

Abstract
A novel biopolymer-based antioxidant, chitosan conjugated with gallic acid (chitosan galloylate, chitosan– GA), is proposed. Electron paramagnetic resonance (EPR) demonstrates a wide range of antioxidant activity for chitosan–GA as evidenced from its reactions with oxidizing free radicals, that is, 1,1-diphenyl-2-picryl-hydrazyl (DPPH), horseradish peroxidase (HRP)–H2O2, carbon-centered alkyl radicals, and hydroxyl radicals. The EPR spectrum of the radical formed on chitosan–GA was attributed to the semiquinone radical of the gallate moiety. The stoichiometry and effective concentration (EC50) of the DPPH free radical with chitosan–GA show that the radical scavenging capacity is maintained even after thermal treatment at 100 °C for an hour. Although the degree of substitution of GA on chitosan was about 15%, its antioxidant capacity, that is, the reaction with carbon-centered and hydroxyl radicals, is comparable to that of GA.
Keywords: Chitosan, Polysaccharide, Gallic acid (GA), Antioxidant, Electron paramagnetic resonance (EPR)
1. Introduction
Chitin and chitosan (Fig. 1a), next to cellulose, together make up the second-most naturally abundant copolysaccharides. These polymers are obtained from the shells of crustaceans, the cuticles of insects, and the cell walls of fungi and yeasts. The polymer skeleton, which consists of pyranose rings of β-(1→4)-2-acetamido-2-deoxy-β-D-glucose (chitin) and β-(1→4)-2-amino-2-deoxy-β-Dglucose (chitosan) linked with a glycosidic linkage, dictates many advantageous properties, for example; biodegradability,1 biocompatibility, 2 bioactivity,3 non-toxicity,4 antimicrobial activities,5 and ion absorption ability.6 Based on those specific properties, chitin and chitosan have received much attention as biomaterials for value-added products, especially foods7 and drugs8 including cosmetics.9
Figure 1.

Chemical structures of (a) chitosan; (b) fully protonated gallic acid; and (c) chitosan–GA.
The development of chitosan and its derivatives is of interest in many areas, especially biomedicine. When considering biological molecules such as lipids, proteins, enzymes, DNA, and RNA, we must be able to recognize how free radical reactions of these substances are involved in human health issues, for example, aging, atherosclerosis, carcinogenesis, and especially in food deterioration. 10–14 Thus, the development and application of either synthetic or natural antioxidants is of high importance in the reduction of unwanted free-radical-mediated oxidations. Recently, chitosan has been exploited as an alternative natural antioxidant as well as for its antibacterial15 and antimutagenetic16 properties. On the basis of its structure and the nature of free-radical reactions, chitosan should form the most stable macro-radicals via the hydroxyl and amino groups;17 however, Alexandrova et al.18 reported that the antioxidant activity of chitosan was essentially zero. Li et al.19 took into account the very high EC50 (1.12 × 106 μgmL−1) of chitosan from EPR studies to point out that inter- and intramolecular hydrogen bonds blunt free-radical reactions. Thus, the development of chitosan for antioxidant activity has to overcome: (i) poor solubility; (ii) chemical inertness based on the strong inter- and intramolecular hydrogen bonds network; and (iii) poor H-atom-donating ability to serve as a good chain-breaking antioxidant.
Accordingly, the introduction of extended hydroxyl or carboxylic groups onto chitosan decreased the EC50 to as low as about 3 × 102 μg mL−1.20 It is important to note that radical scavenging, in most cases, is in an aqueous environment, especially if we consider the use of chitosan in polysaccharide drugs with antioxidant activities. Water-soluble chitosans and their antioxidant activities have been studied. For example, Xing et al.21,22 have reported that a series of chitosan sulfate derivatives with radical scavenging ability for superoxide anion and hydroxyl radicals have EC50 values of around 0.01–0.03 and 1.3–3.3 mg mL−1, respectively, which is 102–105 times lower than that of chitosan. The fact that chitosan shows a strong metal-ion chelating ability suggests that it may be a potential natural product antioxidant, especially when we consider that the deactivation of the catalytic activity of metal ions, especially Fe2+/Fe3+, is a key aspect of effective antioxidant systems. 23,24 Based on this, Xing et al.25 and Guo et al.26 investigated the ferrous ion-chelating effect of chitosan sulfate to determine if deoxyribose oxidation was effectively inhibited. In fact, trace transition metals are also an important factor for prooxidative/antioxidative balance in the certain systems to maximize stability against oxidation.24,27 Trace levels of metals are inevitably present in many foodstuffs27 and cosmetic products28 as well as in thehumanbody.29 These metals represent the main catalysts of oxidation of lipids and other molecules in foods and related products.30 Chitosan may be effective as a chelating agent in providing antioxidant capabilities.
In cases (ii) and (iii) above, it is important to note that chitosan must be functionalized by adding some specific groups in order to enable effective H-atom donation. To our knowledge, this strategy has not yet been reported. Molecules that could be functionalized onto chitosan, either synthetic compounds, such as butylated hydroxyanisole (BHA), and butylated hydroxytoluene (BHT) or natural compounds, such as those found in green tea, rosemary, and tannin, are good candidates.
Gallic acid (GA) or 3,4,5-trihydroxybenzoic acid, a natural phenolic antioxidant from grape seeds, is attractive since the low O–H bond dissociation enthalpy (BDE)31 allows it to serve as both a one- and two-electron reductant. Gallic acid transfers a hydrogen atom,32 yielding a delocalized aroxyl radical,33 that is, a semiquinone free radical,34 which is stable over a wide pH range.35 Belin et al.36 reported that the formation of ester or amide bonds of synthetic gallic acid derivatives seem well adapted to increase the antioxidant efficiency of the galloyl group based on a push–pull effect. This antioxidant effect could prove beneficial to address numerous disease states, including cardiovascular disease. However, other studies have reported the cytotoxic activity of gallic acid.37,38 Additionally, gallic acid has been reported to have both pro-oxidant and antioxidant properties.39
We have succeeded in introducing gallic acid onto chitosan to obtain chitosan gallate (chitosan–GA) (Fig. 1c). This substance shows swelling in water, implying loosely bonded inter- and intramolecular hydrogen bonding due to the bulky gallic acid moiety.40 We propose that this chitosan derivative not only improves the solubility of chitosan, but also provides antioxidant potential via the gallate functional group. In the present work we investigate the ability of chitosan gallate to serve as an antioxidant by studying its reactions with reactive species using electron paramagnetic resonance (EPR) spectroscopy.
2. Experimental
2.1. Chemicals
Chitosan with percent degree of deacetylation (%DD) of 92 (Mv = 9.5 × 105 dalton) was provided from Seafresh Chitosan (Lab) Company Limited, Thailand. Gallic acid, horseradish peroxidase (HRP, Type I), α-(4-pyridyl-1-oxide)-N-tert-butylnitrone (POBN), 4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy (Tempol), and 5,5-dimethyl-1-pyrroline-1-oxide (DMPO) were purchased from Sigma– Aldrich, USA. Zinc metal (dust), hydrogen peroxide solution (30%), sodium hydroxide solution (1 M), ferrous ammonium sulfate (Fe(NH4)2·6H2O), and methanol were bought from Fisher Scientific Co., Fair Lawn, NJ, USA. 2,2′-Azobis(2-amidinopropane) · HCl (AAPH) was from Polysciences, Inc., USA. 1-Ethyl-3-(3′-dimethylaminopropyl) carbodiimide (EDC) and 1,1-diphenyl-2-picryl-hydrazyl (DPPH) were from TCI, Tokyo, Japan. N-Hydroxysuccinimide (NHS) was from Wako, Osaka, Japan. Ethanol and acetic acid (AR grade) were from Lab Scan, Co., Ltd, Thailand. Phosphate-buffered saline (PBS), pH 7.4 was prepared from KH2PO4 (210 mgL−1), Na2HPO4 (407 mgL−1), and NaCl (9000 mg/L). All chemicals were used without further purification.
2.2. Instruments
Fourier-transform infrared spectroscopy (FTIR) analysis was carried out by using a Thermo Nicolet Nexus 670 instrument with 32 scans, and 2 cm−1 resolution. Elemental analysis (EA) was carried out using a Perkin–Elmer 2400 CHN analyzer with a combustion temperature at 950 °C under air atmosphere with O2 as a combustion gas (flow rate 20 mL min−1) and He as a carrier (flow rate 200 mL min−1). 13C Cross-polarization magic-angle spinning nuclear magnetic resonance (13C CP/MAS NMR) spectra were taken on a Bruker DPX-300 at 23 ± 1 °C. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker UXNMR/XWIN-NMR Avance DPX400 at 70 °C using CD3COOD/D2O as a solvent. All EPR spectra were obtained from a Bruker EMX using version 3.2 software. A transverse mode (TM110) cavity and flat cell were used for aqueous solution samples, and a high-sensitivity (HS) cavity was used in the kinetic studies. The magnetic field was calibrated by using the g-value of a solid DPPH standard. Typical EPR spectrometer settings were: receiver gain, 2.5 × 105; microwave power, 10 mW; modulation frequency, 100 kHz; frequency, 9.780 GHz; modulation amplitude, 0.10 G; time constant, 40.96 ms; number of scans, 20; and scan rate, 10 G/21 s. UV radiation was generated by a UV PhotoMax, Model 60100 (Oriel Corp., Stratford, USA) with a 75W Xe bulb. The current density and applied voltage were 70 A and 35 V, respectively.
2.3. Preparation of chitosan conjugated with gallic acid (chitosan–GA)
The conjugation of chitosan with gallic acid in detail was reported previously.40 Here, the optimum conditions were used: Gallic acid (GA, 3 mol equiv to chitosan, 0.510 g) was reacted with reprecipitated chitosan (1 mol, 0.163 g) in the presence of 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide (EDC, 3 mol equiv to GA, 1.725 g) and N-hydroxysuccinimide (NHS, 3 mol equiv to GA, 1.036 g) in EtOH (50 mL). The heterogeneous mixture was left to react at ambient temperature for 24 h. The crude product was filtered and washed thoroughly with EtOH five times to obtain chitosan–GA (Fig. 1).
IR (KBr): 1640 (amide I), 1580 (amide II), 1730 (C=O ester), 3464 (OH), and 895 (pyranose ring). 1H NMR (2% CD3COOD in D2O): δ 7.6 (2H, s, H-a), 2.5 (3H, s, H-Ac), 5.3 (1H, d, H-1), 3.0 (1H, d, H-2), 3.9–4.5 (5H, m, H-3 to H-6 of pyranose ring). 13C NMR: δ 160–180 (C=O ester), 140 (aromatic), 104.6 (C-1 of pyranose ring) 75.5–82.5 (C-3 to C-5 of pyranose ring), 57.3–61.1 (C-2 and C-6 of pyranose ring). Anal. Calcd for (C27H23O16N)0.92-(C21H21O12N)0.08: C, 54.04; H, 3.84; N, 1.62; O, 40.50. Found for GA:chitosan = 3:1 (DS ~15.62%) C, 36.93; H, 6.70; N, 7.07; O, 49.30.
2.4. Formation of semiquinone radicals by air oxidation in alkaline condition
As GA is air-oxidized easily at high pH, NaOH (1 M, 300 μL) was added to chitosan–GA (30 mM, 200 μL) or GA (2.5 mM, 200 μL) at room temperature to generate GA-semiquinone radicals. The solution was placed in a quartz flat cell, and EPR spectra were recorded immediately using a TM110 cavity.
2.5. Formation of semiquinone radicals by horseradish peroxidase–H2O2
To an aqueous solution of chitosan–GA (40 mM, 375 μL), 10 μL of phosphate-buffered saline (PBS, pH 7.4) solution containing H2O2 (20 mM) was added. The mixture was well stirred before adding the buffer solution of HRP (0.04 μgmL−1, 10 μL). PBS buffer was added for a final volume of 500 μL. The mixture was rapidly transferred to a quartz flat cell, and EPR spectra were recorded by using a TM110 cavity. Similarly, GA (20 mM, 50 μL) in PBS was prepared and EPR spectra were recorded.
2.6. Formation of semiquinone radicals by H-atom transfer to carbon-centered radicals
To an aqueous solution of chitosan–GA (40 mM, 300 μL), 25 μL of PBS buffer (pH 7.4) containing AAPH (300 mM) was added. PBS buffer was added for a final volume of 500 μL. The mixture was transferred to a quartz flat cell and placed to a TM110 cavity followed by UV exposure. EPR spectra were recorded during UV exposure at room temperature. Similarly, GA (20 mM, 50 μL) in PBS was prepared and EPR spectra were recorded.
2.7. DPPH radical-scavenging assay
Chitosan–GA (250 μL) solutions with concentrations of 0.09, 0.18, 0.27, 0.37, 0.55, 0.76, and 0.92 mg mL−1 (480–4850 μM) were mixed with methanolic DPPH· solution (DPPH, 200 μM, 750 μL). Each mixture was reacted at room temperature under subdued light and left for 30 min before collecting EPR spectra. Relative percent of DPPH· scavenging capacity was calculated from [(h0 − hc)/h0] × 100, where hc and h0 are the peak heights belonging to the middle peak of DPPH· spectrum of with and without antioxidants, respectively. Similarly, chitosan solution (in 2%v/v HOAc) with the same concentrations as those of chitosan–GA solutions was prepared and studied. In the case of GA, the solutions were prepared at the concentrations of 3.4 × 10−3, 6.8 × 10−3, 8.16 × 10−3, 10.2 × 10−3, 13.6 × 10−3 mg mL−1 (20–80 μM).
2.8. Stoichiometric reaction of GA, chitosan–GA, and the Zn-reduced chitosan–GA with DPPH·
Fully reduced chitosan–GA was prepared by treating chitosan– GA aqueous solution (4840 μM) with zinc powder (7.84 mg, 6 mol equiv to chitosan–GA). The reduced chitosan–GA solutions (250 μL) at 970, 1450, 1940, and 2900 μM were mixed with methanolic DPPH solution (200 μM, 750 μL) and left for 30 min under subdued light at room temperature before transferring to a quartz flat cell to collect EPR spectra using a TM110 cavity. Similarly, GA aqueous solutions (250 μL) at 20, 40, 60, and 80 μM and chitosan solutions (250 μL) at 970, 1450, 1940, and 2900 μM, were prepared and EPR spectra were recorded.
2.9. Carbon-centered radical-scavenging assay
Carbon-centered radicals were generated from AAPH via UV irradiation. POBN was used as a spin-trapping agent.43 A series of chitosan–GA aqueous solutions (0.76 mg mL−1 (4 mM), for 2, 3, 6, 15, 30, 60, 90, 120, 150, and 300 μL) were mixed with PBS buffer (pH 7.4) solution containing AAPH (300 mM, 25 μL) and POBN (500 mM, 25 μL). PBS buffer was added for a final volume of 500 μL. Each mixture was transferred to a quartz flat cell. The EPR spectrum of POBN/R· spin adducts was measured while being exposed to UV radiation directly in the TM110 cavity at room temperature. The relative percent of carbon-centered radical scavenging capacity was calculated from [(h0 − hc)/h0] × 100, where hc and h0 are the peak heights of the low-field line of the POBN/R· spectrum (aN = 15.6 G and aH = 2.6 G) with and without antioxidant, respectively. Similarly, the studies on carbon-centered radical scavenging capacity of GA were carried out by using 0.85 mg mL−1 (5 mM) of GA for 1, 2, 5, 10, 20, 30, 40, 50, 100, and 240 μL.
2.10. Hydroxyl radical-quenching assay via the Fenton reaction
Ferrous ammonium sulfate was dissolved in distilled water, and the pH was adjusted to pH 2.5 by HCl (1 M) to prepare Fe2+ stock solution (100 μM).41 chitosan–GA aqueous solutions (1.89mg mL−1 (10 mM), for 2.5, 5, 10, 25, 50, 75, 100, 150, 250, 250, or 350 μL) were mixed with PBS buffer solution (pH 7.4) containing H2O2 (250 μM, 20 μL) and DMPO (1 M, 5 μL). The Fe2+ stock solution (10 μL) was added into the mixtures, and an additional amount of PBS buffer was added for a total volume of 500 μL. The mixtures were transferred to a quartz flat cell and incubated for 3 min before collecting the EPR spectra. The relative percent of hydroxyl radical-quenching capacity was calculated from [(h0 − hc)/h0] × 100, where hc and h0 are peak heights of the second low-field line of the DMPO/HO· spin adduct (aN = aH = 14.9 G) with and without antioxidants, respectively. Studies on hydroxyl radical-quenching capacity of GA were carried out with the same procedures.
2.11. Thermal stability
Chitosan–GA aqueous solutions (4840 μM) were incubated at 100 °C under aerobic conditions for 30 and 60 min. Chitosan–GA solutions were diluted to 0.09, 0.18, 0.27, 0.37, 0.55, 0.76, and 0.92 mg mL−1 (480–4850 μM). An aliquot (250 μL) of each chitosan– GA concentration was mixed with DPPH methanolic solution (200 μM, 750 μL). The reactions were carried out at room temperature under subdued light and left for 30 min before collecting the EPR spectra. In the case of GA, similar procedures were carried out but using 100 μM of GA incubated at 100 °C and diluted to 1.73 × 10−3, 4× 10−3, 6.8 × 10−3, 8.16 × 10−3, 10.2 × 10−3, 13.6 × 10−3 mg mL−1 (10–80 μM).
3. Results and discussion
3.1. Conjugation of chitosan with GA and structural characterization
The chitosan gallate derivative (chitosan–GA) was prepared according to the previous report.40 The conjugation of chitosan with GA most likely occurs between the carboxylic acid group of GA and the amino group (C-2 of pyranose ring of chitosan) to obtain an amide linkage or the hydroxyl group (C-3 and C-6 of pyranose ring of chitosan) to obtain an ester linkage. Compared to the FTIR spectrum of chitosan, the compound obtained shows the new significant peaks at 1640 and 1730 cm−1, confirming both amide and ester linkages (Fig. 2A).
Figure 2.

(A) FTIR spectra and (B) 13CNMR spectra of (a) chitosan and (b) chitosan–GA.
The 1H NMR spectrum of GA:chitosan showed a new peak at 7.6 ppm belonging to the protons of phenoxyl GA. In addition, 13C CP/MAS NMR confirms the aromatic ring of galloyl group on chitosan at 140 ppm (C=C, aromatic) and at 160.2 ppm (C=O, ester) (Fig. 2B).
When %DS was evaluated by elemental analysis using the C/N ratio, it was found to be ~15.6% (for 3:1 GA:chitosan molar ratio). Based on this degree of substitution, we calculated the average molecular weight per monosaccharide unit of chitosan–GA to be 187.5 g/mol and used this value for preparing chitosan–GA solutions (molar concentrations).
It is important to develop water solubility together along with antioxidant activity; we examined the solubility of the compound by dispersing it in water and observing the transmittance at 500 nm. Chitosan–GA, obtained from the 3:1 GA:chitosan molar ratio, appeared as an almost clear solution having a transmittance above 80%.
3.2. Semiquinone radical formation of chitosan–GA
For the radical-scavenging processes of phenolic antioxidants (ArOH), H-atom donation is the dominant mechanism. This reaction occurs by two separate, but related, pathways to form ArO·. Eq. 1 is the simple single step H-atom-transfer whereas Eq. 2 represents the stepwise electron-transfer/proton-transfer:
| (1) |
| (2) |
The ArO· radical is relatively stable, indicating that ArOH can serve as a good chain-breaking antioxidant.42,43 Semiquinone radicals (GA-semiquinone radicals) are in general less reactive than similar phenoxyl radicals.44,45
In order to determine if the galloyl group of chitosan–GA is capable of forming the GA-semiquinone radical, chitosan–GA and GA were incubated in alkaline aqueous solution (NaOH, pH 13). The EPR spectrum of the GA-semiquinone free radical derived from chitosan–GA (Fig. 3A(a)) shows the same g-value and gives the same three-line EPR spectrum (aH (2) = 1.07 G) with 1:2:1 intensity ratio as the free radical derived from GA (Fig. 3A(b)), as seen previously.46,47
Figure 3.

(A) EPR spectra generated in alkaline solution. (a) Chitosan–GA; and (b) GA in alkaline solution (NaOH, pH 13). (B) EPR spectra of GA radicals formed by the HRP– H2O2 system: (a) GA(4 mM), HRP (0.8 μg/mL), andH2O2 (0.4 mM)in PBS solution (pH 7.4) (g = 2.0054, and ); (b) as in (a), but without HRP and H2O2 (g = 2.0054; (c) as in (a), but without HRP; and (d) as in (a), but without GA (g = 2.0052). (C) EPR spectra of chitosan–GA radicals formed by the HRP–H2O2 system: (a) chitosan–GA (30 mM), HRP (0.8 μg/mL), and H2O2 (0.4 mM) in PBS solution (pH 7.4) (g = 2.0055); (b) as in (a), but without HRP and H2O2 (g = 2.0055); (c) as in (a), but without HRP, and (d) as in (a), but without chitosan–GA (g = 2.0052).
The spectrum of the radicals derived from chitosan–GA shows additional unidentified peaks consistent with hyperfine splitting of a single-spin one-half species, . Although further studies are needed, we suspect that it might come from the functional groups of chitosan, that is, the hydroxyl and amino groups.
To probe for radical formation from chitosan–GA at neutral pH, we used the HRP–H2O2 oxidizing system and GA in various conditions. For example, when GA is subjected to the HRP–H2O2 system at pH 7.4, a second free radical with the same g-value (2.0054) as in the alkaline pH was observed (Fig. 3B(a)). This spectrum shows a doublet of triplets with hyperfine splitting from three protons ( and ).
As shown in Figure 3B(b) and (c), only low-level background EPR spectra are observed in the absence of HRP–H2O2 (b) or HRP (c). When the system contains only HRP–H2O2 (without GA), no signal is observed (Fig. 3B(d)). This confirms that the hyperfine splitting is from GA and not from any other components. The weak EPR signals observed in the absence of HRP–H2O2 are due to the auto-oxidation of GA in atmospheric oxygen at pH >7, as has been observed by Friedman and Jurgens.48
Chitosan–GA with HRP–H2O2 generates an EPR spectrum (Fig. 3C(a)) with the same g-value as that of the GA (Fig. 3B(a)), indicating that chitosan–GA generates GA-semiquinone radical. Figures 3C(b) and (c)) demonstrate that the EPR signal in Figure 3C(a) is from chitosan–GA not from the HRP–H2O2.
Comparing Figure 3C(a) to B(a), it is clear that chitosan–GA gives spectra with a broad line-width and no resolvable hyperfine splittings. This may be due to the fact that the galloyl group is tethered to the chitosan polymer chain; as a result, the isotropic motion of the generated GA radical is restricted.
3.3. Semiquinone radicals formation of chitosan–GA by H-atom transfer to carbon-centered radicals
Lipid peroxidation in a biological system is a good example in which the function of antioxidants is needed.43 In order to investigate the reactivity of chitosan–GA as an antioxidant, the free radicals produced by the azo initiator AAPH were used as a model. AAPH is a hydrophilic compound that produces carbon-centered radicals via thermal decomposition.49 Typically thermal decomposition of AAPH is relied upon to produce a flux of carbon-centered radicals, Eq. 3 without UV light. However, this flux was too low for detecting the free radicals of interest by EPR. We, then, used the UV irradiation (directly while the sample was in the EPR cavity) to increase the flux of radicals derived for AAPH, Eqs. (3)–(6):
| (3) |
| (4) |
| (5) |
| (6) |
POBN is known as an excellent spin trap for carbon-centered radicals, producing persistent spin adducts (Eq. 7).43 Using EPR spin trapping with POBN, the thermal decomposition of AAPH at room temperature yields an EPR spectrum with hyperfine splitting constants (aN = 15.6 G and aH = 2.6 G), consistent with that of a POBN spin adduct of a carbon-centered radical, POBN/R·.49,50 If UV increases the rate of radical production from AAPH, then the formation of POBN spin adducts will be increased. When samples were exposed to UV, the EPR intensity of POBN/R· was increased, reaching a concentration of 10 μM within 200 s. (POBN/R· was quantified using of 3-carboxy-proxyl as a standard.51) When GA was incubated with AAPH and exposed to UV light in the EPR cavity, the EPR spectrum ( and ) obtained (Fig. 4A(a)) was similar to that of the GA radical observed in the HRP–H2O2 system (Fig. 3A(a)). This implies that GA undergoes hydrogen atom (or e−/H+) transfer to repair the free radicals generated by AAPH, leading to a stable GA-derived semiquinone free radical. Only a small doublet spectrum of GA-semiquinone radicals was observed in the absence of UV (Fig. 4A(b)) or AAPH (Fig. 4A(c)). This confirms that the EPR spectrum in Figure 4A(a) is generated from GA. The GA-semiquinone radicals would be formed according to Eqs. (5) and (6):
Figure 4.

EPR spectra generated using AAPH. (A) (a) GA (4 mM) and AAPH (15 mM) in PBS solution (pH 7.4) with UV exposure at room temperature (g = 2.0054, ) ; (b) as in (a), but without UV exposure (g = 2.0054); and (c) as in (a), but without AAPH (g = 2.0054). (B) (b) chitosan–GA (24 mM) and AAPH (15 mM) in PBS solution (pH 7.4) with UV exposure at room temperature (g = 2.0055); (b) as in (a), but without UV exposure; and (c) as in (a), but without AAPH.
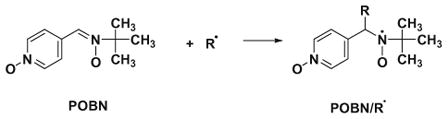 |
(7) |
Chitosan–GA in the presence of the radical-generating system of AAPH/UV gives a broad EPR spectrum (Fig. 4B(a)) with the same g-values (g = 2.0055) as that of chitosan–GA in the HRP–H2O2 system (Fig. 3B(a)). This indicates that chitosan–GA serves as a hydrogen atom donor to repair the oxidizing radicals generated by AAPH (Eqs. (5) and (6)). If any of the components are omitted, only ‘baseline’ spectra are observed (Fig. 4B(b) and (c)). This confirms that: (i) the exposure of chitosan–GA to UV does not induce significant radical formation; and (ii) chitosan–GA exhibits antioxidant activity to repair oxidizing free radicals.
3.4. DPPH radical-scavenging capacity
1,1-Diphenyl-2-picryl-hydrazyl free radical (DPPH·) is a relatively stable free radical; thus, EPR is a useful and practical approach for the evaluation of the antioxidative potential of GA and chitosan–GA.32,52 Antioxidant capacity was evaluated by examining the dose–response of percent of free radical scavenging capacity versus [antioxidant] (Fig. 5). In the case of chitosan, no reduction of DPPH· was observed, even though its concentration was as high as 0.23 mg mL−1 (1200 μM). This may be due in part to the inhibition of radical scavenging because of inter- and intramolecular hydrogen bonding. The scavenging capacity of chitosan– GA for DPPH· increases as its concentration increases, up to 87% when the concentration reached 0.23 mg mL−1 (1200 μM). The EC50 value, which expresses the antioxidant concentration to reduce the radicals by 50%, is a good quantitative indicator of antioxidant capacity. As seen in Figure 5, the EC50 for scavenging of DPPH· by chitosan–GA is 0.14 mg mL−1 (740 μM). For comparison, it should be noted that for sulfated chitosans reported by Xing et al.,25 for example, TSCTS (the sulfated chitosan of C3 and C6 sulfonation) showed a good radical-scavenging ability, 0.025 mg mL−1 (EC50), whereas the other sulfated chitosan derivatives exhibited DPPH· scavenging ability less than 20% at concentrations as high as 0.40 mg mL−1. Taking this into consideration, the introduction of an H-atom donating group, that is, gallic acid, onto chitosan is a good strategy to develop a chitosan derivative with robust antioxidant capacity.
Figure 5.

Scavenging capacity of chitosan–GA (▲), chitosan (■) and GA (●) on DPPH radical. Results are mean ± SD (n = 3).
It is important to verify that the hydroxyl groups of the GA on chitosan were not oxidized to quinones during preparation and storage, as this would lower the apparent DPPH·-radical scavenging ability of chitosan–GA. If quinones were present, the introduction of Zn0 will reduce these quinones to hydroquinones. This would increase the observed stoichiometry for the reaction of chitosan–GA with DPPH·. When chitosan–GA was treated with Zn0, the stoichiometry of the reaction of chitosan–GA with DPPH· was found to be the same, that is, 0.11 (Table 1). This confirms that the OH groups of the GA on chitosan–GA were not oxidized to quinones during synthesis and storage. It can be concluded that the scavenging ability of our chitosan–GA preparation was not reduced due to any prior oxidation of the galloyl groups.
Table 1.
Stoichiometry and EC50 of reaction between DPPH free radicals and chitosan–GA under experimental conditions
| Conditions | Stoichiometrya | EC50 (mg mL−1) |
|---|---|---|
| Room temperature | 0.11 ± 0.01 | 0.137 ± 0.004 |
| Reduced chitosan–GAb | 0.11 ± 0.03 | 0.138 ± 0.007 |
| Incubated for 30 minc | 0.08 ± 0.03 | 0.144 ± 0.009 |
| Incubated for 60 minc | 0.10 ± 0.01 | 0.141 ± 0.007 |
All values reported are mean ± SD, n = 5.
The number of DPPH free radicals scavenged by a molecule of antioxidant.
Antioxidant samples were reacted with Zn0 metal 6 mol equiv to antioxidant.
Antioxidant samples were incubated at 100 °C.
3.5. Carbon-centered radical-scavenging capacity
In free-radical mediated oxidations, especially lipid peroxidation, an oxidant abstracts a hydrogen atom from a carbon–hydrogen bond resulting in the formation of carbon-centered radicals. These carbon-centered radicals react rapidly with oxygen to form peroxyl radicals that propagate the chain reactions of lipid peroxidation. 53 To examine the efficiency of chitosan–GA in repairing carbon-centered radicals, the EPR signal intensities of POBN/R· with various concentrations of chitosan–GA were compared. A comparative study of free GA and chitosan was done. It is important to note that the higher the antioxidant potential of ArOH (Eq. 5), the smaller the EPR signal of POBN/R· (Eq. 7). The EPR signal intensity in the system with chitosan–GA, GA, or chitosan (Fig. 6A(a), (b), or (c)) is smaller than that observed without chitosan– GA, GA, or chitosan (Fig. 6A(d)). This confirms the carbon-centered radical scavenging capacity of chitosan–GA.
Figure 6.

Chitosan–GA scavenges carbon-centered radicals. (A) EPR spectra of POBN/R· spin adduct from: (a) the mixture of chitosan–GA (0.01 mM), POBN (25 μM), AAPH (15 mM), and PBS buffer pH 7.4 with UV exposure; (b) as in (a), but with GA (0.01 mM) instead of chitosan–GA; (c) as in (a), but with chitosan (0.01 mM); (d) as in (a), but without chitosan–GA, GA, and chitosan; (e) as in (a), but without AAPH; and (f) as in (b), but without AAPH. (B) Carbon-centered radicals scavenging capacity of GA (●), chitosan–GA (▲), and chitosan (■). Results are mean ± SD (n = 3).
In order to confirm that UV enhances only the rate of radical generation from AAPH, and not from chitosan–GA or GA, a sample without AAPH was examined. As no spin adduct formation is observed when samples (chitosan–GA or GA) were exposed to UV light (Fig. 6A(e) and (f)), it can be concluded that the carbon-centered radicals trapped by POBN were from AAPH.
Figure 6B shows that the scavenging capacity of chitosan–GA and GA for the carbon-centered radicals generated by AAPH reaches 100% when the concentration approaches 0.45 mg mL−1 (2,400 μM)and 0.19 mg mL−1 (1000 μM), respectively. The EC50 values for chitosan–GA, and GA are about 0.021 mg mL−1 (110 μM) and 0.0019 mg mL−1 (10 μM), respectively. In the case of the original chitosan, the carbon-centered radical scavenging capacity is very low (~10%), even when the concentration is almost 0.5 mg mL−1. In terms of the GA moiety, 110 μM of chitosan–GA would be 16 μM in GA (110 μM × 15% DS); the reducing capacity of chitosan– GA is only 1.6 times less than that of GA. This demonstrates that chitosan–GA is quite an effective radical scavenger when we consider that the substitution of the galloyl group is only about 15%.
In a previous report, chitosan sulfate was shown to have an antioxidant activity for carbon-centered radicals, scavenging 60% when its concentration was about 0.125 mg mL−1,54 whereas to scavenge carbon-centered radicals to the same extent with chitosan– GA required only 0.038 mg mL−1 (200 μM). Thus, the conjugation of antioxidant molecule, that is, GA, on chitosan is an excellent route to enhance the antioxidant activity to chitosan.
3.6. Hydroxyl radical-quenching assay via the Fenton reaction
By preventing the formation of hydroxyl radical complexation of iron by chitosan has been reported to enhance its antioxidant properties.55 Here, we examined the scavenging capacity of chitosan– GA for hydroxyl radicals generated via the Fenton reaction (Eq. 8):
| (8) |
It is important to note that the very high reactivity of HO· and its broad linewidth preclude its direct detection by EPR. Thus, the EPR spin-trapping agent, 5,5-dimethyl-1-pyrroline-1-oxide (DMPO) was used to detect HO·. DMPO forms a long-lived spin adduct with HO· (DMPO/HO·; aN = aH = 14.9 G), allowing determination of the relative amount of HO· in the system, Eq. 9:
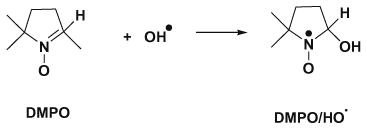 |
(9) |
The decrease in EPR signal intensity for DMPO/HO· with increasing concentration of chitosan–GA, confirms the quenching capacity of chitosan–GA for HO· (Fig. 7). Figure 7 also provides some important information about the mechanism of chitosan as related to the Fenton reaction. That is, the percentage of the HO· quenching capacity is as high as 90% at a GA concentration of 0.19 mg mL−1 (1000 μM), and starts to decrease when the GA concentration is higher than this. In order to verify this phenomenon, an additional experiment was carried out in which the concentration of H2O2 was increased. This produced the expected increase in HO·. Therefore, the percentage of hydroxyl radical scavenging might be expected to decrease. However, at this higher level of H2O2 a similar trend was found for the HO· quenching capacity. This might be due to the pro-oxidant chemistry of GA in the Fenton reaction. As GA is an excellent reducing agent, it can reduce Fe3+ to Fe2+ (Eq. 10), and as a result increase overall formation of HO· (Eq. 9). Gil-Longo and González-Vázquez56 reported that
Figure 7.

Hydroxyl radical-quenching capacity of chitosan–GA (▲), GA with 10 μM H2O2 (●), GA with 50 μMH2O2 (◆), and chitosan (■). Results are mean ± SD (n = 3).
| (10) |
gallic acid can be a pro-oxidant polyphenol in physiological solutions. This may not occur as readily with chitosan–GA as chitosan contains the metal-chelating function, that is, –NH2, that could blunt the catalytic activity of iron.
The capacity to quench HO· increases as the concentration of chitosan–GA increases, reaching 90% at a concentration above 0.38 mg mL−1 (2000 μM); in contrast the capacity to quench HO· by chitosan is minimal (Fig. 7). Chitosan and some derivatives have the ability to chelate metal ions,23,57 especially transition metals. Hence, we suspect that chitosan may form a complex with Fe2+ or Fe3+, resulting in the blunting of HO· formation via the Fenton reaction. Andres and Arthur55 have reported that the chelation of Fe2+ leads to a decrease of hydroxyl radical generation. Here, we suspect that the amino group of chitosan effectively functions as an Fe2+ chelating group by forming a chitosan–Fe2+ complex.58 Moseley et al.59 have shown a propensity for derivatization of chitosan with SO4 − at amino groups. As reported initially by Buettner et al.60 and supported by Decker,27 the unwanted reactions of pro-oxidant metals can be addressed by appropriate chelating agents; these agents can inactivate the catalytic activity of these metals, or physically minimize interactions between pro-oxidant metals and oxidizable targets, for example, lipids. A synergistic effect might also occur as gallic acid enhances the water solubility of chitosan. These reports support our observations on the HO·-scavenging ability of chitosan– GA and suggest that the metal-chelating ability of chitosan as an important factor. This is an interesting issue that will require a detailed investigation to underpin the development of chitosan–GA as a bio-chelating antioxidant.
The EC50 values of chitosan–GA and GA for decreasing [DMPO/HO·] are 0.066 mg mL−1 (350 μM) and 0.009 mg mL−1 (50 μM), respectively (Fig. 7). In comparison, it is important to note that the EC50 in term of GA moiety of chitosan–GA (350 μM of chitosan– GA is 52 μM in GA moieties) is almost equal to that of GA (50 μM). In other words, the antioxidant activity with respect to hydroxyl radical of chitosan–GA is comparable to that of GA, even though the degree of substitution of the galloyl group on chitosan is only 15%.
Guo et al.26 reported that to reduce HO· by 25% required 2.5 mg mL−1 of chitosan Schiff bases; whereas, in our case with chitosan–GA, only 0.057 mg mL−1 (300 μM) is required. Also, the EC50 of sulfated chitosan reported by Xing et al.21 was 3.3 mg mL−1, while chitosan–GA requires only 0.067 mg mL−1. Thus, chitosan– GA has excellent radical-scavenging ability compared to other derivatives.
3.7. Thermal stability
The antioxidant potential, after being subjected to 100 °C for 30 or 60 min, was examined by following the reduction of DPPH·. It was found that the stoichiometries (DPPH·:GA) were 0.08 ± 0.03 and 0.10 ± 0.01, respectively. The EC50 was calculated to be 0.144 ± 0.009 and 0.141 ± 0.007 (mg mL−1), respectively (Table 1). It is clearly seen that before and after incubation at 100 °C for both times, the stoichiometry and the EC50 are almost the same. This suggests the possibility of using chitosan–GA as a natural antioxidant polymer product for applications at higher temperatures.
4. Conclusions
This work demonstrates that by functionalizing chitosan with a radical scavenging group, that is, GA, a novel polysaccharide antioxidant (chitosan gallate) could be created. Even if only 15% of the chitosan units had a GA moiety, the galloyl group could effectively transfer a H-atom, forming stable semiquinone radicals. These stable radicals could be found in all of the oxidizing systems examined. As compared to other chitosan derivatives, such as sulfated chitosan (EC50 = 0.1 mg mL−1 for R·54 and 3.269 mg mL−1 for HO·21), chitosan– GA (EC50 = 0.021 mg mL−1 for R· and 0.066 mg mL−1 for HO·) showed a lower EC50, approximately five times lower for R· and 50 times lower for HO·. Based on the comparative EC50 of chitosan– GA/GA, chitosan–GA exhibited radical scavenging ability on the order of HO· (0.066/0.063 mg mL−1 = 1.05 times) >R· (0.021/0.0127 mg mL−1 = 1.65 times) >DPPH· (0.14/0.01 mg mL−1 = 14 times). The equivalent EC50 of chitosan–GA and GA for the hydroxyl radical-quenching system suggested the synergistic function of chitosan in retarding the pro-oxidation of GA in the Fenton reaction. The equivalent DPPH radical-scavenging ability of chitosan–GA, before and after thermal treatment, confirmed the stability of the galloyl group on chitosan–GA. As both chitosan and GA are natural products, chitosan–GA is expected to provide a novel, practical, naturally desired polysaccharide antioxidant in applications where safety reasons are a main concern, for example in food preservation, cosmetics, and biomedical products.
Oxidative reactions cause damage to lipids and proteins thereby influencing food quality. The use of antioxidants is the most common approach to increase the oxidative stability of foods.27 Actually there is no one antioxidant that can control all the factors involved in the oxidative stability of foods. Thus, multicomponent antioxidant systems are used. Controlling oxidations via free-radical scavengers is often dependent on controlling the contributions of metal catalysts (e.g., pro-oxidant metals.), concentration and activity. Chitosan gallate may be a multifunctional antioxidant having chelating abilities as well as chain-breaking capacity, water solubility, and natural product properties. Moreover, as the efficiency of phenolic free-radical scavengers is also dependent on their volatility and heat stability, the conjugation of GA onto chitosan may facilitate and prolong its stability. This novel type of polysaccharide antioxidant could be a model for developing antioxidant additives for foods as well as for cosmetic and even medical products in which adventitious, catalytic metals are a concern.
Acknowledgments
The authors (W.P.) gratefully acknowledge the Commission on Higher Education, Ministry of Education, the Faculty of Science, Kasetsart University, and the National Research Council of Thailand, for the research funds. The authors acknowledge the EPR Facility, The University of Iowa, Iowa City, IA, USA, for providing the EPR measurements.
Abbreviations
- Chitosan–GA
chitosan gallate
- GA
gallic acid
- HRP
horseradish peroxidase
- POBN
α-(4-pyridyl-1-oxide)-N-tert-butylnitrone
- Tempo
4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy
- DMPO
5,5-dimethyl-1-pyrroline-1-oxide
- AAPH
2,2′-azobis (2-amidinopropane)·HCl
- DPPH
1,1-diphenyl-2-picryl-hydrazyl
- NHS
N-hydroxysuccinimide
- EDC
1-ethyl-3-(3′-dimethylaminopropyl) carbodiimide
- PBS
phosphate-buffered saline
Contributor Information
Wanvimol Pasanphan, Email: wanvimol.p@ku.ac.th.
Suwabun Chirachanchai, Email: csuwabun@chula.ac.th.
References
- 1.Yamamoto H, Amaika M. Macromolecules. 1997;30:3936–3937. [Google Scholar]
- 2.Richardson SC, Kolbe HV, Duncan R. Int J Pharm. 1999;178:231–243. doi: 10.1016/s0378-5173(98)00378-0. [DOI] [PubMed] [Google Scholar]
- 3.Dumitriu S, Popa MI, Cringu A, Stratone A. Colloid Polym Sci. 1989;267:595–599. [Google Scholar]
- 4.Marguerite R. Prog Polym Sci. 2006;31:603–632. [Google Scholar]
- 5.Kendra DF, Hadwiger LA. Exp Mycol. 1984;8:276–281. [Google Scholar]
- 6.Peniche CC, Alwarez LW, Arguelles MW. J Appl Polym Sci. 1987;46:1147–1150. [Google Scholar]
- 7.Knorr D. In: Biotechnology of Marine Polysaccharides. Colwell RR, Pariser ER, Sinskey AJ, editors. Hemisphere; New York: 1985. p. 313. [Google Scholar]
- 8.Kifune K. In: Advances in Chitin and Chitosan. Brine CJ, Sanford PA, Zikakis JP, editors. Elsevier; Essex: 1992. p. 9. [Google Scholar]
- 9.Dong GK, Young J, Changyong C, Sung HR, Seong KK, Mi KJ, Jae WN. Int J Pharm. 2006;319:130–138. [Google Scholar]
- 10.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. VIII Clarendon Press; Oxford: 1989. [Google Scholar]
- 11.Rice-Evans CA, Diplock AT. Free Radical Biol Med. 1993;15:77–96. doi: 10.1016/0891-5849(93)90127-g. [DOI] [PubMed] [Google Scholar]
- 12.Hussain SP, Hofseth LJ, Harris CC. Nat Rev Cancer. 2003;3:276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 13.Brash DE, Havre PA. Proc Natl Acad Sci USA. 2002;99:13969–13971. doi: 10.1073/pnas.232574399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fiorentino A, D’Abrosca B, Pacifico S, Cefarelli G, Uzzo P, Monaco P. Bioorg Med Chem Lett. 2007;17:636–639. doi: 10.1016/j.bmcl.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 15.Sudarshan NR, Hoover DG, Knorr D. Food Biotechnol. 1992;6:257–272. [Google Scholar]
- 16.Kogan G, Skorik YA, Žitnanová I, Križková L, Ďuračková Z, Gomes CAR, Yatluk YG, Krajčovič J. J Toxicol Appl Pharm. 2004;201:303–310. doi: 10.1016/j.taap.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 17.Xue CH, Yu GL, Hirata T, Terao J, Lin H. Biosci, Biotechnol, Biochem. 1998;2:206–209. doi: 10.1271/bbb.62.206. [DOI] [PubMed] [Google Scholar]
- 18.Alexandrova VA, Obukhova GV, Domnina NS, Topchiev DA. Macromolecules Symposium. 1999;144:413–422. [Google Scholar]
- 19.Li WJ, Jiang X, Xue PH, Chen SM. Chin Sci Bull. 2002;11:887. [Google Scholar]
- 20.Sun T, Xie W, Xu P. Carbohydr Polym. 2004;58:379–382. [Google Scholar]
- 21.Xing R, Liu S, Yu H, Guoa Z, Lia Z, Lia P. Carbohydr Polym. 2005;61:148–154. [Google Scholar]
- 22.Xing R, Liu S, Yu H, Zhang Q, Li Z, Li P. Carbohydr Res. 2004;339:2515–2519. doi: 10.1016/j.carres.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 23.Ngah WSW, Ghani SA, Kamari A. Bioresour Technol. 2005;96:443–450. doi: 10.1016/j.biortech.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 24.Buettner GR. J Biochem Biophys Methods. 1988;16:27–40. doi: 10.1016/0165-022x(88)90100-5. [DOI] [PubMed] [Google Scholar]
- 25.Xing R, Yu H, Liu S, Zhang W, Zhang Q, Lia Z, Lia P. Bioorg Med Chem. 2005;13:1387–1392. doi: 10.1016/j.bmc.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 26.Gou Z, Xing R, Liu S, Yu H, Wang P, Li C, Li P. Bioorg Med Chem Lett. 2005;15:4600–4603. doi: 10.1016/j.bmcl.2005.06.095. [DOI] [PubMed] [Google Scholar]
- 27.Decker EA. Trends Food Sci Technol. 1998;9:241–248. [Google Scholar]
- 28.Nnorom IC, Igwe JC, Oji-Nnorom CG. African J Biotechnol. 2005;4:1133–1138. [Google Scholar]
- 29.Halliwell B, Gutteridge JMC. Mol Aspects Med. 1985;8:89–193. doi: 10.1016/0098-2997(85)90001-9. [DOI] [PubMed] [Google Scholar]
- 30.Fomuso LB, Corredig M, Akoh CC. J Agric Food Chem. 2002;50:7114–7119. doi: 10.1021/jf020256j. [DOI] [PubMed] [Google Scholar]
- 31.Ji HF, Zhang HY, Shen L. Bioorg Med Chem Lett. 2006;16:4095–4098. doi: 10.1016/j.bmcl.2006.04.096. [DOI] [PubMed] [Google Scholar]
- 32.López M, Martínez F, Valle CD, Ferrit M, Luque R. Talanta. 2003;60:609–616. doi: 10.1016/S0039-9140(03)00191-7. [DOI] [PubMed] [Google Scholar]
- 33.Jorgensen LV, Madsen HL, Thomsen MK, Dragsted LO, Skibsted LH. Free Radical Res. 1999;30:207–220. doi: 10.1080/10715769900300231. [DOI] [PubMed] [Google Scholar]
- 34.Rice ECA, Miller J, Paganga G. Free Radical Biol Med. 1996;20:933–956. doi: 10.1016/0891-5849(95)02227-9. [DOI] [PubMed] [Google Scholar]
- 35.Huang D, Ou B, Prior RL. J Agric Food Chem. 2005;53:1841–1856. doi: 10.1021/jf030723c. [DOI] [PubMed] [Google Scholar]
- 36.Belin F, Barthelemy P, Ruiz K, Lacombe JM, Pucci B. Helv Chim Acta. 2003;86:247–265. [Google Scholar]
- 37.Qiu X, Takemura G, Koshiji M, Hayakawa Y, Kanoh M, Maruyama R. Heart Vessels. 2000;15:90–99. doi: 10.1007/s003800070038. [DOI] [PubMed] [Google Scholar]
- 38.Nogaki A, Satoh K, Iwasaka K, Takano H, Takahama M, Ida Y. Anticancer Res. 1998;18:3487–3491. [PubMed] [Google Scholar]
- 39.Yen GC, Duh PD, Tsai HL. Food Chem. 2002;79:307–313. [Google Scholar]
- 40.Pasanphan W, Chirachanchai S. Carbohydr Polym. 2008;72:169–177. [Google Scholar]
- 41.Qian SY, Buettner GR. Free Radical Biol Med. 1999;26:1447–1456. doi: 10.1016/s0891-5849(99)00002-7. [DOI] [PubMed] [Google Scholar]
- 42.Madsen HL, Bertelsen G. Trends Food Sci Technol. 1995;6:271–277. [Google Scholar]
- 43.Venkataraman S, Schafer FQ, Buettner GR. Antioxid Redox Signaling. 2004;6:631–638. doi: 10.1089/152308604773934396. [DOI] [PubMed] [Google Scholar]
- 44.Wardman P. J Phys Chem Ref Data. 1989;18:1637–1755. [Google Scholar]
- 45.Kruk I, Aboul EHY, Michalska T, Lichszteld K, Ktadna A. Luminescence. 2005;20:81–89. doi: 10.1002/bio.808. [DOI] [PubMed] [Google Scholar]
- 46.Adams M, Blois MS, Sands RH. J Chem Phys. 1958;28:774. [Google Scholar]
- 47.Oniki T, Takahama U. J Wood Sci. 2004;50:545–547. [Google Scholar]
- 48.Friedman M, Jurgens HS. J Agric Food Chem. 2000;48:2101–2110. doi: 10.1021/jf990489j. [DOI] [PubMed] [Google Scholar]
- 49.Li ASW, Cummings KB, Roethling HP, Buettner GR, Chignell CF. J Magn Reson. 1988;79:140–142. [Google Scholar]
- 50.Buettner GR. Free Radical Biol Med. 1987;3:259–303. doi: 10.1016/s0891-5849(87)80033-3. [DOI] [PubMed] [Google Scholar]
- 51.Venkataraman S, Martin SM, Schafer FQ, Buettner GR. Free Radical Biol Med. 2000;29:580–585. doi: 10.1016/s0891-5849(00)00404-4. [DOI] [PubMed] [Google Scholar]
- 52.Williams WB, Cuvelier ME, Berset C. Lebensm -Wiss Technol. 1995;28:25–30. [Google Scholar]
- 53.Gardner HW. Free Radical Biol Med. 1989;7:65–86. doi: 10.1016/0891-5849(89)90102-0. [DOI] [PubMed] [Google Scholar]
- 54.Huang R, Mendis E, Kima SK. Int J Biol Macromol. 2005;36:120–127. doi: 10.1016/j.ijbiomac.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 55.Andres AC, Arthur IC. Free Radical Biol Med. 2004;36:1303–1316. doi: 10.1016/j.freeradbiomed.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 56.Gil-Longo J, González-Vázquez C. J Nutr Biochem. 2009;xx:xxx–xxx. doi: 10.1016/j.jnutbio.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 57.Burke A, Yilmaz E, Hasirci N, Yilmaz O. J Appl Polym Sci. 2002;84:1185–1192. [Google Scholar]
- 58.Guzman J, Saucedo I, Revilla J, Navarro R, Guibal E. Int J Biol Macromol. 2003;33:57–65. doi: 10.1016/s0141-8130(03)00067-9. [DOI] [PubMed] [Google Scholar]
- 59.Moseley R, Waddington R, Evans P, Halliwell B, Embery G. Biochim Biophys Acta, Gen Sub. 1995;1244:245–252. doi: 10.1016/0304-4165(95)00010-9. [DOI] [PubMed] [Google Scholar]
- 60.Buettner GR, Oberley LW, Leuthauser SWHC. Photochem Photobiol. 1978;28:693–695. doi: 10.1111/j.1751-1097.1978.tb07001.x. [DOI] [PubMed] [Google Scholar]