

Abstract
Duchenne muscular dystrophy is mainly caused by mutations that disrupt the generation of a translatable mRNA transcript. Most such mutations occur in parts of the gene that are not essential for its function and thus might be eliminated from the transcript to permit translation of a partially functional protein that would convert the disease to a milder clinical form. Two such antisense oligonucleotides of different backbone chemistries have been successful when tested on the mdx mouse, targeting exon 23, containing the nonsense mutation. Subsequently we have tested the more effective of these, the morpholino, on the dystrophic dog, where it is necessary to skip 2 exons, again with beneficial results. Currently, results of 2 human trials targeting exon 51 have also yielded promising preliminary results.
Keywords: Duchenne muscular dystrophy, Becker muscular dystrophy, exon skipping, antisense oligonucleotides
Introduction
In many respects, Duchenne muscular dystrophy has been a landmark disease. It was the first to be discovered by the technique of positional cloning, and the culprit gene was identified as very large and complex,1,2 likely to pose a considerable challenge to attempts at replacement gene therapy, particularly in so massive and diffusely distributed a tissue as skeletal muscle. But its large size and modular structure also qualify this gene as a good target for restitution by exon skipping, because most mutations occur in parts of the gene that have proved inessential for retention of some function.3–6 This was substantiated by the detection of deletion mutations that do not disrupt the open reading frame and are associated with milder clinical phenotypes, ranging from the mild Becker muscular dystrophy to near-normal muscular function (Figure 1).7 Such observations provide the best evidence as to which Duchenne muscular dystrophy mutations might benefit from exclusion of exons from the mRNA transcript so as to restore its translatability into a partially functional protein. Among the more urgent tasks for the advancement of the exon-skipping strategy is the assembly of an accurate and comprehensive registry of mutations and the severity of disease with which they are associated.
Figure 1.

A. Diagram of the configuration of exon boundaries in the region of exon 45 of the normal dystrophin gene as represented in the pre-spliced RNA transcript. Flat ends represent coincidence of exon and codon boundaries. Small or large blocks at the boundary denote respectively one or 2 nucleotides beyond the last complete codon. In the normal gene, the bondaries of adjacent exons complement one another to maintain the open reading frame
B. The most common Duchenne mutation, deletion of exon 45, puts 2 incompatible boundaries together in the spliced mRNA, resulting in a shift of open reading frame that generates a downstream nonsense sequence, resulting in premature termination of translation. The consequent lack of dystrophin produces a severe Duchenne pathology.
C. When the deletion includes exon 46, the splicing of exon 44 to 47 leaves the open reading frame undisturbed and so that the mRNA can be translated into a slightly shortened but partly functional protein, resulting in the milder Becker muscular dystrophy phenotype. In some instances, boys with exon 45 deletions spontaneously skip exon 46 from a proportion of the transcripts, resulting in an unexpectedly mild clinical phenotype
Experimental Progress
Exon-skipping Reagents and Experimental Models
To achieve targeted exclusion of specific exons, it is necessary to use oligonucleotide analogues whose sequence is designed to be antisense to sites on the mRNA primary transcript that are involved in the mechanism of splicing of the target exon in question. Because the main aim is to preserve the modified transcript, it is not possible to use normal oligonucleotides, which would trigger destruction of any duplex formed, but modified forms or chemical analogues that are resistant to degradation by endonucleases.
The first available modification was the 2′O-methyl-phosphorothioate backbone, which was tested both on tissue cultures of human Duchenne muscular dystrophy muscle8,9 and on the mdx mouse model of Duchenne muscular dystrophy. This animal carries a nonsense mutation in exon 2310 which, because it is comprised of intact codons, can be excluded from the transcript without altering the open reading frame (Figure 2). Early evidence that this exon could be excluded from a proportion of dystrophin transcripts by treatment with an appropriately designed antisense oligonucleotide was obtained by polymerase chain reaction detection of several transcripts lacking this exon in tissue cultures of mdx mouse muscle cells.3 Subsequently, a sequence for efficiently excluding exon 23 from the transcript was optimized on cultures of immortomouse-mdx (H-2 Kb-tsA58 X mdx) myogenic cells,11 both by polymerase chain reaction identification of the shortened transcript and by use of Western blot to detect restoration of synthesis of the protein.12 To convert this process for practical use requires that the reagent be able to penetrate into the nuclei of muscle muscle fibers and be nontoxic at doses and with administration protocols that generate effective exon skipping in muscles body-wide.
Figure 2.

A. The mdx mouse model of Duchenne muscular dystrophy is attributable to a nonsense mutation in exon 23 that blocks translation beyond this point
B. Because exon 23 is not a frame-shifting exon, it can be skipped to generate a translatable transcript that produces dystrophin.
After the initial demonstration of the principle of antisense oligonucleotide-induced exon skipping in tissue culture, there was a considerable hiatus before any real indication that significant amounts of dystrophin could be generated by skipping a disruptive exon in vivo. This first came from experiments where the 2′O-methyl-phosphorothioate antisense sequences that had been shown to be effective in the tissue cultures of Immortomouse (H-2Kb–tsA58 transgenic mouse) mdx muscle were injected into muscles of the mdx mouse. The efficiency of this procedure was greatly enhanced when the oligonucleotide was mixed with the block co-polymer F127.13 This same mixture was also found to effect some exon skipping in widespread muscles of the body when injected intravenously, but to generate only very insubstantial amounts of dystrophin, and not in all muscles.14 Most worryingly from a therapeutic point of view, there was no significant production of dystrophin in the heart.
A more promising turn of events was the subsequent demonstration that equivalent molarities of the same antisense oligonucleotide sequence on the morpholino backbone induced more robust skipping of the mutant exon 23 on intramuscularly injection and, more importantly when injected intravenously, when it elicited production of potentially therapeutic amounts of dystrophin in widespread muscles of the mdx mouse.15 This finding came as a surprise because morpholino antisense oligonucleotides had previously been found to penetrate tissue cultured muscle cells very poorly. The mechanism whereby, in vivo, they negotiate the vascular barriers and enter a good proportion of muscle fibers remains a mystery, one that is in urgent need of elucidation, for the principle barrier to practical application of antisense oligonucleotides for treatment of Duchenne muscular dystrophy is their limited overall efficiency of entry into muscle. This largely reflects considerable inter-fiber variability: some fibers display close to wild-type levels of dystrophin immunostaining, while close neighbors show no detectable signal above background.
There is also considerable variation between anatomical muscles as to the proportion of fibers that become dystrophin-positive. In general, this variation appears to reflect some intrinsic property of the anatomically distinct muscles, for there is some overall consistency between animals as to which muscles express the largest amounts of dystrophin in response to a given protocol of antisense oligonucleotide administration, although some individual instances of left/right diversity are seen. Our best conjecture at present is that the pathological status of individual muscle fibers or of the local microvasculature are major factors in facilitating antisense oligonucleotide entry. A further disappointment was that, as in the case of the 2′O-methyl-phosphorothioate antisense oligonucleotide, there was no significant evocation of dystrophin production in cardiac muscle, although some hope is given by the demonstration that use of ultrasound and microbubbles enhances exon skipping in skeletal muscle and, to some extent, in the heart.16
Functional Studies in the Dystrophic Dog
To further move the exon skipping strategy toward human trials, we undertook a preclinical study in the canine model of Duchenne muscular dystrophy. The most available mutation in this animal arose in the golden retriever and is designated golden retriever muscular dystrophy, or GRMD.17–19 To facilitate laboratory work, this mutation has been bred onto the beagle background to produce a smaller and more readily handled animal. Apart from providing a major change of scale (>1000-fold) the point mutation in this animal also extends our experience in that restoration of open reading frame requires skipping at least 2 target exons from the transcript (Figure 3). The mutation itself is a single base change in the acceptor splice site of exon 7 that results in exclusion of this exon from the mRNA.19,20 To restore a translatable transcript requires skipping exons 6 and 8.
Figure 3.
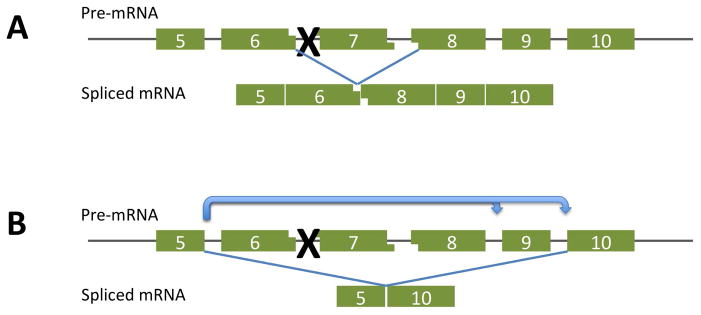
A. In the golden retriever muscular dystrophy dog, a single base mutation in the acceptor splice site of exon 7 prevents this exon from being spliced into the mRNA transcript. The resulting apposition of exons 6 and 8 produces a frame shift that prevents its productive translation.
B. To restore open reading frame to this mutation, a skip is required of at least 2 exons, 6 and 8. When this is provoked by use of oligonucleotides directed against sequences involved in the splicing of these 2 exons, exon 9 is also lost from the transcript. Since this is not a frame-shifting exon, the resulting mRNA, lacking exons 5–10, is translatable into a partially functional dystrophin protein.
Accordingly, antisense oligonucleotide sequences were designed to bind to the internal sequences that enhance splicing of both exons, to the donor splice site of exon 6, and to the acceptor site of exon 8, and were then tested for efficacy in tissue cultures of dog muscle cells.20 To our surprise and transient delight, it turned out that both target exons, plus the non frame-shifting exon 9, were excluded from the mRNA by treatment with either of the anti-exon 6 sequences with the same efficiency as that elicited by the cocktail of all 4 sequences, raising the possibility of skipping several exons with a single antisense oligonucleotide sequence. The sequence targeting the exon 8 splice site removed only exon 8, again together with exon 9, while the sequence designed to target the exon 8 exon splicing enhancer proved ineffectual and was not used in further experiments. When tested by direct injection into leg muscles of the dystrophic dog, the cocktail of the remaining 3 sequences resulted in formation of large regions of dystrophin-positive fibers in a strongly dose-dependent manner. Disappointingly, however, the 2 anti-exon 6 sequences were totally ineffective in vivo, in sharp contrast to their strong activity in tissue culture. This raises questions as to the reliability of in vitro testing as a guide to in vivo activity and is to be borne in mind when screening for sequences against other exons.
Because of the large size of the dog and the high cost of morpholinos, only limited numbers of studies have been conducted on systemic delivery of these antisense oligonucleotides into the beagle/golden retriever muscular dystrophy model. In one initial experiment, a regimen of 5 weekly intravenous infusions into a 5-month-old dog resulted in expression of varying amounts of dystrophin in the body musculature — up to 50% of normal levels in some muscles, with only occasional scattered patches in the heart. Functionally, the treated dog maintained its running speed over the period of treatment, in contrast to its 2 untreated littermates, which both slowed conspicuously during this time. The general condition of this and 2 further treated dogs was scored on a scale comprising 6 functional criteria, according to which all were found to be stabilized by antisense oligonucleotide treatment, while non-treated control dogs deteriorated progressively.20
At present, 2 human trials are underway, both targeting exon 51, skipping of which would theoretically provide treatment for ~17% of patients with Duchenne muscular dystrophy.21,22 One of these uses the 2′O-methyl-phosphorothioate chemistry, the other the morpholino backbone, and the 2 sequences overlap in the exon splicing enhancer region of the exon. Both have reported successful exon skipping on local intramuscularly injection and are currently undergoing further trials with systemic delivery.
Problems and Prospects
Both human trials fall in line with the principles derived from animal experimentation showing that antisense oligonucleotides are able to induce skipping of disruptive exons at a level of efficiency that is adequate to produce significant amounts of dystrophin protein widely dispersed throughout the body musculature.21,22 However, with the existing chemistries, this effect is only borderline useful, particularly as neither chemistry induces significant amounts of dystrophin in the heart, an organ that may become be rendered more susceptible to damage by the restoration of increased activity by restoration of function in the skeletal muscles. Possible solutions to this problem are posed by the addition of cell-penetrating moieties to the morpholino backbone, although these modifications are associated with a slight increase in toxicity.23–25
A major alternative proposal is use of the U7 RNA that is normally involved in splicing of pre-histone RNA but can be modified to carry a complementary to target splice sites in the dystrophin gene.26 Expression plasmids for these modified sequences have been packaged into recombinant adenovirus-associated viral vectors. These readily transduce mature muscle fibers and express the modified U7 transcripts, which bind to their appropriate targets and modify splicing of the associated exons. This approach has the advantage of producing a very long-term effect from a single administration, thus avoiding the problem of frequent re-administration. However, the vectors themselves do excite immune responses; both an antibody response that neutralizes the virus on readministration and, in some tissues, a cell-mediated response. While these problems do not appear insurmountable, they have delayed the application of this technology to human Duchenne muscular dystrophy trials.
The other major question is whether expression of dystrophin in muscles of Duchenne muscular dystrophy boys will merely slow or halt the progress of the disease, or whether there will be some recovery of previously lost function. Experiments on canine muscular dystrophy demonstrated no more than a stabilization of the disease against further progression, but these were short-term studies. One might hold out hope that muscle in which the necrotic and inflammatory processes are halted might exhibit some degree of restoration of normal function.
Acknowledgments
This review is a synthesis of research performed in a number of laboratories, based on a presentation given at the Neurobiology of Disease in Children Symposium: Muscular Dystrophy, in conjunction with the 38th Annual Meeting of the Child Neurology Society, Louisville, Kentucky, October 14, 2009. Supported by grants from the National Institutes of Health (5R13NS040925-09), the National Institutes of Health Office of Rare Diseases Research, the Muscular Dystrophy Association, and the Child Neurology Society. The authors thank Melanie Fridl Ross, MSJ, ELS, for editing this manuscript.
Footnotes
Conflict of interest statement: The author has no financial or personal relationship with organizations that would bias the views expressed.
References
- 1.Koenig M, Hoffman EP, Bertelson CJ, et al. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- 2.Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53(2):219–226. doi: 10.1016/0092-8674(88)90383-2. [DOI] [PubMed] [Google Scholar]
- 3.Dunckley MG, Eperon IC, Dickson G. Modulation of pre-mRNA splicing in the Duchenne muscular dystrophy gene. Biochem Soc Trans. 1996;24(2):276S. doi: 10.1042/bst024276s. [DOI] [PubMed] [Google Scholar]
- 4.Aartsma-Rus A, Bremmer-Bout M, Janson AA, et al. Targeted exon skipping as a potential gene correction therapy for Duchenne muscular dystrophy. Neuromuscul Disord. 2002;12 (Suppl 1):S71–S77. doi: 10.1016/s0960-8966(02)00086-x. [DOI] [PubMed] [Google Scholar]
- 5.Wilton S, Dye DE, Laing NG. Dystrophin gene transcripts skipping the mdx mutation. Muscle Nerve. 1997;20:728–734. doi: 10.1002/(sici)1097-4598(199706)20:6<728::aid-mus10>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 6.Wilton SD, Lloyd F, Carville K, et al. Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides. Neuromuscul Disord. 1999;9(5):330–338. doi: 10.1016/s0960-8966(99)00010-3. [DOI] [PubMed] [Google Scholar]
- 7.Monaco AP, Bertelson CJ, Liechti GS, et al. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. GENOMICS. 1988;2(1):90–95. doi: 10.1016/0888-7543(88)90113-9. [DOI] [PubMed] [Google Scholar]
- 8.Aartsma-Rus A, Janson AA, Kaman WE, et al. Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Hum Mol Genet. 2003;12(8):907–914. doi: 10.1093/hmg/ddg100. [DOI] [PubMed] [Google Scholar]
- 9.van Deutekom JC, Bremmer-Bout M, Janson AA, et al. Antisense-induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum Mol Genet. 2001;10(15):1547–1554. doi: 10.1093/hmg/10.15.1547. [DOI] [PubMed] [Google Scholar]
- 10.Sicinski P, Geng Y, Ryder-Cook AS, et al. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244(4912):1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 11.Morgan JE, Beauchamp JR, Pagel CN, et al. Myogenic cell lines derived from transgenic mice carrying a thermolabile T antigen: a model system for the derivation of tissue-specific and mutation-specific cell lines. Dev Biology. 1994;162:486–498. doi: 10.1006/dbio.1994.1103. [DOI] [PubMed] [Google Scholar]
- 12.Mann CJ, Honeyman K, Cheng AJ, et al. Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc Natl Acad Sci U S A. 2001;98(1):42–47. doi: 10.1073/pnas.011408598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu QL, Mann CJ, Lou F, et al. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat Med. 2003;9(8):1009–1014. doi: 10.1038/nm897. [DOI] [PubMed] [Google Scholar]
- 14.Lu QL, Rabinowitz A, Chen YC, et al. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc Natl Acad Sci U S A. 2005;102(1):198–203. doi: 10.1073/pnas.0406700102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alter J, Lou F, Rabinowitz A, et al. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med. 2006;12(2):175–177. doi: 10.1038/nm1345. [DOI] [PubMed] [Google Scholar]
- 16.Alter J, Sennoga CA, Lopes DM, et al. Microbubble stability is a major determinant of the efficiency of ultrasound and microbubble mediated in vivo gene transfer. Ultrasound Med Biol. 2009;35(6):976–984. doi: 10.1016/j.ultrasmedbio.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 17.Kornegay JN, Tuler SM, Miller DM, Levesque DC. Muscular dystrophy in a litter of golden retriever dogs. Muscle Nerve. 1988;11(10):1056–1064. doi: 10.1002/mus.880111008. [DOI] [PubMed] [Google Scholar]
- 18.Cooper BJ, Winand NJ, Stedman H, et al. The homologue of the Duchenne locus is defective in X-linked muscular dystrophy of dogs. Nature. 1988;334(6178):154–156. doi: 10.1038/334154a0. [DOI] [PubMed] [Google Scholar]
- 19.Sharp NJ, Kornegay JN, Van Camp SD, et al. An error in dystrophin mRNA processing in golden retriever muscular dystrophy, an animal homologue of Duchenne muscular dystrophy. Genomics. 1992;13(1):115–121. doi: 10.1016/0888-7543(92)90210-j. [DOI] [PubMed] [Google Scholar]
- 20.Yokota T, Lu QL, Partridge T, et al. Efficacy of systemic morpholino exon-skipping in duchenne dystrophy dogs. Ann Neurol. 2009;65(6):667–676. doi: 10.1002/ana.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Deutekom JC, Janson AA, Ginjaar IB, et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med. 2007;357(26):2677–2686. doi: 10.1056/NEJMoa073108. [DOI] [PubMed] [Google Scholar]
- 22.Kinali M, Arechavala-Gomeza V, Feng L, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8(10):918–928. doi: 10.1016/S1474-4422(09)70211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moulton HM, Wu B, Jearawiriyapaisarn N, et al. Peptide-morpholino conjugate: a promising therapeutic for Duchenne muscular dystrophy. Ann N Y Acad Sci. 2009;1175:55–60. doi: 10.1111/j.1749-6632.2009.04976.x. (in eng) [DOI] [PubMed] [Google Scholar]
- 24.Wu B, Li Y, Morcos PA, Doran TJ, et al. Octa-guanidine morpholino restores dystrophin expression in cardiac and skeletal muscles and ameliorates pathology in dystrophic mdx mice. Mol Ther. 2009;17(5):864–871. doi: 10.1038/mt.2009.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu B, Moulton HM, Iversen PL, et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc Natl Acad Sci U S A. 2008;105(39):14814–14819. doi: 10.1073/pnas.0805676105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goyenvalle A, Vulin A, Fougerousse F, et al. Rescue of dystrophic muscle through U7 snRNA-mediated exon skipping. Science. 2004;306(5702):1796–1799. doi: 10.1126/science.1104297. [DOI] [PubMed] [Google Scholar]