

Abstract
β-adrenergic receptor (βAR) dysfunction in acute myocardial infarction (MI) is associated with elevated levels of the G protein-coupled receptor kinase-2 (GRK2), which plays a key role in heart failure progression. Inhibition of GRK2 via expression of a peptide βARKct transferred by molecular cardiac surgery with recirculating delivery (MCARD) may be a promising intervention. Five sheep underwent scAAV6-mediated MCARD delivery of βARKct and five received no treatment (control). After a 3 week period, the branch of the circumflex artery (OM1) was ligated. Quantitative PCR data showed intense βARKct expression in the left ventricle (LV). Circumferential fractional shortening was 23.4±7.1% (baseline) vs. −2.9±5.2% (p<0.05) in the control at 10 weeks. In the MCARD-βARKct group this parameter was close to baseline. The same trend was observed with LV wall thickening. Cardiac index fully recovered in the MCARD-βARKct group. LV end-diastolic volume and LV end-diastolic pressure did not differ in both groups. MCARD-mediated βARKct gene expression results in preservation of regional and global systolic function after acute MI without arresting progressive ventricular remodeling.
Keywords: acute myocardial infarction, gene therapy , β-adrenergic receptors , molecular cardiac surgery
Introduction
Ischemic heart disease (IHD) remains a leading cause of morbidity and mortality worldwide. Despite recent advances in modern treatment for IHD, pharmacological therapies reduce symptoms but do not stop and/or reverse the pathophysiologic mechanisms of disease on a molecular level. Thus cardiac-specific gene therapy offers the possibility of improving the cellular abnormalities associated with the pathogenesis of ventricular dysfunction and represents a potential treatment that has recently elicited much attention [1-4]. The ideal gene therapy strategy targets physiologically relevant signaling pathways. One promising molecular target is the βAR signaling system, which plays a pivotal role in cardiac function. During IHD numerous βAR signaling abnormalities occur at the molecular level including decreased βAR density and responsiveness, decreased adenyl cyclase activity, disruption of intracellular Ca2+ cycling and elevated G-protein-coupled receptor kinase 2 (GRK2) activity [5]. Indeed, the role of alterations in cardiac β-adrenergic receptor signaling in small animal models of myocardial infarction is complex. Pathological effects have been demonstrated after chronic βAR activation or as a consequence of overexpressed cardiac β1- or β2ARs [6-8]. In contrast, transgenic rats and rabbit models have demonstrated improved cardiac function following overexpression of the β2AR or expression of a carboxyl-terminal portion of GRK2 (βARKct) which competitively inhibits binding of GRK2 to the Gβγ-protein subunit [9,10]. These potentially conflicting findings need to be reconciled in a relevant preclinical large animal model. Molecular cardiac surgery with recirculating delivery (MCARD) is a novel surgical technique which isolates the cardiac from the systemic circulation, allowing for retrograde infusion of the gene of interest, extended vector residence time in the coronary system, and increased myocardial transcapillary gradient with minimal collateral tissue transgene expression [11]. Here we couple the MCARD delivery technique with a highly promising potential therapeutic transgene, βARKct, delivered using a scAAV6 vector into the coronary vasculature before cardiac ischemia. The aim of the present study was to assess the potential of βARKct pretreatment as a protective mechanism to restore or minimize systolic and diastolic contractile abnormalities in response to acute coronary occlusion; as well as to clarify what happens with βAR signaling alterations in an ovine model of transmural myocardial infarction.
Methods
Animals and study design
All animals received humane care in compliance with the NIH and guidelines established by the Institutional Animal Care and Use Committee of the University of Pennsylvania. Ten Dorsett male sheep (1-1.2 year old, weighing 38.8±3.72 kg) were randomly divided into 2 groups. Five sheep underwent MCARD delivery of βARKct (MCARD-βARKct group) and five received no treatment (control group). After a 3 week period, the first branch of the circumflex artery (OM1) was ligated proximally in all ten sheep. All ten animals survived to euthanasia at 10 weeks (7 weeks post infarct). Baseline studies (normal sheep) were pooled as the common reference group prior to intervention for both (MCARD-βARKct) and the control infarct groups.
Vector design and production
A recombinant self-complementary adeno-associated viral vector, serotype 6, encoding the carboxyl terminus of human β-adrenergic receptor kinase under the control of the human immediate early cytomegalovirus gene promoter with a splice donor/acceptor sequence and polyadenylation signal from the human-globin gene was constructed (scAAV6/CMV/βARKct) [9].
Surgical protocol
Surgical details of the MCARD procedure have been described previously [11]. We briefly discuss the main points. A median sternotomy was performed, and a transepicardial echocardiogram used to evaluate global cardiac function prior to cannulation. After heparin administration, the right carotid artery was cannulated for systemic perfusion. The aortic root vent, superior vena cava (SVC) cannula, inferior vena cava (IVC) cannula and retrograde cardioplegia catheter were placed, and cardiopulmonary bypass was initiated. The right and the left azygous (hemiazygous) veins were ligated. The left ventricle (LV), right ventricle (RV) and aortic root vents were connected via a Y connector to the venous limb of the cardiac venous return circuit. The arterial limb of the cardiac circuit was connected to the coronary sinus catheter. The aorta was cross clamped. Cold crystalloid cardioplegia was delivered antegrade. The heart was isolated by tightening the SVC and IVC snares and cross clamping the pulmonary artery. The virus solution consisting of 1014 genome copies of scAAV6/CMV/βARKct was injected into the retrograde catheter and recirculated for 20 minutes. The coronary circuit was then flushed to wash out residual vector. The aortic cross clamp was removed, and rewarming was initiated. Post-procedure transepicardial echocardiogram assessed maintenance of global cardiac function. As the animal was being weaned from bypass, the first branch of the obtuse marginal artery was identified, and a Prolene snare stitch was loosely applied, advanced exteriorly through the chest wall, and buried in the subcutaneous fascia. The animal was subsequently weaned completely from CPB and closed in standard fashion.
Creation of acute transmural myocardial infarction in control group
One hour prior to creation of infarct, a central venous infusion was started with amiodarone and lidocaine to prevent cardiac arrhythmias. After thoracotomy the proximal portion of the first branch of the circumflex artery was identified and ligated with prolene suture. ECG changes were noted, and the surrounding tissue discoloration confirmed infarct creation. The thoracotomy incision was closed. All animals received critical cardiac postoperative care without incident.
Creation of acute transmural myocardial infarction in MCARD-βARKct group
The anesthesia, monitoring and pharmacologic preparation of these animals was identical to the control group. The subcutaneous space on the left chest wall containing the snare around OM1 was identified and opened, and the snare was tightened, creating myocardial infarction. ECG changes were recorded. The incision was repaired in standard fashion. All animals were survived to euthanasia.
Echocardiography
We performed echocardiography in all animals with a 7-MHz transducer (Acuson, Mountain View, CA). Using two-dimensional, spectral Doppler and color flow Doppler modalities, a comprehensive assessment of cardiac structures and function was obtained. Images were acquired in short and long axis at the high, middle and low papillary muscle levels.
Magnetic resonance imaging (MRI) analysis of cardiac hemodynamics
CINE cardiac MRI series were generated for each animal at baseline and at 10 weeks. LV pressure-gated images were recorded with a Millar catheter (Millar Instruments, Houston, TX) throughout the MRI. LV volumes over the cardiac cycle were obtained with a custom image analysis package developed at the University of Pennsylvania (ImageJ) and validated with Segment Cardiac MRI software. Pressure data from the Millar catheter was loaded into MATLAB 9.0 (MathWorks, Natick, MA) and designed to calculate hemodynamic parameters for 5-10 consecutive pressure cycles [12].
Hemodynamic assessment
To assess regional systolic function by means of CINE cardiac MRI, we have chosen fractional wall shortening and regional left ventricular wall thickening in myocardial infarct zone. Briefly, a software algorithm analyzed the position changes in epicardial and endocardial myocardial points to derive the infarction zone wall calculations. For the global cardiac function, we used cardiac index and stroke volume index. Diastolic function was evaluated through two parameters: left ventricular diastolic volume index and left ventricular end diastolic pressure.
Western Blotting
Frozen cardiac tissue samples acquired post-euthanasia were disrupted by Omni TH Tissue Homogenizer in RIPA lysis buffer (CST, Danvers, MA) supplemented with Complete Protease Inhibitor Cocktail (La Roche, Basel, Switzerland). Protein extract concentrations were quantified by using Bio-Rad Protein Assay (Bio-Rad Lab, Hercules,CA) according to the manufacturer’s protocol. A total of 150 μg of each protein sample was loaded into the wells and resolved in a NuPAGE 12%-BT gel. For Western-blot analysis, proteins were electrotransferred onto .45 μm Nitrocellulose membranes using the Tris/CAPS buffer system at 0.8 mA/cm2 for one hour by a TE70X Semi-dry Blotter. Immunostaining was done using a rabbit polyclonal anti-GRK2 antibody (SCB, CA, USA) as a primary with Peroxidase conjugated affinipure F(ab’)2 Fragment Goat Anti-rabbit IgG antibody as a secondary and mouse monoclonal anti-GAPDH-Peroxidase conjugate antibody (Sigma, MO, USA). Immunodetection was done using Lumi-Light Western Blotting Substrate and developing using Blue Light Autorad Film (Fisher Scientific, Waltham, MA).
Reverse Transcriptase Real Time Quantitative Polymerase Chain Reaction (RTqPCR)
RT Q-PCR was performed using the MyiQ RT PCR Detection System and analyzed using the MyiQ software package (Bio-Rad Lab, CA). The samples were disrupted in liquid nitrogen using mortar and pestle, followed by homogenization via pipetting. Total RNA was isolated using the RNeasy Fibrous Mini Kit (Quiagen,Valencia,CA), according manufacturing protocol. First strand cDNA synthesis was performed by reverse transcription of 1 μg of total RNA with iScript cDNA Synthesis kit as recommended. Real-Time qPCR analysis was performed in optical 96-well plates using the iQ SYBRGreen Supermix (Bio-Rad Lab, CA). To examine the expression of βARKct, two sets of specific primers were used, including: GRK2-forward – 5′-CCCTCTCACCATCTCTGAG-3′, GRK2-reverse -5′-CGGTTGGGGAACAAGTAGAA-3′ and βARKct – forward (5′- ATGCATGGCTACATGTCCAA-3′), βARKct – reverse 5′-ATCTCCTCCATGGTCAGCAG-3′. The level of 18S rRNA was used to calculate normalized expression. The ΔΔCt analysis was performed by using the IQ5 software.
Measurement of physiological parameters
Routine laboratory values were obtained to assess appropriate physiologic function. Studies have been conducted with Antech Diagnostics Lab (Lake Success, NY).
β-adrenergic receptor density and adenyl cyclase activity
Adenyl cyclase activity was estimated indirectly by measuring cAMP and βAR binding assays from myocardial sarcolemmal membranes, as described previously [9].
Histological examination
Cardiac sections were frozen in embedding compound and sectioned into 5 μm slices. Hematoxylin and eosin and Masson’s trichrome staining were performed on microscopic sections and examined for evidence of myocardial injury. For study of cardiomyocyte hypertrophy we performed morphometric analysis of cross-sectional cardiomyocyte area using phosphotungstic acid-hematoxylin staining. At least 5 images were captured for each tissue field.
Statistical Analysis
Data are presented as mean ± SEM. Single factor analysis of variance was used to compare the values for all cardiac mechanics parameters for the three data sets: Baseline, Control (10 weeks), and MCARD-βARKct (10 weeks). Student’s t test was used to test for significance between two groups. Multiple group comparisons for RT-qPCR and Western Blot expression were also analyzed by single-factor ANOVA. For all analyses, p <0.05 was considered significant.
Results
Characterization of animals and assessment of myocardial Infarction
All of the sheep in this series recovered from anesthesia, received critical cardiac postoperative care without incident and were survived to euthanasia at 10 weeks. Average CPB time was 112 ±10.6 min, and cross clamp time 54±8.6 min. In one case in the MCARD-βARKct group, ~20 min after OM1 ligation, the sheep developed ventricular tachycardia with ventricular fibrillation, which was converted successfully to sinus rhythm. The clinical course of all sheep was benign and none had hemodynamic deterioration or clinical signs of congestive heart failure. All animals demonstrated ECG changes consistent with transmural anterolateral infarction with initial ST segment elevation and inverted T waves in leads I, AVL,V2-V5 and subsequently, pathologic Q waves in V2-V6. We assessed myocardial injury through the analysis of stained histological sections of all parts of the atria and ventricles. Among all sheep had been diagnosed transmural MI and our LV scar quantification approach is depicted in (Fig.1). The total infarct zone based on the MRI scan with gadolinium contrast was 6.5±2.2% for the control group and 6.8±0.9% (p>0.05) for the MCARD-βARKct group as a percentage of the total free left ventricular myocardial volume. These quantitative MRI results were confirmed histologically. The mean pre- and postoperative values of all physiological parameters used to assess lung, kidney, liver and pancreatic function were within normal limits. In all animals, echocardiography revealed normal ventricular and valvular function after MCARD procedure.
Figure 1.



A. Baseline cardiac MRI featuring End diastolic (EDD) and End systolic (ESD) dimensions
B. Control group (10 weeks) cardiac MRI featuring End diastolic (EDD and End systolic dimensions with infarct zone (MI, yellow)
C. MCARD-βARKct group (10 weeks) cardiac MRI featuring End diastolic and End systolic dimensions with infarct zone (MI,yellow)
D. Delayed Enhancement MRI: Myocardial Infarct Characterization
E. Histological Confirmation of Myocardial Infarction Size
Effects of MCARD-βARKct on cardiac and collateral organs gene expression in post-MI hearts
Real time quantitative PCR revealed up to over 300 copies of βARKct mRNA for each copy of GRK2 mRNA in the anterior wall of the left ventricle as well as global transcription of βARKct throughout the left ventricle with no expression in the liver and lung (Fig. 2A). These findings showed intense βARKct expression in the LV anterior and lateral wall, less intense but significant expression in septum. Western blot analysis demonstrated strong, absolute expression of βARKct transgene in all parts of LV, and these results fully confirmed the data obtained by RTqPCR. (Fig. 2B). No βARKct was detectable in the liver and lung by either technique, confirming the remarkable cardiac-specificity and the restriction of collateral expression in other organs of the MCARD platform (Fig. 2C).
Figure 2.

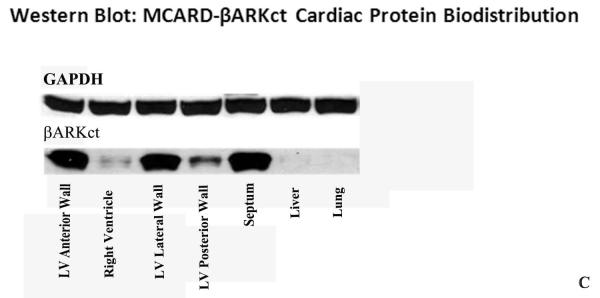
A: RT-qPCR of βARKct expression in the heart and collateral organs in MCARD-βARKct group at 10 weeks.
B: Western blot: βARKct proten biodistribution in the heart and collateral organs in MCARD-βARKct group at 10 weeks. βARKct referenced to GAPDH standard control.
C: Image of Western blot: βARKct proten biodistribution in the heart and collateral organs
Effects of MCARD-βARKct delivery on cardiac cAMP activity and βAR density in post-MI hearts
In the MCARD-βARKct group βAR density in the LV was 352±13 fmol/mg membrane protein, versus 294±21 fmol/mg membrane protein in control group. There was a trend toward higher βAR density after βARKct delivery compared with controls, although these data were not statistically significant. We also did not find a significant difference in cAMP activity between the two groups (119.6±11 vs. 108±19, p>0.05).
Effects of MCARD-βARKct delivery on regional and global LV systolic function in post-MI hearts
Circumferential segmental fractional shortening of the myocardial infarct area was 23.4±7.1% for normal subjects (baseline) vs. −2.9±5.2% (p<0.05) in the control group at 10 weeks. In the MCARD-βARKct group at 10 weeks this parameter was very close to baseline (16.6±5.5%, p>0.05), but was significantly higher than in the control group (p<0.05) (Fig. 3A). The same trend was observed with respect to regional ventricular wall thickening. There was a significant difference between control group vs. MCARD-βARKct group at 10 weeks (-0.42±0.6 mm vs. 1.79±0.4 mm, p<0.05) and control group vs. baseline (-042±0.6 mm vs. 2.5±0.8 mm, p<0.05), but no significant difference between the MCARD-βARKct group and baseline (p>0.05) (Fig. 3B). Cardiac index was significantly decreased in the control group at 10 weeks (1811±170 ml/min·m2) compared to baseline (2402±167 ml/min·m2 , p<0.05). However in the MCARD-βARKct group this parameter did not differ from the baseline (2356±160 ml/min·m2, p<0.05) (Fig. 4A). A similar trend was observed in the stroke volume index, which was significantly reduced in control group at 10 weeks compared to baseline (18.8±2.5 ml/m2 vs. 23.4±1.3 ml/m2, p<0.05), but not in the MCARD-βARKct group (25.4±1.6 ml/m2, p>0.05) (Fig. 4B).
Figure 3.

A: Left ventricular fractional wall shortening in the myocardial infarct area at baseline, control group (10 weeks) and MCARD-βARKct group (10 weeks)
* p<0.05 control vs.baseline; † p<0.05 MCARD-βARKct group vs. control group.
B: Left ventricular transmural wall thickening of the myocardial infarct area at a baseline, control group (10 weeks) and MCARD-βARKct groups (10 weeks)
* p<0.05 baseline vs. control group;
† p<0.05 MCARD-βARKct group vs. control group
Figure 4.

A: Cardiac index at baseline, control group (10 weeks) and MCARD-βARKct group (10 weeks)
* p<0.05 baseline vs. control group;
† p<0.05 MCARD-βARKct group vs. control group
B: Stroke volume index at baseline, control group (10 weeks) and MCARD-βARKct group (10 weeks)
* p<0.05 baseline vs. control group;
† p<0.05 MCARD-βARKct group vs. control group
Effects of MCARD-βARKct delivery on LV dimensions in post-MI hearts
We assessed the effect of βARKct gene expression on remodeling (Table 1). LV end-diastolic volume index in MCARD-βARKct treated sheep was not significantly different from the control group at 10 weeks (48.3±2.8 ml/m2 vs. 42.2±4.5 ml/m2, p>0.05) but in both groups it was significantly increased compared to baseline (37.8±2.0, p<0.05) (Fig. 5A). The same trends were observed with LV end-diastolic pressure. In MCARD-βARKct animals LV end-diastolic pressure was unchanged compared with the control (9.50±0.4 mmHg vs. 10.08 ± 2.2 mmHg, p > 0.05), however it increased significantly by 43,1% and 29.2 % (p<0.05) compared to baseline in each group respectively (Fig. 5B).
Table 1.
Summary of Hemodynamic data
|
Baseline
|
10 Weeks Post MI
|
||
|---|---|---|---|
| Control | MCARD- βARKct | ||
| Global Parameter: | |||
| COi[mL/min ·m2] | 2402±167 | 1811±120* | 2356±160† |
| EDVi[mL] | 38±2 | 42±5* | 51±3* |
| ESVi[mL] | 15±1 | 23±3* | 25±3* |
| SVi[mL/m 2] | 23±1 | 19±3* | 25±2† |
| EF[%] | 62±2 | 44±2* | 52±5* |
| Infarct Region Parameter: | |||
| LV Wall Fractional Wall Shortening[%] | 23±7 | −3±5* | 16±5† |
| LV Wall Thickening [cm] | 2.5±0.8 | ±0.4±0.6* | 1.7±0.5† |
p<0.05 between Baseline and either 10 Week Post MI Control OR MCARD-BARKct Groups
p<0.05 between Control and MCARD-BARKct Groups at 10 Weeks Post MI
Figure 5.

A: Left ventricular end diastolic volume index at baseline, control group (10 weeks) and MCARD-βARKct group (10 weeks)
* p<0.05 baseline vs. control group and MCARD-βARKct group.
B: Left ventricular end diastolic pressure at baseline, control group (10 weeks) and MCARD-βARKct group (10 weeks)
* p<0.05 baseline vs. control group and MCARD-βARKct group.
Effects of MCARD-βARKct delivery on cardiomyocyte hypertrophy at the border zone in post-MI hearts
Morphometric analysis has demonstrated decreased cardiomyocyte hypertrophy in gene-treated animals. A significant difference (p<0.05) was found between the cardiomyocyte cross-sectional area at the border zone for the MCARD-βARKct (110.6±16.4 μm2) and the control group (132.5±23.8 μm2) (Fig.6).
Figure 6.

Average cross sectional cardiac myocyte area in the infarct borderzone
* p<0.05 control group (10 weeks) vs. MCARD-βARKct group (10 weeks)
Discussion
The present study differs from others in several important ways. First, in addition to showing improvement in regional and global contractility, we demonstrate the failure of BARKct expression to attenuate adverse ventricular remodeling in a large animal model, which contrasts with the results reported in rodents [9]. It is likely due to differences in molecular signaling responsible for pathological hypertrophy (remodeling) between large and small animals [13]. This finding is particularly important because current thinking about heart failure has a balanced view of the relative importance of systolic and diastolic dysfunction [14, 15]. The therapeutic strategy used in this study was pretreatment of the left ventricle by means of gene transfer of a GRK2 inhibitor 3 weeks before infarct creation. Although this study is not fully clinically relevant it allowed us to achieve an adequate interval for βARKct expression before acute cardiac ischemia. Western blot analysis demonstrated robust expression of βARKct in the LV myocardium and RTqPCR confirmed robust biodistribution of vector genomes and transcription of βARKct transgene throughout the left ventricle with over 300 copies of the βARKct transgene mRNA for every copy of GRK2 mRNA in both the LV anterior and lateral walls. There was no detectable expression of βARKct in the liver and lung, documenting the exquisite cardiac-specificity of the MCARD procedure. The mean pre- and postoperative values of indices of major organ function were within normal limits. Thus it was demonstrated that MCARD is a safe, highly efficient cardiac-specific therapeutic gene delivery platform.
Circumferential fractional shortening and transmural ventricular wall thickening represent important parameters of regional myocardial systolic function [16, 17]. Recent developments in hardware and software with MRI under real-time imaging allowed us to investigate these parameters in the sheep. The mechanical adaptations following acute transmural MI include LV remodeling and are characterized by increases in LV volumes and progressive worsening of cardiac contractility and impaired relaxation. Using a normal heart sheep model we previously reported that MCARD/βARKct delivery results in robust gene expression and enhances cardiac contractility and lusitropy [18]. The present study for the first time demonstrates that LV-specific, MCARD/AAV6-mediated gene transfer of the βARKct peptide in a model of acute transmural myocardial infarction can significantly preserve regional and global systolic function at 10 weeks but does not slow the progression of ventricular remodeling.
Proposed molecular targets of gene therapy for IHD including both chronic and acute ischemia include those designed to promote angiogenesis and limit reperfusion injury and apoptosis [19]. In this study we have chosen to target the βAR signaling in a large animal model. It is known that the beta-adrenergic receptor system is significantly altered after MI, secondary to various membrane and cytoplasmic biochemical changes, accounting for βAR downregulation and global myocardial desensitization [5]. The activation of βARs increases Ca2+-flux across the sarcolemmal membrane and sarcoplasmic reticulum through their interactions with G-proteins, activation of adenyl cyclase and cAMP-dependent protein kinase and phosphorylation of the target sites thereby increasing cardiac myocyte systolic contractility [20]. The same pathway also occur during ischemia [5, 20]. Results of this study indicated that in contrast to the rescue of several rodent models of heart failure the higher-level expression of BARKct in our model was sufficient for enhancement of systolic function but less for diastolic performance and was not capable of rescuing the progression of cardiac remodeling. The exact mechanisms contributing to ventricular dilatation after ischemia are poorly understood although it is thought that the impairment of isovolumetric ventricular relaxation and decreased compliance of the left ventricle leads to elongation of the cardiac cell, extracellular matrix remodeling, and altered intracellular signaling pathways [14,15]. Thus our finding of persistent and progressive post infarction ventricular dilatation despite robust βARKct expression suggests that additional molecular targets may need to be exploited to limit remodeling in larger animal models. Nonetheless, given, the encouraging results that have been observed in rodent models of heart failure [9], isolated myocytes and transgenic animal models with βARKct gene expression [5], one can readily understand the rationale for coupling the βARKct transgene with the highly efficient and cardiac-specific MCARD gene delivery platform. Although our results largely confirm the potential of βARKct gene expression to preserve systolic function in a large animal post infarction model as was previously observed in rodents, we found discordant results with respect to the preservation of diastolic function and the arrest of maladaptive remodeling [5]. These data underscore the necessity for large animal studies using efficient gene delivery strategies, such as MCARD, to assess the potential benefits of gene therapy in the preclinical setting as a critical adjunct to ongoing clinical trials.
There are several limitations to this study that warrant further investigation. First, we used βARKct gene delivery prior to MI creation and therefore this approach does not have a direct clinical correlate. We also did not compare our data with the MCARD-empty virus delivery, due to the high expense and relatively low yield of this control group since improved myocardial function after cardiopulmonary bypass would be unlikely. Furthermore we did not evaluate a potential dose response of βARKct vector/gene construct.
In summary we found that acute transmural myocardial infarction after ligation of the first branch of circumflex artery in ovine model combined with decreased circumferential fractional shortening and transmural ventricular wall thickening and increased LV end-diastolic volume index and LV end-diastolic pressure. MCARD resulted in robust gene expression with no detectable extracardiac expression and no detrimental effects on major organ function. Additionally, MCARD-mediated βARKct gene expression results in significant preservation of regional and global systolic function after acute MI.
Acknowledgements
This work was sponsored in part by the National Heart, Lung, and Blood Institute (1-R01-HL083078-01A2) and the Gene Therapy Resource Program (GTRP) of the National Heart, Lung and Blood Institute NIH P30-DKO47757. We would like to extend acknowledgments to Charles Yarnall, Alice Isidro and Michael Petrov for their excellent technical assistance. Also we thank Ted Plappert for completing echocardiography studies and James Pilla for MRI studies as well as Natalia Zinchenko and Jane Ingram for performing histological staining. Anthony Carty, JanLee Jensen, James Marx, Jennifer Kirsch and Allison Czarnecki deserve thanks as well for excellent care and handling of the animals in the Biomedical Research Building and Glenolden Vivarium-University of Pennsylvania.
Abbreviations
- βAR
β-adrenergic receptor
- MI
Myocardial infarction
- GRK2
G protein-coupled receptor kinase-2
- βARKct
Carboxyl-terminal portion of GRK2
- MCARD
Molecular cardiac surgery with recirculating delivery
- MRI
Magnetic resonance imaging
- RTqPCR
Reverse Transcriptase Real Time Quantitative Polymerase Chain Reaction
References
- 1.Nayak L, Rosengart TK. Gene therapy for heart failure. Semin Thorac Cardiovasc Surg. 2005;17:343–347. doi: 10.1053/j.semtcvs.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Emani S, Ramlawi B, Sodha NR, Li J, Bianchi C, Sellke FW. Increased vascular permeability after cardiopulmonary bypass in patients with diabetes is associated with increased expression of vascular endothelial growth factor and hepatocyte growth factor. J Thorac Cardiovasc Surg. 2009;138:185–191. doi: 10.1016/j.jtcvs.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bulcao CF, Pandalai PK, D’Souza KM, Merrill WH, Akhter SA. Uncoupling of myocardial beta-adrenergic receptor signaling during coronary artery bypass grafting: the role of GRK2. Ann Thorac Surg. 2008;86:1189–1194. doi: 10.1016/j.athoracsur.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kido M, Sullivan CC, Deutsch R, Jamieson SW, Thistlethwaite PA. Gene transfer of a TIE2 receptor antagonist prevents pulmonary hypertension in rodents. J Thorac Cardiovasc Surg. 2005;129:268–276. doi: 10.1016/j.jtcvs.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 5.Koch WJ, Lefkowitz RJ, Rockman HA. Functional consequences of altering myocardial adrenergic receptor signaling. Annu Rev Physiol. 2000;62:237–260. doi: 10.1146/annurev.physiol.62.1.237. [DOI] [PubMed] [Google Scholar]
- 6.Kudei RK, Iwase M, Uechi M, Vatner DE, Oka N, Ishikawa Y, Shannon RP, Bishop SP, Vatner SF. Effects of chronic beta-adrenergic receptor stimulation in mice. J Mol Cell Cardiol. 1997;29:2735–2746. doi: 10.1006/jmcc.1997.0508. [DOI] [PubMed] [Google Scholar]
- 7.Engelhardt S, Hein L, Wiesmann F, Lohse MJ. Progressive hypertrophy and heart failure in beta 1-adrenergic receptor transgenic mice. Proc Natl Acad Sci U S A. 1999;96:7059–7064. doi: 10.1073/pnas.96.12.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liggett SB, Tepe NM, Lorenz JN, Canning AM, Jantz TD, Mitarai S, Yatani A, Dorn GW. Early and delayed consequences of beta (2)-adrenergic receptor overexpression in mouse hearts: critical role for expression level. Circulation. 2000;101:1707–1714. doi: 10.1161/01.cir.101.14.1707. [DOI] [PubMed] [Google Scholar]
- 9.Rengo G, Lymperopoulos A, Zincarelli C, Donniacuo M, Soltys S, Rabinowitz JE, Koch WJ. Myocardial adeno-associated virus serotype 6-BARKct gene therapy improves cardiac function and normalizes the neurohormonal axis in chronic heart failure. Circulation. 2009;119:89–98. doi: 10.1161/CIRCULATIONAHA.108.803999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tevaearai HT, Walton GB, Keys JR, Koch WJ, Eckhart AD. Acute ischemic cardiac dysfunction is attenuated via gene transfer of a peptide inhibitor of the β-adrenergic receptor kinase (βARK1) J Gene Med. 2005;7:1172–1177. doi: 10.1002/jgm.770. [DOI] [PubMed] [Google Scholar]
- 11.White JD, Thesier DM, Swain JD, Katz MG, Tomasulo KE, Henderson A, Wang L, Yarnall C, Fargnoli A, Sumaroka M, Isidro A, Petrov M, Holt D, Nolen-Walston R, Koch WJ, Stedman HH, Rabinowitz JE, Bridges CR. Myocardial gene delivery using molecular cardiac surgery with recombinant adeno-associated virus vectors in vivo. Gene Ther. 2011;18:546–552. doi: 10.1038/gt.2010.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuehne T, Yilmaz S, Steendijk P, Moore P, Groenink M, Saaed M, Weber O, Higgins CB, Ewert P, Fleck E, Nagel E, Schulze-Neick I, Lange P. Magnetic resonance imaging analysis of right ventricular pressure-volume loops. Circulation. 2004;110:2010–6. doi: 10.1161/01.CIR.0000143138.02493.DD. [DOI] [PubMed] [Google Scholar]
- 13.Katz MG, Fargnoli AS, Tomasulo CE, Pritchette LA, Bridges CR. Model-specific selection of molecular targets for heart failure gene therapy. J Gene Med. 2011;13:573–586. doi: 10.1002/jgm.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krum H, Abraham WT. Heart failure. Lancet. 2009;373:941–955. doi: 10.1016/S0140-6736(09)60236-1. [DOI] [PubMed] [Google Scholar]
- 15.Vanhecke TE, Kim R, Raheem SZ, McCullough PA. Myocardial ischemia in patients with diastolic dysfunction and heart failure. Curr Cardiol Rep. 2010;12:216–222. doi: 10.1007/s11886-010-0101-1. [DOI] [PubMed] [Google Scholar]
- 16.Gomez AM, Guatimosim S, Dilly KW, Vassort G, Lederer WJ. Heart failure after myocardial infarction: altered excitation-contraction coupling. Circulation. 2001;104:688–693. doi: 10.1161/hc3201.092285. [DOI] [PubMed] [Google Scholar]
- 17.Sjaastad I, Wasserstrom A, Sejersted OM. Heart failure-a challenge to our current concepts of excitation-contraction coupling. J Physiol. 2003;546:33–47. doi: 10.1113/jphysiol.2002.034728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Katz MG, Fargnoli AS, Swain JD, Tomasulo CE, Ciccarelli M, Huang ZM, Rabinowitz JE, Bridges CR. AAV6-βARKct gene delivery mediated by molecular cardiac surgery with recirculating delivery (MCARD) in sheep results in robust gene expression and increased adrenergic reserve. J Thorac Cardiovasc Surg. 2012;143:720–726. doi: 10.1016/j.jtcvs.2011.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lavu M, Gundewar S, Lefer DJ. Gene therapy for ischemic heart disease. J Mol Cell Cardiol. 2010;50:742–750. doi: 10.1016/j.yjmcc.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Dhalla NS. Modification of β-adrenoceptor signal transduction pathway by genetic manipulation and heart failure. Mol Cell Biochem. 2000;214:131–155. doi: 10.1023/a:1007131925048. [DOI] [PubMed] [Google Scholar]