizers allow us to construct separate mechanistic descriptions of their action (Figure 4D). For anion-based stabilization, we can classify adducts into three categories based on both their protein binding affinity and ability to dissociate from proteins complexes following activation in the gas-phase. It is important to point out that these three anion classes, while related, are not the same as those presented in Figure 4A. The class that includes Cl−, tartrate2− and NO3− exhibits a strong stabilizing influence on protein structure (green track, Figure 4D). They canbind in large numbers and readily dissociate from the protein surface after relatively minimal activation. The observed dissociation of anion-based adducts corresponding to [H-“anion”] type neutrals acts to carry away excess rotational and vibrational energy from gas-phase protein ions, thus abating any dramatic internal energy increases for the protein and allowing it to retain a compact, native-like structure.34 In contrast, anions that do not bind (HCO3− and F−), nor dissociate from gas-phase protein ions (I− and ClO4−) do not provide significant structural stabilization (blue and yellow tracks respectively, Figure 4D). As shown in Figures 4D and 4F, the gas-phase acidity of the conjugate acid form of the anion (equivalent to anion proton affinity) correlates well with these three classes of stabilizing anions, and in turn with their relative binding affinities. This correlation is apparent when singly-charged anions are considered, where anions with low gas-phase acidity fall into the yellow track, those with intermediate affinity in the green track, and those with the highest values are in the blue track. Multiply-charged anions are more-difficult to place a priori due to likely multi-dentate protein interactions.
Whereas anions studied to date perform optimally as stabilizers when relying entirely on the dissociative-cooling process described above, optimal cationic stabilizers are those that remain bound to the protein assembly, even following extensive activation in the gas phase (red track, Figure 4D). These highly-stabilizing cations correlate with those that have larger charge-per-unit-area values, such as Mg2+ and Ca2+ shown in Figure 4E. The higher surface charge density of these cations, much in excess of any anions tested, gives these adducts access to modes of stabilization that rely either upon multidentate interactions within the protein, enabling them to tether regions of its structure, or by replacing highly-mobile proton charge carriers with less-mobile cationic charge carriers that restrict charge mobility and frustrate the Coulombic unfolding of subunits within the complex.36 Note that anions with low gas-phase acidity (e.g., I− and ClO4−), though having higher protein binding strengths when compared to the other singly-charged anions studied here, do not reach the charge-per-unit-area values of stabilizing cations (Mg2+ and Ca2+). Armed with this mechanistic knowledge, we have endeavored to use tailored anion/cation pairs for protein stabilization in order to make use of both stabilization mechanisms simultaneously.
4. Drastic differences of protein stabilization by cationic/anionic Hofmeister series in solution and in the gas phase
On first inspection, drastic differences are apparent between the rank order determined by our data in the gas-phase (Figure 4A) and the well-known Hofmeister series of cations and anions describing their influence upon protein stability in solution: TMA+ > NH4+ > Rb+ > K+ > Na+ > Li+ > Ca2+ > Mg2+ > Ba2+ and SO42− > HPO4− > F− > OAc− > Citrate3− > HCO3− > Cl− > NO3− > I− > ClO4− > SCN−. For example, NO3−, SCN− and Cl− are all protein stabilizers in the gas-phase, but act as structure destabilizers in bulk solvent. Similarly, cations stabilize gas-phase proteins following a nearly reversed order relative to most measurements of protein stability carried out in solution. Multiple reports have shown that Hofmeister-type protein stabilization in solution depends upon ion hydration and the ability of ions to alter water surface tension, along with direct ion-protein binding.3, 15, 78 In our stability measurements of desolvated proteins, the influence of both bulk solvent and local hydration layers are absent, and our data are instead dominated by overall protein-counterion binding affinity, adduct dissociation, and Coulombic effects incumbent upon protein unfolding in the gas-phase (Figure 4D). Thus, it is likely that the lack of protein solvation contributes substantially to the differences we observe between the stabilizing influence of Hofmeister salts in the gas-phase and in solution.
In spite of the clear differences between protein stabilization in the gas-phase and in solution noted above, it is potentially instructive to mine our current dataset for any information that may suggest critical links between our data and those collected in the condensed phase. To attempt this, we first note the strong correlation between gas-phase acidity and pKa measurements acquired in solution for the anions studied here (Figure 4F). As stated above, anion proton affinity seems likely to govern the relative amount of anions bound to gas-phase proteins in our data (Figure 4D). We also note that the surface charge density of the cations studied here have a high correlation to their Lewis acid strengths in solution. Mirroring the above anion-based correlation, we found cation charge-per-unit-area to be an able predictor of protein-cation complex stability (Figure 4E). Both correlations, while not representing demonstrable proof, provide some evidence that the relative binding affinities observed for protein-counterion complexes in our gas-phase measurements may mirror those in solution.
To further probe the potential correlations between our gas-phase data and protein-salt interactions in solution, we used MS to quantify the anions bound to proteins for a broad range of interacting pairs, as previous data had found a strong correlation between perchlorate binding observed by MS and the number of surface accessible basic sites on a given protein.21 It is important to note that all the data and analysis shown in Figure 4 is derived by normalizing the stabilization effects observed to the number of adducts bound to protein ions in the first instance. Figure 5A shows MS data for 7+ CYC monomers generated from solutions containing a range of anion additives. Unlike MS data for more-massive multiprotein complexes, where only average numbers of [H-“anion”] type adducts can be extracted from the average mass shifts observed relative to control (Figure 5B), the mass resolving power achieved in our CYC experiments is sufficient to resolve individual bound populations for the anion-protein complexes observed. The total number of anions bound to each protein is indicated in the blue histograms in Figures 5C–F, under which the black column represents the number of basic sites already occupied by charge carriers based on the ion charges state observed. Our data show that for anions known to have strong protein interactions (including nitrate, chloride, iodide and perchlorate anions), the quantity of binding observed by MS, when added to those sites occupied by unpaired charge carriers, correlates reasonably well the maximum expected number of binders based on the known surface assessable binding sites in solution. We attribute those cases where we observe excess binding (values exceeding the black dashed line) to anion condensation during the nESI process. Taken together with protein-ligand binding studies carried out where intensity values in MS measurements can be directly correlated to binding strengths between proteins and small molecules,79–82 it is highly likely that our data represent a direct measure of protein-anion binding affinities highly correlated to those in solution. As such, it is likely that our observations, and the rank orders of stabilizing/destabilizing salts extracted from our data, serve primarily to highlight the critical importance of protein solvation in the Hofmiester stabilization of proteins in solution.
Figure 5.

Correlations between our gas-phase data and protein-salt interactions in solution. A) nESI-MS data of the 7+ charge state of CYC (10 μM) generated from solutions containing 100 mM ammonium acetate (control) and a series of solutions containing both 100 mM ammonium acetate and 1 mM salts (ammonium-based salts with different anions) reveal a distribution of adducts (indicated as blue peaks) resolved by MS without collisional activation. Peaks corresponding to adducted ions arising from sodium, potassium, and sodium + potassium-binding are shown in black, and observed to dominate in our control (acetate), fluoride, and bicarbonate datasets. B) nESI-MS data for tetrameric ConA (10 μM) obtained from a solution containing 100 mM ammonium acetate (control) and a series of solutions containing both 100 mM ammonium acetate and 5 mM salts (ammonium-based salts with different anions). Each spectrum was obtained using identical instrumental conditions without collisional activation. The centroid m/z values corresponding to the 19+ charge state of protein-anion complexes are indicated by blue dashed lines. C)–F) Histogram plots showing the approximate number of residual anions bound to protein complex ions (shown as blue columns) stacked on the number of sites occupied by unpaired positive charges (shown as black columns), for CYC, TTR, avidin, and ConA, respectively. The black dashed line represents the number of surface solvent-accessible basic sites, as determined by the DEPTH program. 90
The rank order describing the stabilizing influence of protein salt interactions in the albescence of solvent also exhibits reasonable agreement with a number of studies that have observed so-called ‘reversed’ Hofmeister series, primarily for positively charged proteins at low salt concentrations.30–32 Spectroscopic data indicates that these inversed datasets result from the differing strengths of anion associations with positively charged protein surfaces, are well-correlated with the sizes of hydrated anions, and are related directly to the hydration free energy of stabilizing anions.83 While our gas-phase data correlate more-strongly to these ‘reversed’ Hofmeister series, a direct mechanistic correlation between the two rank orders is unlikely given what we currently understand of our gas-phase data. It is likely that continuing studies that highlight differences between gas-phase and ‘reversed’ Hofmeister series may further-pinpoint the role of anion hydration in solution-phase protein stabilization.
5. Developing the combined effects by adding the salt with the stabilizing cations and anions
In our previous experiments, we found that the stabilization mechanism accessed by cations and anions are not mutually exclusive, and can therefore be accessed simultaneously to enhance the gas-phase stability of protein structure through tailored salts. Data demonstrate that Ca2+, Mg2+, tartrate2−, Cl−, citrate2−, and NO3− are strongly stabilizing for protein structure in the gas-phase, and their combination leads to 8 potential salts that may be useful for IM-MS measurements of native-like protein structure. However, if we consider the binding of both free anions and cations in solution a prerequisite for the enhanced stabilization of proteins in the gas phase, then salts containing tartrate and citrate must be excluded from our list, as they can act as strong chelators for suppressing protein-metal interactions during nESI.84 Consequently, our preliminary list of highly-stabilizing salt additives is reduced to four potential choices: Ca(NO3)2, CaCl2, Mg(NO3)2 and MgCl2.
In this report, we use Ca(NO3)2 to demonstrate such combined stabilizing effects for gas-phase protein structure. To ensure that both Ca2+ and NO3− can simultaneously bind to protein complexes and are subsequently carried into the gas-phase, we performed preliminary experiments on CYC monomers, where we are able to use MS to resolve individual protein-bound populations. Data for 7+ CYC is shown in Figure 6A, where monomer ions are generated from four different buffer compositions (control/100% 100 mM NH4OAc, 100 mM NH4OAc with added NH4NO3, 100 mM NH4OAc with added Ca(OAc)2, 100 mM NH4OAc with added Ca(NO3)2). The resolved adduct populations corresponding to [H-’NO3−’] type adducts (blue) together with Ca2+ adducts (red) can be observed in our CYC monomer dataset following incubation with Ca(NO3)2. Following these proof-of-principle experiments, we extended our data to include a number of tetrameric protein complexes prepared under the same conditions. In these data, we observe large shifts in ion mass, indicative of simultaneous Ca2+ and NO3− binding when compared with negative (samples containing pure ammonium acetate buffer) and positive (samples containing either NH4NO3 or Ca(OAc)2 additives) control data(Figure 6B).
Figure 6.

The combined influence of tailored Hofmeister anions and cations on gas-phase protein stability. A) nESI-MS data of the 7+ charge state for CYC obtained from ammonium acetate-based solutions containing 10 μM CYC and no added salt (control), 400 μM ammonium nitrate, 200 μM calcium acetate, or 200 μM calcium nitrate. The resolved populations of cation and anion additives are denoted by red and blue colors, respectively. The other peaks (black) except for the apo-CYC correspond to adducts arising from sodium or potassium-binding. B) nESI-MS data for TTR (5 μM) generated from ammonium acetate-based solutions with no added salt (control, black), 4 mM ammonium nitrate (blue), 2 mM calcium acetate (red), or 2 mM calcium nitrate (purple). C) & D) Histogram plots of collision energy (eV*) required to dissociate or unfold 50% of the population of monomeric CYC, tetrameric avidin, ConA, and TTR are shown for the four buffer conditions mentioned above. E) A CIU fingerprint contour plots are shown for 14+ TTR generated from ammonium acetate-based solutions with no added salt (control), 4 mM ammonium nitrate, 2 mM calcium acetate, or 2 mM calcium nitrate, where ion trap collision voltage is charted against IM drift time, and the ion intensities are denoted by a color-coded axis (blues correlated to low ion intensity, whereas reds indicate high ion intensity). The conformational forms for the tetramer (I, II, II′ or III) are labeled as described in the text, and the two most compact forms of the protein observed are highlighted (white box). The inset figures show the IM arrival time distributions of 14+ TTR ions acquired at the indicated trap collision voltages, with the peak centroids corresponding to the intermediate structural families labeled.
In order to investigate the stabilizing effects of the complexes created above, we performed CIU and CID stability measurements on monomer (CYC) and tetramer ions (TTR, avidin and ConA) created from solutions containing added Ca(NO3)2. Figures 6C and 6D show histogram plots of the normalized collision energy (eV*) at which Itet and If for these ions decrease to 50% of their original values as a function of buffer composition. Generally, we observe that cations are stronger stabilizers of gas-phase protein structure than anions, as they act by remaining bound to the assembly at relatively high internal temperatures. Most importantly, it is clear from our data that the simultaneous presence of both stabilizing cations and anions causes a significant increase in protein complex stability relative our control datasets, resulting in 8% and 10% average increases in gas-phase protein quaternary and tertiary structure stabilities respectively.
To further validate the joint stabilization provided by combined cations and anions, and provide insight into the stabilization mechanism at work, we constructed CIU unfolding ‘fingerprints’ for protein complex ions derived from solutions containing added Ca(NO3)2 as well as control samples containing its constituent anion and cation components. Changes in the protein ion tertiary/secondary structures are induced during the CIU process, leading to several structural ensembles that are stable on the millisecond timescale and can be resolved in both IM drift time and collision energy. For clarity, CIU fingerprint data is projected as a contour plot (Figure 6E) where intensities for the features observed are denoted by a color-based axis. Careful analysis of CIU fingerprint data allows the nature of protein stabilization to be identified by noting the conformational features that are stabilized (elongated on the collision energy axis) relative to control data.34, 56, 60, 85
Control fingerprints for the TTR tetramer reveal three major conformational families (I, II, III) observed under the conditions used for our experiments here. These features are easily resolved in drift time and have distinct patterns as a function of collision voltage (Figure 6E). For example, fingerprint data acquired for TTR with added NO3− shows that the most compact conformer (I) is observed at substantially higher collision voltages when compared to those ions generated from pure ammonium acetate solutions, indicating that the stabilization observed in our experiments is due primarily to the enhanced stability of this compact conformer. In contrast, TTR incubated with added Ca2+ displays a substantially different CIU fingerprint, with both the most compact and partially unfolded forms of the complex being stabilized. As discussed above, the differences in the fingerprints recorded for Ca2+-bound and NO3−-bound TTR are indicative of the separate stabilization mechanisms operative for these two adduct populations. Specifically, Ca2+ stabilizes the complex through tight binding, such that intermediately unfolded conformers of the protein are partially stabilized. In contrast, NO3− stabilizes the complex through dissociative cooling, and therefore cannot, by definition, stabilize partially unfolded protein conformers. Additionally, a new intermediate unfolded species (II′, inset Figure 6E) emerges in the Ca2+ fingerprint. This observation is also consistent with our mechanism, in which strongly-bound cations modulate the collisional unfolding process through tethering flexible regions within proteins or by limiting charge migration by replacing highly mobile proton charge carriers. Critically, fingerprint data collected from ions incubated with Ca(NO3)2 displays elements from both the fingerprints of its constituent components, exhibiting a highly stabilized compact (I) state, a stabilized II state, and the appearance of a II′ state. Thus, our CIU fingerprint data supports the joint stabilization of protein complexes through simultaneous binding of both Ca2+ and NO3− adducts, resulting in enhanced protein stability when compared to their individual effects.
6. The Future of Gas-phase Protein Structure Measurements and Their Potential Impact on Our Understanding of Hofmeister Stabilization
Measurements of desolvated protein stability have a clear place in supporting the emergent field of gas-phase structural biology, which seeks to use solvent-free structural information to develop native-state protein models.86 This challenging endeavor hinges on a strong correlation between solution and gas-phase protein structures, and such a correlation has been measured for a broad array of proteins and their complexes.62 At the same time, a number of cases and specific structural elements within proteins have been observed to substantially rearrange in the absence of solvent.65, 87, 88 Stabilized proteins and multiprotein complexes could potentially avoid many of these effects, facilitating the use of gas-phase measurements in the construction of native-state protein models. It is also likely that the mechanistic aspects of anion and cation stabilization discussed here have significant implications for the use of MS in the analysis of membrane-bound protein complexes. In these experiments, detergent micelles are used as stabilizing agents to transport hydrophobic proteins and protein complexes into the gas-phase from aqueous solutions by nESI, and collisional activation is used to remove bound detergent from the protein ions following desolvation.58, 89 Understanding the mechanistic principles that govern the action of the relatively simplified stabilizing additives studied here will likely provide an enhanced ability to utilize IM and MS data for membrane protein complexes by avoiding unwanted unfolded protein conformations, thus enabling the rapid determination of protein topologies critical in our understanding of multiple cellular processes and disease states.59
The direct implications of the gas-phase data shown in this report for Hofmeister stabilization in solution hinge on both the drastically different stabilization mechanisms observed, and the similarities detected in the numbers of bound anions in our experiments to those expected to associate with the protein in solution. IM-MS data clearly show that anions and cations can stabilize protein complexes through separate mechanisms, and that these modes of action are unique to the gas-phase. Furthermore, it is likely that the number of bound anions and cations observed by MS in our data bear a strong correlation to the bound populations present in solution. As such, the dramatically different rank orders observed in gas-phase experiments when compared with solution can be taken as evidence for the critical importance of solvation effects in the mechanism of Hofmeister stabilization in solution. Finally, in addition to providing enhanced stabilizing additives for the gas-phase measurement of native-like protein structures, it is clear that continuing measurements of protein-counterion complexes will provide a useful tool for quantifying bound cation and anion populations in support of solution-phase measurements of Hofmeister protein stabilization.
Acknowledgments
This work is supported by the National Institutes of Health (1-R01-GM-095832-01) and by University of Michigan startup funds.
References
- 1.Robinson CV, Sali A, Baumeister W. Nature. 2007;450:973–982. doi: 10.1038/nature06523. [DOI] [PubMed] [Google Scholar]
- 2.Vrbka L, Jungwirth P, Bauduin P, Touraud D, Kunz W. Journal of Physical Chemistry B. 2006;110:7036–7043. doi: 10.1021/jp0567624. [DOI] [PubMed] [Google Scholar]
- 3.Zhang YJ, Furyk S, Bergbreiter DE, Cremer PS. Journal of the American Chemical Society. 2005;127:14505–14510. doi: 10.1021/ja0546424. [DOI] [PubMed] [Google Scholar]
- 4.Baldwin RL. Biophysical Journal. 1996;71:2056–2063. doi: 10.1016/S0006-3495(96)79404-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kunz W. Current Opinion in Colloid & Interface Science. 2010;15:34–39. doi: 10.1016/j.cocis.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collins KD. Methods. 2004;34:300–311. doi: 10.1016/j.ymeth.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 7.Kunz W, Henle J, Ninham BW. Current Opinion in Colloid & Interface Science. 2004;9:19–37. [Google Scholar]
- 8.Collins KD, Washabaugh MW. Quarterly Reviews of Biophysics. 1985;18:323–422. doi: 10.1017/s0033583500005369. [DOI] [PubMed] [Google Scholar]
- 9.Vanzi F, Madan B, Sharp K. Journal of the American Chemical Society. 1998;120:10748–10753. [Google Scholar]
- 10.Omta AW, Kropman MF, Woutersen S, Bakker HJ. Science. 2003;301:347–349. doi: 10.1126/science.1084801. [DOI] [PubMed] [Google Scholar]
- 11.Kropman MF, Bakker HJ. Journal of the American Chemical Society. 2004;126:9135–9141. doi: 10.1021/ja039147r. [DOI] [PubMed] [Google Scholar]
- 12.Batchelor JD, Olteanu A, Tripathy A, Pielak GJ. Journal of the American Chemical Society. 2004;126:1958–1961. doi: 10.1021/ja039335h. [DOI] [PubMed] [Google Scholar]
- 13.Bostrom M, Williams DRM, Ninham BW. Physical Review Letters. 2001:87. doi: 10.1103/PhysRevLett.87.168103. [DOI] [PubMed] [Google Scholar]
- 14.Smith JD, Saykally RJ, Geissler PL. Journal of the American Chemical Society. 2007;129:13847–13856. doi: 10.1021/ja071933z. [DOI] [PubMed] [Google Scholar]
- 15.Zhang YJ, Cremer PS. In: Annual Review of Physical Chemistry. Leone SR, Cremer PS, Groves JT, Johnson MA, Richmond G, editors. Vol. 61. 2010. pp. 63–83. [DOI] [PubMed] [Google Scholar]
- 16.Lund M, Vacha R, Jungwirth P. Langmuir. 2008;24:3387–3391. doi: 10.1021/la7034104. [DOI] [PubMed] [Google Scholar]
- 17.Lund M, Vrbka L, Jungwirth P. Journal of the American Chemical Society. 2008;130:11582. doi: 10.1021/ja803274p. [DOI] [PubMed] [Google Scholar]
- 18.Prakash H, Kansara BT, Mazumdar S. International Journal of Mass Spectrometry. 2010;289:84–91. [Google Scholar]
- 19.Stephenson JL, McLuckey SA. Analytical Chemistry. 1997;69:281–285. doi: 10.1021/ac961119m. [DOI] [PubMed] [Google Scholar]
- 20.Verkerk UH, Kebarle P. Journal of the American Society for Mass Spectrometry. 2005;16:1325–1341. doi: 10.1016/j.jasms.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 21.Flick TG, Merenbloom SI, Williams ER. Analytical Chemistry. 2011;83:2210–2214. doi: 10.1021/ac1031012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verkerk UH, Peschke M, Kebarle P. Journal of Mass Spectrometry. 2003;38:618–631. doi: 10.1002/jms.475. [DOI] [PubMed] [Google Scholar]
- 23.Felitsyn N, Peschke M, Kebarle P. International Journal of Mass Spectrometry. 2002;219:39–62. [Google Scholar]
- 24.Timofeev O, Zhu MM, Gross ML. International Journal of Mass Spectrometry. 2004;231:113–117. [Google Scholar]
- 25.Kebarle P, Verkerk UH. Mass Spectrometry Reviews. 2009;28:898–917. doi: 10.1002/mas.20247. [DOI] [PubMed] [Google Scholar]
- 26.Pegram LM, Record MT. Journal of Physical Chemistry B. 2007;111:5411–5417. doi: 10.1021/jp070245z. [DOI] [PubMed] [Google Scholar]
- 27.Pegram LM, Record MT. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:14278–14281. doi: 10.1073/pnas.0606256103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pegram LM, Record MT. Journal of Physical Chemistry B. 2008;112:9428–9436. doi: 10.1021/jp800816a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pegram LM, Record MT., Jr Chemical Physics Letters. 2008;467:1–8. doi: 10.1016/j.cplett.2008.10.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang YJ, Cremer PS. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:15249–15253. doi: 10.1073/pnas.0907616106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bostroem M, Parsons DF, Salis A, Ninham BW, Monduzzi M. Langmuir. 2011;27:9504–9511. doi: 10.1021/la202023r. [DOI] [PubMed] [Google Scholar]
- 32.Bostrom M, Tavares FW, Finet S, Skouri-Panet F, Tardieu A, Ninham BW. Biophysical Chemistry. 2005;117:217–224. doi: 10.1016/j.bpc.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 33.Flick TG, Merenbloom SI, Williams ER. Journal of the American Society for Mass Spectrometry. 2011;22:1968–1977. doi: 10.1007/s13361-011-0218-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han LJ, Hyung SJ, Mayers JJS, Ruotolo BT. Journal of the American Chemical Society. 2011;133:11358–11367. doi: 10.1021/ja203527a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Merenbloom SI, Flick TG, Daly MP, Williams ER. Journal of the American Society for Mass Spectrometry. 2011;22:1978–1990. doi: 10.1007/s13361-011-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han L, Hyung SJ, Ruotolo BT. Angew Chem Int Ed Engl. 2012 doi: 10.1002/anie.201109127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prell JS, Flick TG, Oomens J, Berden G, Williams ER. Journal of Physical Chemistry A. 2010;114:854–860. doi: 10.1021/jp909366a. [DOI] [PubMed] [Google Scholar]
- 38.Bush MF, Oomens J, Saykally RJ, Williams ER. Journal of the American Chemical Society. 2008;130:6463–6471. doi: 10.1021/ja711343q. [DOI] [PubMed] [Google Scholar]
- 39.Dunbar RC, Hopkinson AC, Oomens J, Siu CK, Siu KWM, Steill JD, Verkerk UH, Zhao JF. Journal of Physical Chemistry B. 2009;113:10403–10408. doi: 10.1021/jp903064w. [DOI] [PubMed] [Google Scholar]
- 40.Dunbar RC, Polfer NC, Oomens J. Journal of the American Chemical Society. 2007;129:14562. doi: 10.1021/ja076131i. [DOI] [PubMed] [Google Scholar]
- 41.O’Brien JT, Prell JS, Steill JD, Oomens J, Williams ER. Journal of Physical Chemistry A. 2008;112:10823–10830. doi: 10.1021/jp805787e. [DOI] [PubMed] [Google Scholar]
- 42.Bush MF, O’Brien JT, Prell JS, Saykally RJ, Williams ER. Journal of the American Chemical Society. 2007;129:1612–1622. doi: 10.1021/ja066335j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Armentrout PB, Rodgers MT, Oomens J, Steill JD. Journal of Physical Chemistry A. 2008;112:2248–2257. doi: 10.1021/jp710885a. [DOI] [PubMed] [Google Scholar]
- 44.Walters RS, Pillai ED, Duncan MA. Journal of the American Chemical Society. 2005;127:16599–16610. doi: 10.1021/ja0542587. [DOI] [PubMed] [Google Scholar]
- 45.Shin JW, Hammer NI, Diken EG, Johnson MA, Walters RS, Jaeger TD, Duncan MA, Christie RA, Jordan KD. Science. 2004;304:1137–1140. doi: 10.1126/science.1096466. [DOI] [PubMed] [Google Scholar]
- 46.Miller DJ, Lisy JM. Journal of Chemical Physics. 2006:124. [Google Scholar]
- 47.Bush MF, O’Brien JT, Prell JS, Wu CC, Saylkally RJ, Williams ER. Journal of the American Chemical Society. 2009;131:13270–13277. doi: 10.1021/ja901011x. [DOI] [PubMed] [Google Scholar]
- 48.Iino T, Ohashi K, Inoue K, Judai K, Nishi N, Sekiya H. Journal of Chemical Physics. 2007:126. doi: 10.1063/1.2730830. [DOI] [PubMed] [Google Scholar]
- 49.Choi JH, Kuwata KT, Cao YB, Okumura M. The Journal of Physical Chemistry A. 1998;102:503–507. [Google Scholar]
- 50.Robertson WH, Johnson MA. Annual Review of Physical Chemistry. 2003;54:173–213. doi: 10.1146/annurev.physchem.54.011002.103801. [DOI] [PubMed] [Google Scholar]
- 51.Bush MF, Saykally RJ, Williams ER. Journal of the American Chemical Society. 2007;129:2220. doi: 10.1021/ja068357b. [DOI] [PubMed] [Google Scholar]
- 52.Wyttenbach T, Bowers MT. Modern Mass Spectrometry. 2003;225:207–232. [Google Scholar]
- 53.Loo JA, Berhane B, Kaddis CS, Wooding KM, Xie YM, Kaufman SL, Chernushevich IV. Journal of the American Society for Mass Spectrometry. 2005;16:998–1008. doi: 10.1016/j.jasms.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 54.Freeke J, Robinson CV, Ruotolo BT. International Journal of Mass Spectrometry. 2010;298:91–98. [Google Scholar]
- 55.Merenbloom SI, Flick TG, Daly MP, Williams ER. Journal of the American Society for Mass Spectrometry. 2011:1–13. doi: 10.1007/s13361-011-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Freeke J, Bush MF, Robinson CV, Ruotolo BT. Chemical Physics Letters. 2012;524:1–9. [Google Scholar]
- 57.Carulla N, Zhou M, Giralt E, Robinson CV, Dobson CM. Acc Chem Res. 2010;43:1072–1079. doi: 10.1021/ar9002784. [DOI] [PubMed] [Google Scholar]
- 58.Barrera NP, Isaacson SC, Zhou M, Bavro VN, Welch A, Schaedler TA, Seeger MA, Miguel RN, Korkhov VM, van Veen HW, Venter H, Walmsley AR, Tate CG, Robinson CV. Nature Methods. 2009;6:585–U549. doi: 10.1038/nmeth.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou M, Morgner N, Barrera NP, Politis A, Isaacson SC, Matak-Vinković D, Murata T, Bernal RA, Stock D, Robinson CV. Science. 2011;334:380–385. doi: 10.1126/science.1210148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hyung SJ, Robinson CV, Ruotolo BT. Chemistry & Biology. 2009;16:382–390. doi: 10.1016/j.chembiol.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 61.Hopper JTS, Oldham NJ. Journal of the American Society for Mass Spectrometry. 2009;20:1851–1858. doi: 10.1016/j.jasms.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 62.Benesch JLP, Ruotolo BT. Current Opinion in Structural Biology. 2011;21:641–649. doi: 10.1016/j.sbi.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bush MF, Hall Z, Giles K, Hoyes J, Robinson CV, Ruotolo BT. Analytical Chemistry. 2010;82:9557–9565. doi: 10.1021/ac1022953. [DOI] [PubMed] [Google Scholar]
- 64.Leary JA, Schenauer MR, Stefanescu R, Andaya A, Ruotolo BT, Robinson CV, Thalassinos K, Scrivens JH, Sokabe M, Hershey JWB. Journal of the American Society for Mass Spectrometry. 2009;20:1699–1706. doi: 10.1016/j.jasms.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 65.Politis A, Park AY, Hyung SJ, Barsky D, Ruotolo BT, Robinson CV. Plos One. 2010:5. doi: 10.1371/journal.pone.0012080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ruotolo BT, Benesch JLP, Sandercock AM, Hyung SJ, Robinson CV. Nature Protocols. 2008;3:1139–1152. doi: 10.1038/nprot.2008.78. [DOI] [PubMed] [Google Scholar]
- 67.Zhong Y, Hyung S-J, Ruotolo BT. Expert Review of Proteomics. 2012;9:47–58. doi: 10.1586/epr.11.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Giles K, Pringle SD, Worthington KR, Little D, Wildgoose JL, Bateman RH. Rapid Communications in Mass Spectrometry. 2004;18:2401–2414. doi: 10.1002/rcm.1641. [DOI] [PubMed] [Google Scholar]
- 69.Giles K, Williams JP, Campuzano I. Rapid Communications in Mass Spectrometry. 2011;25:1559–1566. doi: 10.1002/rcm.5013. [DOI] [PubMed] [Google Scholar]
- 70.Zhong YY, Hyung SJ, Ruotolo BT. Analyst. 2011;136:3534–3541. doi: 10.1039/c0an00987c. [DOI] [PubMed] [Google Scholar]
- 71.Hernandez H, Robinson CV. Nat Protoc. 2007;2:715–726. doi: 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]
- 72.Felitsyn N, Kitova EN, Klassen JS. Analytical Chemistry. 2001;73:4647–4661. doi: 10.1021/ac0103975. [DOI] [PubMed] [Google Scholar]
- 73.Jurchen JC, Williams ER. Journal of the American Chemical Society. 2003;125:2817–2826. doi: 10.1021/ja0211508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li JW, Taraszka JA, Counterman AE, Clemmer DE. International Journal of Mass Spectrometry. 1999;185:37–47. [Google Scholar]
- 75.Shelimov KB, Clemmer DE, Hudgins RR, Jarrold MF. Journal of the American Chemical Society. 1997;119:2240–2248. [Google Scholar]
- 76.Valentine SJ, Anderson JG, Ellington AD, Clemmer DE. Journal of Physical Chemistry B. 1997;101:3891–3900. [Google Scholar]
- 77.Shvartsburg AA, Li FM, Tang KQ, Smith RD. Analytical Chemistry. 2007;79:1523–1528. doi: 10.1021/ac061306c. [DOI] [PubMed] [Google Scholar]
- 78.Zhang Y, Furyk S, Sagle LB, Cho Y, Bergbreiter DE, Cremer PS. Journal of Physical Chemistry C. 2007;111:8916–8924. doi: 10.1021/jp0690603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Loo JA. International Journal of Mass Spectrometry. 2000;200:175–186. [Google Scholar]
- 80.Liu L, Bagal D, Kitova EN, Schnier PD, Klassen JS. Journal of the American Chemical Society. 2009;131:15980. doi: 10.1021/ja9060454. [DOI] [PubMed] [Google Scholar]
- 81.Deng L, Sun N, Kitova EN, Klassen JS. Analytical Chemistry. 2010;82:2170–2174. doi: 10.1021/ac902633d. [DOI] [PubMed] [Google Scholar]
- 82.Kitova EN, Soya N, Klassen JS. Analytical Chemistry. 2011;83:5160–5167. doi: 10.1021/ac200244u. [DOI] [PubMed] [Google Scholar]
- 83.Marcus Y. Ion properties. Wiley Online Library; 1997. [Google Scholar]
- 84.Pan JX, Xu K, Yang XD, Choy WY, Konermann L. Analytical Chemistry. 2009;81:5008–5015. doi: 10.1021/ac900423x. [DOI] [PubMed] [Google Scholar]
- 85.Pagel K, Hyung SJ, Ruotolo BT, Robinson CV. Analytical Chemistry. 2010;82:5363–5372. doi: 10.1021/ac101121r. [DOI] [PubMed] [Google Scholar]
- 86.Hyung SJ, Ruotolo BT. Proteomics. 2012 In press. [Google Scholar]
- 87.Hogan CJ, Ruotolo BT, Robinson CV, de la Mora JF. Journal of Physical Chemistry B. 2011;115:3614–3621. doi: 10.1021/jp109172k. [DOI] [PubMed] [Google Scholar]
- 88.Jurneczko E, Barran PE. Analyst. 2011;136:20–28. doi: 10.1039/c0an00373e. [DOI] [PubMed] [Google Scholar]
- 89.Barrera NP, Di Bartolo N, Booth PJ, Robinson CV. Science. 2008;321:243–246. doi: 10.1126/science.1159292. [DOI] [PubMed] [Google Scholar]
- 90.Tan KP, Varadarajan R, Madhusudhan MS. Nucleic Acids Research. 2011;39:W242–W248. doi: 10.1093/nar/gkr356. [DOI] [PMC free article] [PubMed] [Google Scholar]