

Abstract
Phosphorylation of histone variant H2AX at serine 139, named γH2AX, has been widely used as a sensitive marker for DNA double-strand breaks (DSBs). γH2AX is required for the accumulation of many DNA damage response (DDR) proteins at DSBs. Thus it is believed to be the principal signaling protein involved in DDR and to play an important role in DNA repair. However, only mild defects in DNA damage signaling and DNA repair were observed in H2AX deficient cells and animals. Such findings prompted us and others to explore H2AX-independent mechanisms in DNA damage response. Here, we will review recent advances in our understanding of H2AX dependent and independent DNA damage signaling and repair pathways in mammalian cells.
Keywords: H2AX, MRE11/RAD50/NBS1, DNA damage, DNA repair, NHEJ, HR
1. Introduction
The genome, which contains nuclear DNA, is continuously challenged by a variety of genotoxic stresses. These insults lead to ruptures of sugar-phosphate DNA backbone which can generate single-stranded DNA (ssDNA) breaks and double-stranded DNA breaks (DSBs). DSBs are the most lethal type of DNA damage, and their inefficient or inaccurate repair can create mutations and chromosomal translocations that induce genomic instability and ultimately cancer development [1,2]. Many events are responsible for generating DSBs: ionizing radiation or treatment with radiomimetic drugs [3,4], reactive oxygen species, drugs and DNA modifications that induce replication and/or transcription stress [5–8], and ultraviolet radiation in S phase cells [9,10]. In addition, DSBs can arise during normal physiological processes, such as V(D)J recombination, class switch recombination, and meiosis [11–13]. Integration following retroviral infection also induces DSBs [16]. Similarly, telomere shortening would reveal unprotected double-stranded ends [14,15]. To deal with DNA double-stranded breaks, cells are equipped with two major repair pathways, the non-homologous end-joining (NHEJ) pathway and the homologous recombination (HR) pathway. In the NHEJ pathway, DSB ends are simply joined directly or joined after limited processing. Therefore, NHEJ occurs rapidly and is used throughout the cell cycle. On the other hand, HR mainly takes place during late S and G2 phases since it uses a sister homologue as a template for repair [17].
In addition to these repair pathways, cells also possess evolutionarily conserved pathways that are collectively known as DNA damage response (DDR). DDR enables the cell to sense DNA damage, propagate DNA damage signals, and activate signaling cascades that subsequently evoke a multitude of cellular responses, including cell-cycle checkpoints to slow down or stall damaged cells until the resolution of lesions. If unrepaired DSBs persist, cells will undergo either apoptosis or senescence to prevent the duplication and partitioning of damaged DNA into daughter cells [18].
Over the past decades, many studies have contributed and led to the outline of the molecular framework of DDR pathways. Histone variant H2AX is a key DDR component. It becomes rapidly (i.e., within minutes) phosphorylated at its carboxyl terminus to form so-called γH2AX at DSB sites [19]. Interestingly, many DNA damage response and DNA repair proteins also accumulate around the sites of DSBs, where they can be visualized as DNA damage-induced foci. The focus formation of many of these DNA damage repair proteins requires H2AX, thus implying that H2AX plays an important role at early stage of DNA damage response. However, there is evidence that supports the existence of H2AX-independent DNA damage response and repair pathways. Here, we will review these recent findings and propose a revised model that integrates both the H2AX-dependent and H2AX-independent DNA damage response and repair pathways in mammalian systems.
2. H2AX-dependent DNA damage signaling pathways
H2AX is a member of histone H2A family, which is one of the five types of histones that package and organize eukaryotic DNA into chromatin. The basic composition of chromatin is the nucleosome. Each nucleosome consists of eight histone molecules, two from each of the four core histones (H2A, H2B, H3, and H4) to form an octamer, which is wrapped by approximately 146 bp of DNA. The linker histone, H1, interacts with linker DNA between nucleosomes and functions in compacting chromatin into higher order structures. H2AX is actually a rare histone variant that is distributed throughout mammalian chromatin. It makes up ~10% of total H2A molecules in normal human fibroblasts, with this percentage varying among different cell types [3,19,20]. Substantial studies have placed H2AX at the center stage of DDR. H2AX modifications, including phosphorylation, ubiquitylation, and acetylation are of great importance in DDR pathways. They are critical not only for recruiting downstream DNA damage and repair proteins, but also for the amplification of DNA damage signals.
2.1 H2AX phosphorylation and function in response to DSBs
In response to DSBs, the conserved C-terminal tail of H2AX becomes rapidly phosphorylated at serine-139 by PI3-K like kinases, including ATM, ATR and DNA-PKcs. ATM and DNA-PKcs display functional redundancy in phosphorylating H2AX following ionizing radiation, while ATR is more important for H2AX phosphorylation in response to DNA damage that would slow or stall replication forks [19]. The phosphorylated H2AX, named γH2AX, is one of the first proteins involved in DNA damage response (DDR) pathways. It is required for DNA damage signal amplification and subsequent accumulation of numerous DDR proteins at DSBs sites to form so called ionizing radiation induced foci (IRIF) [21–25] (also see Figure 1 for a modified model of the DNA damage signaling cascade). Given this important function of γH2AX in DDR, it is not surprising that several protein phosphatases, PP2A, PP4, PP6, and Wip1, have been shown to dephosphorylate γH2AX and negatively regulate H2AX functions [26–30].
Figure 1. H2AX-dependent DNA damage response and repair pathways.
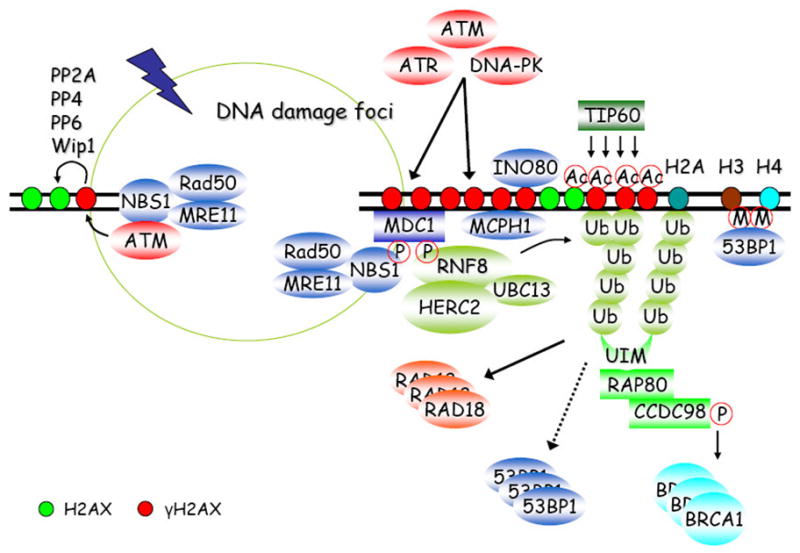
The MRN-mediated ATM activation at DSB sites initiates a series of phosphorylation events, including the generation of γH2AX (phosphorylation of H2AX at serine 139). γH2AX can then recruit MDC1, MCPH1 and INO80 via direct protein-protein interactions. MDC1 serves as a platform for NBS1 and RNF8 docking in a phosphorylation-dependent manner. RNF8 initiates a DNA damage-induced ubiquitylation cascade which is required for further tethering 53BP1, RAD18, BRCA1, and RAP80-containing complex at damage sites. TIP60 can catalyze H2AX acetylation to promote its ubiquitylation. In addition, γH2AX can be negatively regulated via dephosphorylation by a number of phosphatases, such as PP2A, PP4, PP6 and Wip1.
MDC1 (Mediator of Damage Checkpoint protein 1) works very closely with γH2AX in DDR, since it is required for almost all of the γH2AX-dependent focus formation events following DNA damage. In response to DSBs, MDC1 binds directly to γH2AX through its BRCT domains [31,32]. The phosphorylation of six SDTDXD/E repeats near the N terminus of MDC1 serves to recruit NBS1 (Nijmegen Breakage Syndrome 1) and to regulate the intra-S-phase checkpoint in response to DNA damage [33–36]. In addition, MDC1 also recruits E3 ubiquitin ligase RNF8 in a phosphorylation-dependent manner, with the latter being responsible for tethering 53BP1 (p53-Binding Protein 1), BRCA1 (Breast and Ovarian Cancer Susceptibility Protein 1), and RAP80 (Receptor-Associated Protein 80)-containing complexes at damage sites [37–41]. More importantly, a signal amplification loop that includes the factors H2AX, MDC1, and probably NBS1 activates and/or retains ATM at DSB sites and leads to the spreading of H2AX phosphorylation up to megabase regions surrounding DSBs [42]. This long-range γH2AX/MDC1 localization adjacent to DSBs serves as a landing site for the accumulation of other DDR proteins.
Besides MDC1, MCPH1 (also known as BRIT1) is also recruited to DSB sites via its direct interaction with γH2AX, as is mediated by the C-terminal BRCT domains of MCPH1[43]. MCPH1 plays roles in chromatin condensation, chromatin remodeling, HR repair, and cell-cycle checkpoints via its association with multiple DNA damage signaling and repair proteins [44–47].
Moreover, H2AX phosphorylated by other kinases, but not ATM, plays multiple roles during apoptosis and necrosis [48]. DNA-PKcs phosphorylates H2AX at serine-139 during apoptotic DNA fragmentation while ATM is dispensable for this process [49–51]. Ultraviolet (UV) A irradiation can strongly induce H2AX phosphorylation via c-Jun N-terminal kinase (JNK) and phosphorylation of H2AX by JNK was associated with induction of apoptosis [52]. Another group reported that serum starvation induces H2AX phosphorylation to regulate apoptosis via p38 MAPK (mitogen-activated protein kinase) pathway [53]. Taken together, these results suggest that while ATM is involved in H2AX phosphorylation in irradiated cells, DNA-PK and other kinases have an important role in H2AX phosphorylation during apoptosis, which is also supported by the fact that apoptosis induces degradation of ATM [54], therefore making DNA-PK or some other kinases the predominant kinase responsible for γH2AX induction during apoptotic DNA fragmentation. Intriguingly, recent studies indicate that phosphorylation of tyrosine 142 of H2AX prevents the recruitment of repair complexes, while promoting the binding of pro-apoptotic factors to γH2AX. These observations therefore provide a plausible model for H2AX in regulation of apoptotic versus repair response following DNA damage [55,56].
Human chromosomes are capped and stabilized by telomeres. Telomeres assist in maintaining genomic integrity by preventing chromosome ends from being recognized as double-strand damage ends and protecting against chromosome-chromosome fusions and rearrangements. Telomeres are protected by capping structures consisting of core protein complexes that bind with sequence specificity to telomeric DNA [57]. As mentioned above, the shortening or uncapping of telomeres will reveal unprotected double-stranded ends and trigger a DNA damage response, in which damage response proteins, such as γH2AX, will accumulate at the dysfunctional telomeres [58,59]. In mTR−/− cells (cells lacking the RNA component of telomerase), the damage marker γH2AX has been shown to localize to the telomere in late generation, thereby suggesting that critically shortened telomeres are recognized directly as DNA breaks [60]. It has also been reported that subtelomeric regions in budding yeast are constitutively modified by γH2AX, suggesting that telomeres can be recognized as a form of DSB damage [61]. While telomeres can be targeted by γH2AX, H2AX is neither necessary for the normal mitotic telomere maintenance, nor required for the chromosome fusions caused by dysfunctional telomeres. Nevertheless, H2AX appears to have a critical role in controlling telomere movement during meiosis [62].
2.2 H2AX acetylation and ubiquitylation in response to DSBs
Phosphorylation is not the only kind of modification occurred on H2AX. Recent studies have revealed dynamic regulations of the synthesis, recognition, and hydrolysis of ubiquitin chains at DNA damage sites [63,64]. The formation of ubiquitin chains at DSB sites is believed to be initiated by a ring-finger-containing nuclear factor called RNF8 [37–39]. RNF8 consists of an N-terminal FHA domain and a C-terminal RING finger that has E3 ubiquitin ligase activity. RNF8 is recruited to DSB sites in a γH2AX-MDC1-dependent manner via a direct interaction between phosphorylated MDC1 and RNF8 FHA domain. Indeed, biochemical and genetic experiments recently confirmed that at damage sites, RNF8 works together with an E2 ubiquitin-conjugating enzyme, UBC13, to ubiquitylate histones H2A, H2B, and γH2AX [37,39,65–68] by conjugating primarily Lys63-linked ubiquitin chains. However, RNF8 only initiates the formation of ubiquitin chains at the DSB sites. Another E3 ligase, the RIDDLE syndrome protein RNF168, recognizes limited ubiquitin chains synthesized by RNF8 through its two motif-interacting-with-ubiquitin (MIU) domains. RNF168 then associates with UBC13 and maintains and/or further elongates polyubiquitin chains at DSBs [66,67].
Just to add a bit more complexity to this damage-induced ubiquitylation cascade, a recent study has shown that HERC2, a large HECT domain-containing protein, interacts with RNF8 in response to ionizing radiation and facilitates the assembly of ubiquitin-conjugating enzyme UBC13 with RNF8, thereby promoting DNA damage-induced formation of ubiquitin chains [69]. Moreover, two recent papers highlight the importance of SUMO modification in the DNA damage-induced ubiquitylation cascade. RNF8/HERC2 mediated ubiquitylation of H2A is facilitated by PIAS4-dependent SUMOylation [70]. BRCA1 can also be modified by SUMO2/3 in a PIAS1-dependent manner, which increases its E3 ubiquitin ligase activity [71].
Although H2AX is ubiquitylated by RNF8 and RNF168, mutating all of the lysines in H2AX did not decrease ubiquitylated H2A foci formation, nor did it impair the recruitment of downstream DDR factors. Thus, although γH2AX initiates this ubiquitylation cascade and is a bona fide substrate of RNF8 and RNF168, there are other substrates such as H2A that are also ubiquitylated at DSB sites [37,38,40,66,67]. It will be interesting to determine in the future as whether or not different substrates of RNF8 and RNF168 would carry out distinct functions in DDR.
Another aspect of this ubiquitylation-dependent signaling cascade that remains to be addressed is the mechanism by which ubiquitylation chains cause the recruitment of various DNA damage repair proteins. This process is most likely mediated in part by proteins containing ubiquitin-binding domains. For example, the recruitment of BRCA1 involves a protein complex that contains the ubiquitin-binding subunit RAP80. RAP80 targets BRCA1 to DNA breaks through its ability to act as a molecular bridge between poly-ubiquitylated histones and CCDC98, the latter directly binds and recruits BRCA1 [72–74]. Once recruited, BRCA1 and its binding partner BARD1 can activate the intra-S phase and G2/M cell-cycle checkpoints, as well as participate in homologous recombination repair [75,76]. BRCA1/BARD1 function as a heterodimeric E3 ubiquitin ligase capable of catalyzing the formation of K6-linked poly-ubiquitin chains. As previously stated, PIAS1-dependent SUMOylation of BRCA1 can increase its E3 ubiquitin ligase activity, which may also play a role in DDR [71]. However, the specific substrates of BRCA1 and how BRCA1-dependent ubiquitylation of these substrates regulates their functions in response to DNA damage remain unclear. More recently, another E3 ubiquitin ligase, RAD18, was reported to be recruited to sites of DNA breaks in an RNF8-dependent manner, where it then interacts with RAD51C and promotes homologous recombination [77]. In this case, the recruitment of RAD18 requires the UBZ (ubiquitin-binding zinc finger) domain of RAD18, which directly binds to ubiquitin chains [77].
There are many other DDR proteins that depend on RNF8 and RNF168 for their accumulation at DSB sites. However, the manner of their recruitment by this ubiquitylation cascade remains to be resolved. For instance, the damage-induced focus localization of p53-binding protein 1 (53BP1) requires RNF8 and RNF168. There is no evidence that 53BP1 can bind directly to ubiquitin. Rather, it is postulated that 53BP1 may recognize methylated histones surrounding DSBs, that may become exposed following histone ubiquitylation [78–80]. Further experimentation is required to ascertain whether this is indeed the case or if 53BP1 localization requires another yet-to-be-identified mediator that has ubiquitin-binding activity.
In addition to being ubiquitylated, histones are also acetylated following DNA damage. For example, histone acetyltransferase TIP60 participates in DDR [81,82], partially by promoting histone H4 acetylation and the accumulation of repair proteins, including RAD51, at DSB sites [83]. Tip60 also acetylates and activates ATM at DSB sites [84]. With specific regard to H2AX, TIP60 has been shown to catalyze H2AX acetylation, independently of its phosphorylation status, in response to ionizing irradiation [85]. Furthermore, acetylation of H2AX by TIP60 is proposed to be pre-requisite for H2AX ubiquitylation [85]. This acetylation-dependent H2AX ubiquitylation by TIP60-UBC13 complex may result in the release of H2AX from damaged chromatin, thereby enhancing chromatin dynamics and allowing the access of repair proteins to DSB sites. Such complex crosstalks involving phosphorylation, acetylation, ubiquitylaton, and SUMOylation of H2AX and other DDR proteins illustrate a highly coordinated network that is activated in response to DNA damage. The details regarding the interplays among these posttranslational modifications warrant further investigation.
2.3 γH2AX and chromatin remodeling in response to DSBs
The highly compacted structure of chromatin acts as a natural barrier against access to DNA during transcription, DNA damage repair, and recombination. Abundant evidence suggests that many chromatin remodeling complexes are involved in DDR and DNA repair [86]. In response to DNA damage, chromatin structure can be altered by several mechanisms, including the action by ATP-dependent chromatin remodeling complexes, the incorporation or removal of histone variants into or from nucleosomes, and covalent histone modifications [87]. As we mentioned above, the primary role of H2AX in DDR is to facilitate the access of repair proteins to DSB sites. In the same capacity, H2AX also participates in DNA damage induced chromatin remodeling by promoting the recruitment of remodeling complexes and/or other histone modifying protein complexes, one of which being the INO80 complex.
The INO80 complex is a multi-subunit, ATP-dependent chromatin remodeling complex, whose ability is to regulate transcriptional processes is well established [88]. Recent studies reveal that the INO80 complex also has a crucial function in many other cellular processes, including DNA repair [88,89]. In yeast, the INO80 complex is recruited to HO endonuclease-induced DSB through its specific interaction with damage-induced phosphorylated H2A (equivalent to γH2AX in human). This interaction requires Nhp10, an HMG-like subunit in the INO80 complex. Loss of Nhp10 or γH2AX results in reduced INO80 recruitment to DSB sites. Moreover, yeast INO80 mutants are hypersensitive to DNA-damaging agents [90,91]. In this way, it is clear that H2AX not only participates in the accumulation of numerous DDR and DNA repair factors to DSB sites, but also facilitates chromatin modification and remodeling at and near DSB sites to further promote DNA damage response and repair processes.
3. Histone H2AX-dependent and independent DNA repair pathways
Although we have extensively discussed H2AX-dependent DDR signaling pathways, it is worth pointing out that H2AX (in particular its most important modification, γH2AX) is neither required for the initial localization of some DDR proteins such as MRN (MRE11-RAD50-NBS1) complex, 53BP1, and BRCA1 to DSB sites, nor required for the accumulation of other DDR proteins, such as WRN, BLM, and more recently CENP-A at DNA damage sites [92–94]. More importantly, H2AX-deficient cells show only mild defects in DNA damage checkpoint control and DNA repair, suggesting that H2AX assists in, but is not critical for DNA damage checkpoint activation and DNA repair processes [95,96]. The mild defects could be explained by the fact that H2AX is not required for the initial MRN and/or ATM localization at DSB sites, which is likely to be the essential step involved in DNA damage checkpoint control.
As formerly discussed, the functions of H2AX in DNA repair likely depend on its ability to accumulate many DNA damage response and repair proteins at or near the sites of DNA breaks. It is reasonable to speculate that such accumulation of DDR and repair proteins at or near DSB sites increase the local concentration of these proteins and thus effectively promote DSB repair. It is also possible that γH2AX and the associated proteins that it helps to accumulate may assist in holding broken ends together, thereby allowing time for DNA repair and minimizing the risk of misrepair [23,24,97]. This may be the reason that H2AX is more involved in the repair of free DSBs formed by ionizing radiation, but is largely dispensable for V(D)J recombination and for retroviral postintegration repair, which require additional factors like RAG1/2 and intergrases [16,22,62,98]. Since γH2AX predominantly forms in response to DSBs, we will next discuss the involvement of H2AX in the two main DSB repair pathways, named NHEJ and HR.
3.1 Non-homologous end joining (NHEJ)
As the name implies, NHEJ pathway repairs DSBs without using the information from homologous sequences. DNA breaks are sensed by Ku70/80 heterodimer, which in turn recruits DNA-dependent protein kinase catalytic subunit DNA-PKcs, and assembles the Ku/DNA-PK complex to activate its kinase activity. DNA-PK functions as a regulatory component in NHEJ, by potentially facilitating and regulating the processing of DNA ends. DNA-PK complex also helps to recruit other NHEJ components such as XRCC4, DNA ligase IV, XLF, and Artemis, which carry out end rejoining reaction. NHEJ is an ancient DNA repair mechanism that can be traced back to haploid organisms in which a homologous DNA copy was not available. Hence, early in evolution, NHEJ served to provide a survival advantage for these organisms and their ancestors. Even in higher eukaryotes, NHEJ is often used due to its ability to repair breaks quickly and at any time in the cell cycle. Moreover, NHEJ is also critically important for the repair of physiological DSBs created during V(D)J recombination and class switch recombination. Therefore, patients harboring mutations in NHEJ components are not only sensitive to ionizing radiation, but also suffer severe immunodeficiency [99].
One piece of the evidence supporting H2AX-independent NHEJ is that even proteins, such as 53BP1, BRCA1, and MRE11, that require H2AX for stable accumulation at DSB sites, can still be recruited to DSB sites in H2AX null cells [95,96]. This observation suggests that H2AX-dependent accumulation of such repair proteins is one, but not the only, way that these proteins localize to DSB sites. Furthermore, MRN complex is known to play a role in NHEJ. MRN complex binds directly to DSBs independently of H2AX [95,96] and the NHEJ function of MRE11 occurs independent of H2AX [100]. The MRN complex, as the initial sensor, carries out at least two distinct functions following DNA damage. One is to promote DNA repair by itself or by transient localization of several DNA damage repair proteins in the absence of H2AX [96]. The other is to activate ATM and ATM-dependent checkpoints [101–103]. The separate functions of MRN in DNA damage response is strongly supported by two recent publications [104,105], which demonstrate while the nuclease activity of MRE11 is essential to initiate DNA repair, it is largely dispensable for ATM activation.
53BP1 has also been implicated as a facilitator of NHEJ [106], especially during the S phase of the cell cycle [107]. Specifically, 53BP1-deficient mice and cells display significant deficits in class-switch recombination and long-range V(D)J recombination [108–110]. So far, the molecular mechanism of damage-induced 53BP1 focus formation remains a puzzle. There are at least four ways that 53BP1 may be recruited and accumulate at DSB sites: (1) 53BP1 binds to methylated lysine 79 of histone H3 or dimethylated lysine 20 of histone H4 near or at DSBs [78,79]; (2) The GAR motif of 53BP1 is arginine methylated by PRMT1 and is necessary for 53BP1 DNA binding activity [111]; (3) RNA mediated 53BP1 and DSB sites interaction [112]; (4) 53BP1 interacts with MRN complex and requires MRN for transient tethering at DSB sites [96,113]. While some of these methods of recruitment or accumulation of 53BP1 at DSB may still be H2AX dependent or at least facilitated by H2AX, we have shown that the transient recruitment of 53BP1 by MRN complex can occur in H2AX null cells [96,113]. This observation, in addition to the mild class-switch recombination defect observed in H2AX-deficient mice, suggests that 53BP1 can function in DNA repair independently of H2AX.
Although BRCA1 is best known for its involvement in HR repair, there is some indication that BRCA1 may also contribute to NHEJ. In terms of mechanisms, a recent study showed that BRCA1 is involved in NHEJ via its interaction with Ku80 through its N terminus [114]. The accumulation of N or C termini of BRCA1 at DSBs exhibits distinct kinetics. In vivo experiments showed that the N-terminal BRCA1 fragment accumulates and dissociates rapidly from laser-induced DSBs, while the C-terminal fragment accumulates slowly at DSBs and remains at these sites. These data suggest that the recruitment of BRCA1 via its N terminus occurs independently of H2AX and most likely contributes to NHEJ. In contrast, the accumulation of BRCA1 via its C terminus mainly contributes in HR [114].
NHEJ and HR factors are believed to be independently recruited to DSB sites and may even antagonize each other. As revealed by experiments using laser-induced DSBs, the retention of NHEJ factors at DSB sites is transient, whereas HR factors persist at these unrepaired lesions [115,116]. This may reflect the difference in speed of NHEJ versus HR repair processes. The spatio-temporal relationship between NHEJ and DDR activation is not clear and subject to much speculation. NHEJ at DSB sites may occur so rapidly that DDR, including ATM activation and H2AX phosphorylation, does not even take place before the lesion is repaired. As a rare histone variant, H2AX is only present, on average, in one of every five or ten human nucleosomes, where H2A is the predominant form in histone octamers [99]. In this way, the majority of DSBs should occur several nucleosomes away from the nearest octamer containing an H2AX molecule. Given the fact that the classic NHEJ probably requires less than 30 bp on each side of the DNA break [99], and that most H2AXs locate several nucleosomes away from the DSBs, it is hard to imagine that H2AX phosphorylation would be critical for NHEJ.
Nevertheless, several studies suggested that γH2AX might have some impact on NHEJ [117,118]. Most of these evidences are based on immunofluorescent co-localization studies in which the damage sites detected likely contained a mixture of HR and NHEJ events. Since γH2AX is involved in the accumulation of DNA repair proteins, such as 53BP1 and MRN complex at DSB sites, and these proteins are known to play a role in some specialized NHEJ processes, we believe that γH2AX can contribute to certain types of NHEJ. We further postulate that although H2AX is likely dispensable for classic NHEJ repair, it plays an accessory role in specific NHEJ processes that require the stable accumulation of several DNA damage repair proteins, including MRN, 53BP1, and BRCA1 (see Figure 2 for a modified model of H2AX-independent repair pathways).
Figure 2. H2AX-independent DSB recognition and repair pathways.
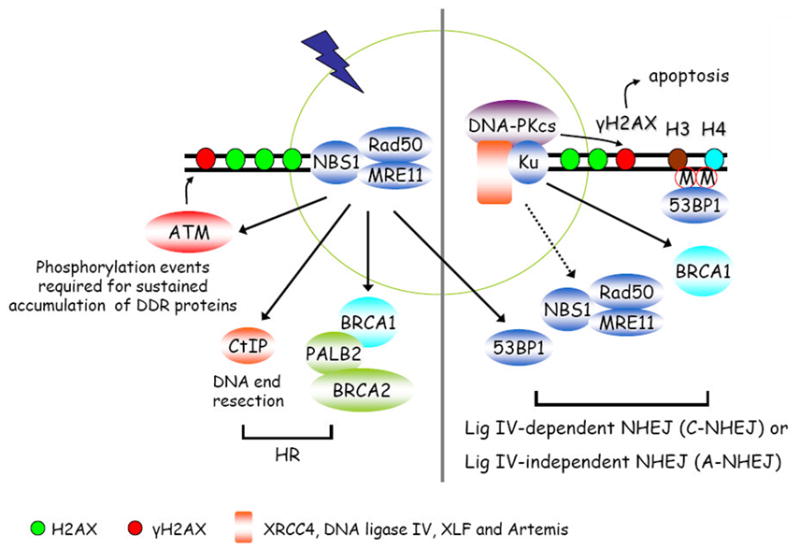
The MRN complex or Ku70/80 heterodimer recognizes and binds directly to broken DNA ends. The MRN complex recruits CtIP and BRCA1/PALB2/BRCA2 to initiate its preferred homologous recombination (HR). At the same time, MRN complex also recruits and activates ATM, which is involved in the initiation a serial of phosphorylation events including γH2AX. As presented in Figure 1, γH2AX and H2AX-dependent signal transduction events are required for the sustained accumulation of many DNA damage response and repair proteins at DSB sites to facilitate DNA repair processes. If the broken DNA ends are recognized by Ku70/80, it then recruits DNA-PKcs and other NHEJ components to carry out end joining reactions. NHEJ is now suggested to be mediated by two sub-pathways, the lig IV-dependent classic NHEJ pathway (C-NHEJ) and the lig IV-independent alternative end joining pathway (A-NHEJ). It is worth pointing out that the recruitment of MRN complex, 53BP1, and BRCA1 to DSB sites may facilitate NHEJ, especially the A-NHEJ pathway.
3.2 Homologous recombination (HR)
In HR, the information on the sister chromatid is usually used for the repair of a broken chromatid. In this case, DSB is sensed and recognized by MRN complex, which can be recruited to the DSB site to generate single-stranded DNA (ssDNA) regions via end resection. Once the DNA ends are resected, RPA binds efficiently to ssDNA and with the help of some mediators such as BRCA2, RAD51 can then replace RPA and form nucleoprotein filaments to invade the homologous template and create D-loop and Holliday junction. This process eventually primes DNA synthesis to copy and ultimately restore genetic information that was disrupted by DSB [17].
Similar to its role in NHEJ, H2AX only modulates HR repair efficiency [95,98,119–121]. In the absence of H2AX, MRN complex can recognize DSB sites and initiate DNA end resection and HR repair. Moreover, in H2AX-deficient cells, MRN complex is initially involved in the transient recruitment of other signaling and repair factors, such as BRCA1 and 53BP1, at DSB sites [96]. This highlights a critical role of MRN complex, and perhaps CtIP (MRN-CtIP axis), at early stage of DNA damage response [122] (Figure 2).
The generation of ssDNAs, especially RPA-coated ssDNAs, is believed to be an intermediate step for HR repair [123,124]. It has been shown that depletion of NBS1, MRE11, or CtIP can greatly impair RPA foci formation (the readout for ssDNA generation) in response to DSBs. Nevertheless H2AX, MDC1, or ATM deficiency exhibits seemingly normal RPA foci formation [96]. Consistently, DNA damage-induced RPA foci formation has been shown to be independent of γH2AX, given that the PI3 kinase inhibitor Wortmannin can block DNA damage-induced γH2AX, but not RPA foci formation [125]. In addition, recruitment of HR repair protein RAD51 to damage sites has long been known to function independently of H2AX phosphorylation status [22]. Moreover, deficiencies in any of the components in the H2AX-MDC1-RNF8-RAP80 pathway do not abolish PALB2-dependent RAD51 foci formation [126]. CCDC98, a localizer of BRCA1 to DNA damage, plays downstream of the H2AX-MDC1 axis [41,74,127]. However cells depleted of CCDC98 or RAP80 exhibit mild HR defects [74], again implicating that although RAP80-CCDC98-mediated localization of BRCA1 has a role in the DNA damage response, they are not essential for HR repair.
The fact that any deficiency in the H2AX-MDC1 axis only leads to mild DSB repair defects suggests that γH2AX might regulate the repair of selected DSBs or assist specific repair sub-pathways [128]. H2AX may perform an accessory function in HR repair via its ability to stabilize and accumulate DNA damage repair proteins at or near DSB sites. The moderate role of the H2AX-MDC1 axis in HR repair is also supported by mouse genetic studies. Depletion of components directly or indirectly involved in HR pathway such as, ATR [129,130], MRN complex [131–133], CtIP [134], BRCA1 [135], BRCA2 [136], and RAD51 [137], in mice all result in embryonic lethality, suggesting that the intact HR pathway is critical important during embryogenesis. On the other hand, H2AX−/− mice exhibit relatively mild phenotypes with some degree of genomic instability [22]. In fact, mice lacking other factors involved in the H2AX-MDC1 axis, such as ATM [138–140], and MDC1 [31], all display increased genomic instability and cancer susceptibility; nevertheless these null mice are viable. Again, such observations indicate that an HR-mediated DSB repair pathway can occur, albeit at lower efficiency, in cells or animals deficient in any of the components involved in the H2AX-mediated DNA damage-signaling cascade.
4. Summary and perspective
This review has concentrated on H2AX and discussed its role in DNA damage response and DNA repair. We would like to use two cartoons to summarize this review: the H2AX-dependent DNA damage response and repair pathways (as shown in Figure 1) and the H2AX-independent DSB recognition and repair pathways (Figure 2). The H2AX-dependent stable accumulation phase has been extensively studied. However, the H2AX-independent initial recognition of DNA breaks and the selection or the commitment of NHEJ versus HR pathways in DSB repair remain largely unknown. Future studies should focus on the interplay between Ku70/80 and MRN complexes, given that these are the two protein complexes that recognize DNA breaks and initiate respectively NHEJ and HR repair. How their abilities to bind to DSB are regulated by the structure of DNA breaks, the action of various DSB modifying enzymes, and the different cell cycle phases will help us to appreciate the fascinating coordination of these repair processes in the maintaining of genomic stability.
Acknowledgments
We apologize to those colleagues whose work has not been cited due to space limitation. This work was supported by grants from the National Institutes of Health (CA089239, CA092312, CA100109 to J.C.). J.C is also a recipient of an Era of Hope Scholar award from the Department of Defense (W81XWH-05-1-0470) and a member of M.D. Anderson Cancer Center (CA016672).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jeggo PA, Lobrich M. DNA double-strand breaks: their cellular and clinical impact? Oncogene. 2007;26:7717–9. doi: 10.1038/sj.onc.1210868. [DOI] [PubMed] [Google Scholar]
- 2.McKinnon PJ, Caldecott KW. DNA strand break repair and human genetic disease. Annu Rev Genomics Hum Genet. 2007;8:37–55. doi: 10.1146/annurev.genom.7.080505.115648. [DOI] [PubMed] [Google Scholar]
- 3.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 4.Ismail IH, Wadhra TI, Hammarsten O. An optimized method for detecting gamma-H2AX in blood cells reveals a significant interindividual variation in the gamma-H2AX response among humans. Nucleic Acids Res. 2007;35:e36. doi: 10.1093/nar/gkl1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takahashi A, Ohnishi T. Does gammaH2AX foci formation depend on the presence of DNA double strand breaks? Cancer Lett. 2005;229:171–9. doi: 10.1016/j.canlet.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 6.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 7.Guirouilh-Barbat J, Redon C, Pommier Y. Transcription-coupled DNA double-strand breaks are mediated via the nucleotide excision repair and the Mre11-Rad50-Nbs1 complex. Mol Biol Cell. 2008;19:3969–81. doi: 10.1091/mbc.E08-02-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan J, Ghosal G, Chen J. The annealing helicase HARP protects stalled replication forks. Genes Dev. 2009;23:2394–9. doi: 10.1101/gad.1836409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc Natl Acad Sci U S A. 2006;103:9891–6. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–62. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 11.Edry E, Melamed D. Class switch recombination: a friend and a foe. Clin Immunol. 2007;123:244–51. doi: 10.1016/j.clim.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 12.Soulas-Sprauel P, Rivera-Munoz P, Malivert L, Le Guyader G, Abramowski V, Revy P, de Villartay JP. V(D)J and immunoglobulin class switch recombinations: a paradigm to study the regulation of DNA end-joining. Oncogene. 2007;26:7780–91. doi: 10.1038/sj.onc.1210875. [DOI] [PubMed] [Google Scholar]
- 13.Chicheportiche A, Bernardino-Sgherri J, de Massy B, Dutrillaux B. Characterization of Spo11-dependent and independent phospho-H2AX foci during meiotic prophase I in the male mouse. J Cell Sci. 2007;120:1733–42. doi: 10.1242/jcs.004945. [DOI] [PubMed] [Google Scholar]
- 14.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–56. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura AJ, Chiang YJ, Hathcock KS, Horikawa I, Sedelnikova OA, Hodes RJ, Bonner WM. Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenetics Chromatin. 2008;1:6. doi: 10.1186/1756-8935-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daniel R, et al. Histone H2AX is phosphorylated at sites of retroviral DNA integration but is dispensable for postintegration repair. J Biol Chem. 2004;279:45810–4. doi: 10.1074/jbc.M407886200. [DOI] [PubMed] [Google Scholar]
- 17.Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010;11:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.d’Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8:512–22. doi: 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- 19.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–67. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartova E, Krejci J, Harnicarova A, Galiova G, Kozubek S. Histone modifications and nuclear architecture: a review. J Histochem Cytochem. 2008;56:711–21. doi: 10.1369/jhc.2008.951251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10:886–95. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 22.Celeste A, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–7. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Celeste A, et al. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell. 2003;114:371–83. doi: 10.1016/s0092-8674(03)00567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bassing CH, et al. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 2003;114:359–70. doi: 10.1016/s0092-8674(03)00566-x. [DOI] [PubMed] [Google Scholar]
- 25.Huen MS, Chen J. Assembly of checkpoint and repair machineries at DNA damage sites. Trends Biochem Sci. 2010;35:101–8. doi: 10.1016/j.tibs.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J. gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol Cell. 2005;20:801–9. doi: 10.1016/j.molcel.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 27.Chowdhury D, et al. A PP4-phosphatase complex dephosphorylates gamma-H2AX generated during DNA replication. Mol Cell. 2008;31:33–46. doi: 10.1016/j.molcel.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakada S, Chen GI, Gingras AC, Durocher D. PP4 is a gamma H2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO Rep. 2008;9:1019–26. doi: 10.1038/embor.2008.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Douglas P, Zhong J, Ye R, Moorhead GB, Xu X, Lees-Miller SP. Protein phosphatase 6 interacts with the DNA-dependent protein kinase catalytic subunit and dephosphorylates gamma-H2AX. Mol Cell Biol. 2010;30:1368–81. doi: 10.1128/MCB.00741-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Macurek L, Lindqvist A, Voets O, Kool J, Vos HR, Medema RH. Wip1 phosphatase is associated with chromatin and dephosphorylates gammaH2AX to promote checkpoint inhibition. Oncogene. 2010 doi: 10.1038/onc.2009.501. [DOI] [PubMed] [Google Scholar]
- 31.Lou Z, et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell. 2006;21:187–200. doi: 10.1016/j.molcel.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 32.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–26. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 33.Wu L, Luo K, Lou Z, Chen J. MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. Proc Natl Acad Sci U S A. 2008;105:11200–5. doi: 10.1073/pnas.0802885105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spycher C, Miller ES, Townsend K, Pavic L, Morrice NA, Janscak P, Stewart GS, Stucki M. Constitutive phosphorylation of MDC1 physically links the MRE11-RAD50-NBS1 complex to damaged chromatin. J Cell Biol. 2008;181:227–40. doi: 10.1083/jcb.200709008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Melander F, Bekker-Jensen S, Falck J, Bartek J, Mailand N, Lukas J. Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. J Cell Biol. 2008;181:213–26. doi: 10.1083/jcb.200708210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chapman JR, Jackson SP. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep. 2008;9:795–801. doi: 10.1038/embor.2008.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–14. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kolas NK, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–40. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 40.Wang B, Elledge SJ. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc Natl Acad Sci U S A. 2007;104:20759–63. doi: 10.1073/pnas.0710061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198–202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stucki M, Jackson SP. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 2006;5:534–43. doi: 10.1016/j.dnarep.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 43.Wood JL, Singh N, Mer G, Chen J. MCPH1 functions in an H2AX-dependent but MDC1-independent pathway in response to DNA damage. J Biol Chem. 2007;282:35416–23. doi: 10.1074/jbc.M705245200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wood JL, Liang Y, Li K, Chen J. Microcephalin/MCPH1 associates with the Condensin II complex to function in homologous recombination repair. J Biol Chem. 2008;283:29586–92. doi: 10.1074/jbc.M804080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peng G, et al. BRIT1/MCPH1 links chromatin remodelling to DNA damage response. Nat Cell Biol. 2009;11:865–72. doi: 10.1038/ncb1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alderton GK, Galbiati L, Griffith E, Surinya KH, Neitzel H, Jackson AP, Jeggo PA, O’Driscoll M. Regulation of mitotic entry by microcephalin and its overlap with ATR signalling. Nat Cell Biol. 2006;8:725–33. doi: 10.1038/ncb1431. [DOI] [PubMed] [Google Scholar]
- 47.Chaplet M, Rai R, Jackson-Bernitsas D, Li K, Lin SY. BRIT1/MCPH1: a guardian of genome and an enemy of tumors. Cell Cycle. 2006;5:2579–83. doi: 10.4161/cc.5.22.3471. [DOI] [PubMed] [Google Scholar]
- 48.Artus C, et al. AIF promotes chromatinolysis and caspase-independent programmed necrosis by interacting with histone H2AX. EMBO J. 2010 doi: 10.1038/emboj.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem. 2000;275:9390–5. doi: 10.1074/jbc.275.13.9390. [DOI] [PubMed] [Google Scholar]
- 50.Mukherjee B, Kessinger C, Kobayashi J, Chen BP, Chen DJ, Chatterjee A, Burma S. DNA-PK phosphorylates histone H2AX during apoptotic DNA fragmentation in mammalian cells. DNA Repair (Amst) 2006;5:575–90. doi: 10.1016/j.dnarep.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 51.Solier S, Sordet O, Kohn KW, Pommier Y. Death receptor-induced activation of the Chk2- and histone H2AX-associated DNA damage response pathways. Mol Cell Biol. 2009;29:68–82. doi: 10.1128/MCB.00581-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu C, Zhu F, Cho YY, Tang F, Zykova T, Ma WY, Bode AM, Dong Z. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol Cell. 2006;23:121–32. doi: 10.1016/j.molcel.2006.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu C, Shi Y, Wang Z, Song Z, Zhu M, Cai Q, Chen T. Serum starvation induces H2AX phosphorylation to regulate apoptosis via p38 MAPK pathway. FEBS Lett. 2008;582:2703–8. doi: 10.1016/j.febslet.2008.06.051. [DOI] [PubMed] [Google Scholar]
- 54.Smith GC, d’Adda di Fagagna F, Lakin ND, Jackson SP. Cleavage and inactivation of ATM during apoptosis. Mol Cell Biol. 1999;19:6076–84. doi: 10.1128/mcb.19.9.6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009;458:591–6. doi: 10.1038/nature07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiao A, et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature. 2009;457:57–62. doi: 10.1038/nature07668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–34. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 58.Blackburn EH. Switching and signaling at the telomere. Cell. 2001;106:661–73. doi: 10.1016/s0092-8674(01)00492-5. [DOI] [PubMed] [Google Scholar]
- 59.Wright WE, Shay JW. Telomere-binding factors and general DNA repair. Nat Genet. 2005;37:116–8. doi: 10.1038/ng0205-116. [DOI] [PubMed] [Google Scholar]
- 60.Hao LY, Strong MA, Greider CW. Phosphorylation of H2AX at short telomeres in T cells and fibroblasts. J Biol Chem. 2004;279:45148–54. doi: 10.1074/jbc.M403924200. [DOI] [PubMed] [Google Scholar]
- 61.Kim JA, Kruhlak M, Dotiwala F, Nussenzweig A, Haber JE. Heterochromatin is refractory to gamma-H2AX modification in yeast and mammals. J Cell Biol. 2007;178:209–18. doi: 10.1083/jcb.200612031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fernandez-Capetillo O, et al. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol. 2002;4:993–7. doi: 10.1038/ncb884. [DOI] [PubMed] [Google Scholar]
- 63.Messick TE, Greenberg RA. The ubiquitin landscape at DNA double-strand breaks. J Cell Biol. 2009;187:319–26. doi: 10.1083/jcb.200908074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van Attikum H, Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009;19:207–17. doi: 10.1016/j.tcb.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 65.Huen MS, Huang J, Yuan J, Yamamoto M, Akira S, Ashley C, Xiao W, Chen J. Noncanonical E2 variant-independent function of UBC13 in promoting checkpoint protein assembly. Mol Cell Biol. 2008;28:6104–12. doi: 10.1128/MCB.00987-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stewart GS, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136:420–34. doi: 10.1016/j.cell.2008.12.042. [DOI] [PubMed] [Google Scholar]
- 67.Doil C, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435–46. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 68.Wu J, Huen MS, Lu LY, Ye L, Dou Y, Ljungman M, Chen J, Yu X. Histone ubiquitination associates with BRCA1-dependent DNA damage response. Mol Cell Biol. 2009;29:849–60. doi: 10.1128/MCB.01302-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bekker-Jensen S, Rendtlew Danielsen J, Fugger K, Gromova I, Nerstedt A, Bartek J, Lukas J, Mailand N. HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat Cell Biol. 2010;12:80–6. doi: 10.1038/ncb2008. sup pp 1–12. [DOI] [PubMed] [Google Scholar]
- 70.Galanty Y, Belotserkovskaya R, Coates J, Polo S, Miller KM, Jackson SP. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature. 2009;462:935–9. doi: 10.1038/nature08657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morris JR, et al. The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature. 2009;462:886–90. doi: 10.1038/nature08593. [DOI] [PubMed] [Google Scholar]
- 72.Kim H, Huang J, Chen J. CCDC98 is a BRCA1-BRCT domain-binding protein involved in the DNA damage response. Nat Struct Mol Biol. 2007;14:710–5. doi: 10.1038/nsmb1277. [DOI] [PubMed] [Google Scholar]
- 73.Liu Z, Wu J, Yu X. CCDC98 targets BRCA1 to DNA damage sites. Nat Struct Mol Biol. 2007;14:716–20. doi: 10.1038/nsmb1279. [DOI] [PubMed] [Google Scholar]
- 74.Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194–8. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Deng CX. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006;34:1416–26. doi: 10.1093/nar/gkl010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Durant ST, Nickoloff JA. Good timing in the cell cycle for precise DNA repair by BRCA1. Cell Cycle. 2005;4:1216–22. doi: 10.4161/cc.4.9.2027. [DOI] [PubMed] [Google Scholar]
- 77.Huang J, Huen MS, Kim H, Leung CC, Glover JN, Yu X, Chen J. RAD18 transmits DNA damage signalling to elicit homologous recombination repair. Nat Cell Biol. 2009;11:592–603. doi: 10.1038/ncb1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huyen Y, et al. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432:406–11. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- 79.Sanders SL, Portoso M, Mata J, Bahler J, Allshire RC, Kouzarides T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004;119:603–14. doi: 10.1016/j.cell.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 80.Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–73. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bird AW, et al. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature. 2002;419:411–5. doi: 10.1038/nature01035. [DOI] [PubMed] [Google Scholar]
- 82.Ikura T, et al. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. 2000;102:463–73. doi: 10.1016/s0092-8674(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 83.Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, Herceg Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol. 2006;8:91–9. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- 84.Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A. 2005;102:13182–7. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ikura T, et al. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol Cell Biol. 2007;27:7028–40. doi: 10.1128/MCB.00579-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pandita TK, Richardson C. Chromatin remodeling finds its place in the DNA double-strand break response. Nucleic Acids Res. 2009;37:1363–77. doi: 10.1093/nar/gkn1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–7. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 88.Conaway RC, Conaway JW. The INO80 chromatin remodeling complex in transcription, replication and repair. Trends Biochem Sci. 2009;34:71–7. doi: 10.1016/j.tibs.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 89.Morrison AJ, Shen X. Chromatin remodelling beyond transcription: the INO80 and SWR1 complexes. Nat Rev Mol Cell Biol. 2009;10:373–84. doi: 10.1038/nrm2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Morrison AJ, Highland J, Krogan NJ, Arbel-Eden A, Greenblatt JF, Haber JE, Shen X. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell. 2004;119:767–75. doi: 10.1016/j.cell.2004.11.037. [DOI] [PubMed] [Google Scholar]
- 91.van Attikum H, Fritsch O, Hohn B, Gasser SM. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell. 2004;119:777–88. doi: 10.1016/j.cell.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 92.Lan L, Nakajima S, Komatsu K, Nussenzweig A, Shimamoto A, Oshima J, Yasui A. Accumulation of Werner protein at DNA double-strand breaks in human cells. J Cell Sci. 2005;118:4153–62. doi: 10.1242/jcs.02544. [DOI] [PubMed] [Google Scholar]
- 93.Karmakar P, et al. BLM is an early responder to DNA double-strand breaks. Biochem Biophys Res Commun. 2006;348:62–9. doi: 10.1016/j.bbrc.2006.07.037. [DOI] [PubMed] [Google Scholar]
- 94.Zeitlin SG, Baker NM, Chapados BR, Soutoglou E, Wang JY, Berns MW, Cleveland DW. Double-strand DNA breaks recruit the centromeric histone CENP-A. Proc Natl Acad Sci U S A. 2009;106:15762–7. doi: 10.1073/pnas.0908233106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Celeste A, et al. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol. 2003;5:675–9. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- 96.Yuan J, Chen J. MRE11-RAD50-NBS1 complex dictates DNA repair independent of H2AX. J Biol Chem. 2010;285:1097–104. doi: 10.1074/jbc.M109.078436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bassing CH, Alt FW. H2AX may function as an anchor to hold broken chromosomal DNA ends in close proximity. Cell Cycle. 2004;3:149–53. doi: 10.4161/cc.3.2.689. [DOI] [PubMed] [Google Scholar]
- 98.Bassing CH, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci U S A. 2002;99:8173–8. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lieber MR. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu Rev Biochem. 2010;79 doi: 10.1146/annurev.biochem.052308.093131. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol. 2009;16:814–8. doi: 10.1038/nsmb.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jazayeri A, Balestrini A, Garner E, Haber JE, Costanzo V. Mre11-Rad50-Nbs1-dependent processing of DNA breaks generates oligonucleotides that stimulate ATM activity. Embo J. 2008;27:1953–62. doi: 10.1038/emboj.2008.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 2004;304:93–6. doi: 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- 103.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–4. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 104.Williams RS, et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Buis J, et al. Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell. 2008;135:85–96. doi: 10.1016/j.cell.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Adams MM, Carpenter PB. Tying the loose ends together in DNA double strand break repair with 53BP1. Cell Div. 2006;1:19. doi: 10.1186/1747-1028-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Orsburn B, Escudero B, Prakash M, Gesheva S, Liu G, Huso DL, Franco S. Differential requirement for H2AX and 53BP1 in organismal development and genome maintenance in the absence of PARP1. Mol Cell Biol. 2010 doi: 10.1128/MCB.00091-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Difilippantonio S, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–33. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol. 2004;5:481–7. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- 110.Ward IM, et al. 53BP1 is required for class switch recombination. J Cell Biol. 2004;165:459–64. doi: 10.1083/jcb.200403021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Boisvert FM, Rhie A, Richard S, Doherty AJ. The GAR motif of 53BP1 is arginine methylated by PRMT1 and is necessary for 53BP1 DNA binding activity. Cell Cycle. 2005;4:1834–41. doi: 10.4161/cc.4.12.2250. [DOI] [PubMed] [Google Scholar]
- 112.Pryde F, Khalili S, Robertson K, Selfridge J, Ritchie AM, Melton DW, Jullien D, Adachi Y. 53BP1 exchanges slowly at the sites of DNA damage and appears to require RNA for its association with chromatin. J Cell Sci. 2005;118:2043–55. doi: 10.1242/jcs.02336. [DOI] [PubMed] [Google Scholar]
- 113.Lee JH, Goodarzi AA, Jeggo PA, Paull TT. 53BP1 promotes ATM activity through direct interactions with the MRN complex. EMBO J. 2010;29:574–85. doi: 10.1038/emboj.2009.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wei L, Lan L, Hong Z, Yasui A, Ishioka C, Chiba N. Rapid recruitment of BRCA1 to DNA double-strand breaks is dependent on its association with Ku80. Mol Cell Biol. 2008;28:7380–93. doi: 10.1128/MCB.01075-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, Lukas J. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol. 2006;173:195–206. doi: 10.1083/jcb.200510130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kim JS, Krasieva TB, Kurumizaka H, Chen DJ, Taylor AM, Yokomori K. Independent and sequential recruitment of NHEJ and HR factors to DNA damage sites in mammalian cells. J Cell Biol. 2005;170:341–7. doi: 10.1083/jcb.200411083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–6. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- 118.Yin B, et al. Histone H2AX stabilizes broken DNA strands to suppress chromosome breaks and translocations during V(D)J recombination. J Exp Med. 2009;206:2625–39. doi: 10.1084/jem.20091320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Petersen S, et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–5. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Reina-San-Martin B, Difilippantonio S, Hanitsch L, Masilamani RF, Nussenzweig A, Nussenzweig MC. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J Exp Med. 2003;197:1767–78. doi: 10.1084/jem.20030569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Xie A, Puget N, Shim I, Odate S, Jarzyna I, Bassing CH, Alt FW, Scully R. Control of sister chromatid recombination by histone H2AX. Mol Cell. 2004;16:1017–25. doi: 10.1016/j.molcel.2004.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yuan J, Chen J. N terminus of CtIP is critical for homologous recombination-mediated double-strand break repair. J Biol Chem. 2009;284:31746–52. doi: 10.1074/jbc.M109.023424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.New JH, Sugiyama T, Zaitseva E, Kowalczykowski SC. Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein A. Nature. 1998;391:407–10. doi: 10.1038/34950. [DOI] [PubMed] [Google Scholar]
- 124.Sugiyama T, Kantake N, Wu Y, Kowalczykowski SC. Rad52-mediated DNA annealing after Rad51-mediated DNA strand exchange promotes second ssDNA capture. Embo J. 2006;25:5539–48. doi: 10.1038/sj.emboj.7601412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liu JS, Kuo SR, Melendy T. DNA damage-induced RPA focalization is independent of gamma-H2AX and RPA hyper-phosphorylation. J Cell Biochem. 2006;99:1452–62. doi: 10.1002/jcb.21066. [DOI] [PubMed] [Google Scholar]
- 126.Sy SM, Huen MS, Zhu Y, Chen J. PALB2 regulates recombinational repair through chromatin association and oligomerization. J Biol Chem. 2009;284:18302–10. doi: 10.1074/jbc.M109.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316:1202–5. doi: 10.1126/science.1139621. [DOI] [PubMed] [Google Scholar]
- 128.Riballo E, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell. 2004;16:715–24. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 129.Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. [PMC free article] [PubMed] [Google Scholar]
- 130.de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, Lehmann AR, Hoeijmakers JH. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol. 2000;10:479–82. doi: 10.1016/s0960-9822(00)00447-4. [DOI] [PubMed] [Google Scholar]
- 131.Zhu J, Petersen S, Tessarollo L, Nussenzweig A. Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr Biol. 2001;11:105–9. doi: 10.1016/s0960-9822(01)00019-7. [DOI] [PubMed] [Google Scholar]
- 132.Xiao Y, Weaver DT. Conditional gene targeted deletion by Cre recombinase demonstrates the requirement for the double-strand break repair Mre11 protein in murine embryonic stem cells. Nucleic Acids Res. 1997;25:2985–91. doi: 10.1093/nar/25.15.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Luo G, Yao MS, Bender CF, Mills M, Bladl AR, Bradley A, Petrini JH. Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc Natl Acad Sci U S A. 1999;96:7376–81. doi: 10.1073/pnas.96.13.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chen PL, et al. Inactivation of CtIP leads to early embryonic lethality mediated by G1 restraint and to tumorigenesis by haploid insufficiency. Mol Cell Biol. 2005;25:3535–42. doi: 10.1128/MCB.25.9.3535-3542.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ludwig T, Chapman DL, Papaioannou VE, Efstratiadis A. Targeted mutations of breast cancer susceptibility gene homologs in mice: lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes Dev. 1997;11:1226–41. doi: 10.1101/gad.11.10.1226. [DOI] [PubMed] [Google Scholar]
- 136.Sharan SK, et al. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature. 1997;386:804–10. doi: 10.1038/386804a0. [DOI] [PubMed] [Google Scholar]
- 137.Tsuzuki T, et al. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc Natl Acad Sci U S A. 1996;93:6236–40. doi: 10.1073/pnas.93.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Barlow C, et al. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–71. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 139.Elson A, Wang Y, Daugherty CJ, Morton CC, Zhou F, Campos-Torres J, Leder P. Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci U S A. 1996;93:13084–9. doi: 10.1073/pnas.93.23.13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 1996;10:2411–22. doi: 10.1101/gad.10.19.2411. [DOI] [PubMed] [Google Scholar]