


Abstract
Apolipoprotein B mRNA-editing, enzyme-catalytic, polypeptide-like 3G (i.e., APOBEC3G or A3G) is an evolutionarily conserved cytosine deaminase that potently restricts human immunodeficiency virus type 1 (HIV-1), retrotransposons and other viruses. A3G has a nucleotide target site specificity for cytosine dinucleotides, though only certain cytosine dinucleotides are ‘hotspots’ for cytosine deamination, and others experience little or no editing by A3G. The factors that define these critical A3G hotspots are not fully understood. To investigate how A3G hotspots are defined, we used an in vitro fluorescence resonance energy transfer-based oligonucleotide assay to probe the site specificity of A3G. Our findings strongly suggest that the target single-stranded DNA (ssDNA) secondary structure as well as the bases directly 3′ and 5′ of the cytosine dinucleotide are critically important A3G recognition. For instance, A3G cannot readily deaminate a cytosine dinucleotide in ssDNA stem structures or in nucleotide base loops composed of three bases. Single-stranded nucleotide loops up to seven bases in length were poor targets for A3G activity unless cytosine residues flanked the cytosine dinucleotide. Furthermore, we observed that A3G favors adenines, cytosines and thymines flanking the cytosine dinucleotide target in unstructured regions of ssDNA. Low cytosine deaminase activity was detected when guanines flanked the cytosine dinucleotide. Taken together, our findings provide the first demonstration that A3G cytosine deamination hotspots are defined by both the sequence context of the cytosine dinucleotide target as well as the ssDNA secondary structure. This knowledge can be used to better trace the origins of mutations to A3G activity, and illuminate its impact on processes such as HIV-1 genetic variation.
INTRODUCTION
Apolipoprotein B mRNA-editing, enzyme-catalytic, polypeptide-like 3G (APOBEC3G or A3G) is an important host restriction factor that can inhibit human immunodeficiency virus type 1 (HIV-1) and other viruses via cytosine deamination of viral genomic DNA (1). In the presence of the HIV-1 Vif protein, the activity of APOBEC3G is attenuated and the residual deamination activity of A3G may contribute to the high mutation rate of HIV-1, virus evolution and antiretroviral drug resistance (2–4). However, when the activity of Vif is moderated or extinguished, A3G highly restricts viral replication (1,5,6). This restriction largely results from the high level of deamination during HIV-1 reverse transcription, which can lead to degradation of DNA with abasic residues, a decrease in the specificity of plus-strand initiation, and accumulation of lethal G-to-A mutations on the plus strand (i.e., hypermutation) (7–9). Although it is known that A3G acts exclusively on single-stranded DNA (ssDNA) and acts preferentially at specific sites in a sequence of DNA, termed ‘hotspots,’ the factors that create these critical restriction hotspots are not fully understood (10,11). A3G requires a cytosine dinucleotide context on ssDNA and studies have shown that A3G tends to favor CCCA or T/CCC sequences (12,13). Distinct restriction hotspots on the viral genome often occur outside this four base context, however, as A3G can deaminate at a variety of other sites (14). Interestingly, many CCCA or T/CCC sites are not edited by A3G (3).
As indicated above, published observations to date indicate that hotspot specificity for A3G must be determined by more than the bases immediately 3′ and 5′ of the required cytosine dinucleotide sequence. Specific sequences far upstream or downstream from a hotspot also cannot be necessary for recognition, since A3G can deaminate oligos as small as 16 or 13 nt efficiently in vitro (15,16). However, it is formally possible that certain distal sequences could play some role in large ssDNAs in vivo. Given that cytosine residues in small oligos can be efficiently deaminated in a variety sequence contexts, some other feature of ssDNA in cells is likely protecting otherwise favorable sites from deamination. For example, DNA-binding proteins could make some deamination sites in accessible to A3G. In the case of an HIV-1 infection, the HIV-1 nucleocapsid (NC) protein is a known DNA-binding protein that has important functions in the HIV-1 life cycle (17–22). However, HIV-1 NC and A3G binding is non-competitive on target oligonucleotides, suggesting that NC protein may not prevent access of A3G to a particular target site, and actually could enhance A3G binding (16). Another possibility is that certain HIV-1 ssDNA regions may be protected from A3G due to secondary structure folding that occurs during the reverse transcription process. A3G does not act on dsDNA templates, and therefore ssDNA secondary structure (e.g., stem structures) could act as an accessibility barrier for A3G (23). Also, cytosine bases in small loop structures may also be inaccessible targets, particularly if the proper contacts between enzyme and substrate are no longer in alignment due to physical constraints.
In this study, we have investigated the nature of A3G target sites, in particular the impact of nucleotide bases adjacent to the cytosine dinucleotide target as well as the influence of secondary structure in ssDNAs. Here we demonstrate that by systematic nucleotide base changes on either side of the cytosine dinucleotide target that certain bases are preferred by A3G in order to be optimal targets for cytosine deamination. We also observed that DNA stems represent poor targets for A3G, and can protect an otherwise desirable target sequence from cytosine deamination. Small loop structures can also protect potential A3G target sequences from cytosine deamination. Taken together, our findings provide the first demonstration that A3G cytosine deamination hotspots are defined by both the sequence context of the cytosine dinucleotide target as well as the ssDNA secondary structure. Such observations provide further information for predicting the locations of cytosine deamination by A3G, which is of particular importance in tracing the origins of HIV-1 genetic variation in vivo.
MATERIALS AND METHODS
Preparation of cell lysates
A cell line stably expressing A3G, 293-A3G clone10 (3) was cultured in DMEM supplemented with 10% FetalClone3 (FC3, Hyclone) serum with 1% penicillin/streptomycin (Invitrogen), and 225 µg/ml neomycin (Invitrogen). The parental 293 cells were maintained in DMEM with 10% FC3, 1% penicillin/streptomycin and 225 µg/ml neomycin. Cells were incubated at 37°C in 5% CO2. Cell lysates for in vitro assays of A3G activity were prepared as previously described (15). Briefly, 5 × 106 cells (293 or 293-A3G clone 10) were centrifuged at 1000 rpm for 5 min. Cells were resuspended in 250 µl of lysis buffer (0.626% NP40, 10 mM tris-acetate pH 7.4, 50 mM potassium acetate, 100 mM NaCl and 10 mM EDTA) and 50 µl of protease inhibitor cocktail (Cat. # P2714-1BTL, Sigma-Aldrich), and incubated on ice for 15 min. Cell lysates were centrifuged at 1000 rpm for 2 min, and the supernatants transferred to a pre-chilled tube and centrifuged at 16 000 rpm for 10 min. The clarified cell supernatants were then transferred to a new pre-chilled tube and stored at −80°C prior to use.
Oligonucleotide design and synthesis
ssDNA oligonucleotide secondary structures were predicted by using mFold with the default DNA settings (http://mfold.rna.albany.edu/?q=mfold/DNA-Folding-Form) (24). The oligonucleotides selected for synthesis had only a single predicted structure and were synthesized dual-labeled with TAMRA and FAM fluorophores (Sigma-Aldrich). Table 1 indicates the oligonucleotides used in this study.
Table 1.
Oligonucleotide sequences used in the analysis of the influence of nucleotide sequence and ssDNA secondary structure on the in vitro activity of APOBEC3G
| Oligonucleotide | Sequence 5′-3′ | Oligonucleotide | Sequence 5′-3′ |
|---|---|---|---|
| AccA Open Set 1 | ATTGAACCAGAATGATGTCATTGAATATG | AccC Open | AAACCCCGAGAGAGATCGGACTAAG |
| ΔG = −1.90 | ΔG = −3.35 | ||
| CccC Open Set 1 | ATTGACCCCGAATGATGTCATTGAATATG | TccA Open | AATCCACGAGACAGATCGTACTAAG |
| ΔG = −1.90 | ΔG = −2.36 | ||
| TccT Open Set 1 | ATTGATCCTGAATGATGTCATTGAATATG | TccA Stem | AATCCACGAGACAGATCGTGGAAAG |
| ΔG = −1.90 | ΔG = −6.67 | ||
| GccG Open Set 1 | ATTGAGCCGGAATGATGTCATTGAATATG | TccC Open | AATCCCCGAGAGAGATCGGACTAAG |
| ΔG = −1.90 | ΔG = −3.35 | ||
| AccA Stem Set 1 | ATTGAACCAGAATGATGTCTGGGAATATG | TccG Open | AATCCGCGAGAGAGATCGCATCAAG |
| ΔG = −3.77 | ΔG = −3.47 | ||
| CccC Stem Set 1 | ATTGACCCCGAATGATGTCGGGGAATATG | 3 Loop AccA | ATTGATGCTGACCATCAGCTAATATG |
| ΔG = −7.37 | ΔG = −3.85 | ||
| TccT Stem Set 1 | ATTGATCCTGAATGATGTCAGGTAATATG | 3 Loop CccC | ATTGATGCTGCCCCGCAGCTAATATG |
| ΔG = −3.30 | ΔG = −4.84 | ||
| GccG Stem Set 1 | ATTGAGCCGGAATGATGTCCGGGAATATG | 4 Loop AccA | ATTGATGCTGAACCATCAGCTAATATG |
| ΔG = −4.81 | ΔG = −4.60 | ||
| AccA Open Set 2 | AAACCACGAGAGAGATCGTACTAAG | 4 Loop CccC | ATTGATGCTGACCCCTCAGCTAATATG |
| ΔG = −2.36 | ΔG = −4.10 | ||
| CccC Open Set 2 | AACCCCCGAGAGAGATCGGATGAAG | 5 Loop AccA | ATTGATGCTGAACCAGTCAGCTAATATG |
| ΔG = −3.35 | ΔG = −4.60 | ||
| TccT Open Set 2 | AATCCTCGAGAGATATCTATAAAAG | 5 Loop CccC | ATTGATGCTGAACCCCTCAGCTAATATG |
| ΔG = −2.23 | ΔG = −4.40 | ||
| GccG Open Set 2 | AAGCCGCGAGAGAGATCGCATCAAG | 6 Loop AccA | ATTGATGCTGAAACCAGTCAGCTAATATG |
| ΔG = −3.47 | ΔG = −3.90 | ||
| AccA Stem Set 2 | AAACCACGAGAGAGATCGTGGTAAG | 6 Loop CccC | ATTGATGCTGAACCCCGTCAGCTAATATG |
| ΔG = −7.18 | ΔG = −3.90 | ||
| CccC Stem Set 2 | AACCCCCGAGAGAGATCGGGGGAAG | 7 Loop CccC | ATTGATGCTGAGACCCCGTCAGCTAATATG |
| ΔG = −9.31 | ΔG = −3.50 | ||
| TccT Stem Set 2 | AATCCTCGAGAGAGATCGAGGAAAG | 7 Loop AccA | ATTGATGCTGAGAACCAGTCAGCTAATATG |
| ΔG = −6.36 | ΔG = −3.50 | ||
| GccG Stem Set 2 | AAGCCGCGAGAGAGATCGCGGCAAG | 8 Loop AccA | ATTGATGCTGAGAACCAGATCAGCTAATATG |
| ΔG = −9.96 | ΔG = −3.70 | ||
| GccA Open | AAGCCACAAGAGAGATCTTGCTAAG | 8 Loop CccC | ATTGATGCTGAGACCCCGATCAGCTAATATG |
| ΔG = −4.42 | ΔG = −3.70 | ||
| GccT Open | AAGCCTAAAGAGAGATCTTTGAAAG | 8 Loop TccT | ATTGATGCTGAGATCCTGATCAGCTAATATG |
| ΔG = −3.94 | ΔG = −3.70 | ||
| GccC Open | AAGCCCGAAGAGAGATTCGAATAAG | 8 Loop GccG | ATTGATGCTGAGAGCCGGATCAGCTAATATG |
| ΔG = −2.51 | ΔG = −3.70 | ||
| GccC Stem | AAGCCCGAAGAGAGATTCGGGCAAG | 9 Loop AccA | ATTGATGCTGAGAACCAAGATCAGCTAATATG |
| ΔG = −8.39 | ΔG = −3.70 | ||
| AccG Open | AAACCGCGAGAGAGATCGCACTAAG | 10 Loop AccA | ATTGATGCTGACGAACCAAGATCAGCTAATATG |
| ΔG = −3.47 | ΔG = −3.70 | ||
| AccT Open | AAACCTCGAGACAGATCGAACTAAG | dU Open | AAACUACGAGAGAGATCGTGCTAAG |
| ΔG = −1.46 | ΔG = −2.67 | ||
| CccC Bulge | AATGAAGCCCCGAGCAACTCGGCTTCTATG | dU Stem | ATTGATCCUTGAATGATGTCAGGGGATATG |
| ΔG = −5.30 | ΔG = −1.72 | ||
| dU Bulge | AATGAAGCCUCGAGCAACTCGGCTTCTATG | dU 3 Loop | ATTGATGCTGACUATCAGCTAATATG |
| ΔG = −3.85 | |||
| ΔG = −5.30 |
A3G FRET assay
A fluorescence resonance energy transfer (FRET) based assay was used to detect cytosine deaminase activity of A3G using DNA oligonucleotides as a substrate using previously described assays with minor modifications (15,25). Cell lysates were diluted 3:2 in lysis buffer, and 20 µl of the diluted lysates were used per assay using 96 white-welled assay plates (Bio-Rad). A separate solution of 20 pmoles of oligonucleotide, 10 µg RNase A and 0.04 U uracil DNA glycosylase (UDG) were mixed together in 50 mM Tris pH 7.4, 10 mM EDTA buffer and adjusted to a total volume of 50 µl, then transferred to the assay well. The assay plate was then incubated at 37°C for 5 h. Next, 30 µl of 2 M Tris-acetate, pH 7.9 was added to each well and the plate was incubated at 95°C for 2 min and at 4°C for 2.5 min with a CFX96 real-time PCR system (Bio-Rad). The fluorescence was then measured at 4°C. The endpoint fluorescence from the parental 293 cell lysate was subtracted from all experimental samples in order to calculate a relative change in fluorescence due to A3G activity. Experiments were conducted with three independent replicates. For assays involving HIV-1 NC protein (purified NC protein graciously provided by Dr Rob Gorelick, SAIC, Frederick, MD), experiments were conducted in the presence of HIV-1 NC protein (5 nt per NC) for 1 h on ice. Following incubation, cell lysates were prepared as described above, added to each well for 5 h at 37°C, and then fluorescence measured.
Restriction enzyme FRET assay
To further validate the predicted structures of the FRET oligos, select oligos were digested with restriction enzymes. Regions of the oligo in a stem secondary structure would be cut by the specific restriction enzyme and would release FRET signal. Briefly, 20 pmoles of GccG set 2 open or stem oligo and 5U of Aci I (NEB) were added to a solution of 1× Buffer 3 (NEB) in 100 ul total volume. For GccG set 1 open or stem oligo, 20 pmoles were added with 2U of MspI (NEB) to a solution of 1× Buffer 4(NEB) in 100 ul total volume. Mixes were added to white-welled 96-well plates (Biorad) and incubated at 37°C for 30 min in a C1000 thermal cycler with a CFX96 real-time system (Biorad). Subsequently, the temperature was adjusted to 4°C for 30 s and the plate was read. Experiments were conducted with three independent replicates.
RESULTS
Sequence context as well as ssDNA secondary structure define A3G cytosine deaminase hotspots
Two sets of ssDNA oligonucleotides were initially tested in which the cytosine dinucleotide was located either in an open (unstructured) location or was located within a structured stem; representative examples of the oligonucleotide structures are shown in Figure 1A. The oligonucleotide design strategy helped to minimize sequence changes between the oligonucleotide pairs and distal to the deamination site. However, some additional nucleotide changes were required for some of the oligonucleotides tested in order for the CC dinucleotide to be in the correct structural location within the most stable structure (Table 1 and Supplementary Figure S1). In particular, the CccC Stem Set 1 has an extra base pair in the stem, though the oligonucleotide sequence is consistent with what is shown in Figure 1A. For the TccT Open Set 2 oligonucleotide, the number of loop bases was reduced from 6 to 4 bases, and the number of bases involved in the stem decreased from 7 to 3 bases. Finally, for the 5 Loop CccC oligonucleotide, the bottom base in the ‘5 loop’ in Figure 2A was changed to a C residue rather than a G residue (Figure 2A). The specific predicted mFold structures using the default settings for these three oligonucleotides are indicated in Supplementary Figure S1. Other oligonucleotides were tested in which bases on either side of the cytosine dinucleotide, which represent the base locations that are most critical for A3G recognition (11).
Figure 1.

Nucleotide sequence context and ssDNA secondary structure help to define A3G cytosine deaminase hotspots. (A) Oligonucleotides containing the cytosine dinucleotide targeted by A3G dual-labeled with TAMRA and FAM fluorophores. The red colored ‘CC’ dinucleotide bases represent the A3G target site. The blue colored ‘X’ bases represent the positions at which nucleotide bases were changed. The ‘open’ oligonucleotides are defined as the oligonucleotides in which the target cytosine dinucleotide is located in the unstructured region of the ssDNA, and the ‘stem’ oligonucleotides are defined as the oligonucleotides in which the target cytosine dinucleotide is located within the stem structure. (B) The change in relative fluorescence units (ΔRFU) was calculated for each experiment by subtracting the RFU from the control 293 cell lysates (baseline negative control) from the 293 cell lysates that stably express A3G. The error bars represent the standard deviation from three independent experiments. The positive control for these experiments was an oligonucleotide previously reported to be cleaved by A3G in an oligonucleotide-based FRET assay (15). (C) The ΔRFU was calculated as described above. The average and standard deviation from three independent experiments is shown.
Figure 2.

A3G cytosine deaminase activity against a target cytosine dinucloetide is influenced by location in ssDNA base loops but not in a DNA bulge. (A) Oligonucleotides used to investigate the influence of ssDNA loop size on A3G activity are shown. The red colored ‘CC’ dinucleotide bases represent the A3G target site. The blue colored ‘X’ bases represent the positions at which nucleotide bases were changed. (B) The change in relative fluorescence units (ΔRFU) was calculated for each experiment by subtracting the RFU from the control 293 cell lysates (baseline negative control) from the 293 cell lysates that stably express A3G. The x-axis indicates the number of nucleotide bases in the ssDNA loop. The error bars represent the standard deviation from three independent experiments.
Cell lysates prepared from 293-expressing A3G cells or 293 parental cells were used to incubate with each oligonucleotide along with UDG and RNase A as described in the Materials and Methods section. In the presence of A3G cytosine deaminase activity, creation of a uracil base would occur resulting in an abasic site following uracil base excision by UDG. Base hydrolysis of the abasic site would release a FAM signal from the FRET pair.
Figure 1B demonstrates that the bases on either side of the cytosine dinucleotide are important for A3G activity when located in a non-structured region. In particular, we observed that adenine, cytosine or thymine bases on either side of the cytosine dinucleotide increased A3G activity (P < 0.05) whereas guanine bases on either side of the dinucleotide had a reduced but significant effect (P < 0.05). These results indicate that adenine, cytosine or thymine bases on either side of the cytosine dinucleotide enhance A3G activity and guanine bases limit A3G activity.
We further explored the nature of the nucleotide bases on either side of the cytosine dinucleotide by investigating the base preference on 5′ or 3′ side of the dinucleotide site (Figure 1C). In particular, when the 5′ base was a guanine, there was little activity detected when the 3′ base was an adenine or thymine, but a high A3G signal was observed when the 3′ base was a cytosine. When the 5′ base was an adenine, there was moderate A3G activity unless the 3′ base was a cytosine. Finally, when the 5′ base was a thymine, A3G activity was moderate to relatively high when the 3′ base was either a guanine or an adenine, and activity was enhanced if the 3′ base was a cytosine.
Significantly reduced A3G activity was observed when cytosine dinucleotides were located within an oligonucleotide stem, indicating that A3G can have difficulty in accessing target bases located in regions in which secondary structure exists, in any sequence context (Figure 1C). Taken together, these observations indicate that both the nucleotide base on either side of the cytosine dinucleotide as well as their location in secondary structure can define A3G hotspots.
Structural constraints in DNA loop bases can limit A3G hotspots
Given our observation that ssDNA secondary structure can attenuate A3G activity, we further investigated how the location cytosine dinucleotides in ssDNA loop bases could impact A3G activity. To do this, we tested oligonucleotides in which the cytosine dinucleotide was in either a stem bulge or in a ssDNA loop that ranged from 3 bases in size up to 10 bases in size (Figure 2A). A3G activity was not affected by the cytosine dinucleotide located in a stem bulge, but had low activity when the dinucleotide was located in a 3 nt base loop where either adenine or cytosine was flanking the cytosine dinucleotide (Figure 2B). This indicates that 3 nt base loops can protect A3G hotspots. Interestingly, when cytosine bases flanked the cytosine dinucleotide, high A3G activity was observed within 4–8 nt base loops, but not when adenines flanked the cytosine dinucleotide. Low activity was detected with adenines flanked the cytosine dinucleotide in the 4 nt base loops, and moderate A3G activity detected when the cytosine dinucleotide was located in 5–7 nt base loops with adenine bases flanking (Figure 2B). Higher activity was observed in 8–10 base loops with adenines flanking. This indicates that nucleotide base loop structures can be protected cytosine dinucleotides when flanked by adenine bases in seven base or smaller loops. When cytosine bases flank the cytosine dinucleotide, protection is observed only in a 3 nt base loop. Moderate or low A3G activity was observed with thymine or guanine bases flanking the cytosine dinucleotide, respectively. This observation complements the observations made in the absence of secondary structure.
Since, the binding of HIV-1 NC and A3G is non-competitive on target oligonucleotides, NC protein may not prevent access of A3G to a particular target site, and may enhance A3G binding (16). It is also formally possible that NC may protect certain HIV-1 ssDNA regions due to secondary structure folding that occurs during reverse transcription. Since A3G does not act on dsDNA templates, ssDNA secondary structure (e.g., stem structures) could act as an accessibility barrier for A3G (23). It is also conceivable that cytosine bases in small loop structures may be inaccessible due to physical constraints. To test for potential affects of HIV-1 NC on A3G activity, we selected an oligonucleotide pair in which there was a clearly significant difference in the FRET signal observed when the CC dinucleotide target was located in either a non-structured or structured region (i.e., AccA set 2 open and stem oligonucleotides; Table 1). A HIV-1 NC concentration (i.e., 5 nt per NC) was chosen that is physiologically relevant based upon what is predicted in the virus particle (26,27). As indicated in Figure 3, the addition of NC was found to have no effect on A3G activity when the CC dinucleotide was either in the AccA set 2 open or stem oligonucleotide.
Figure 3.
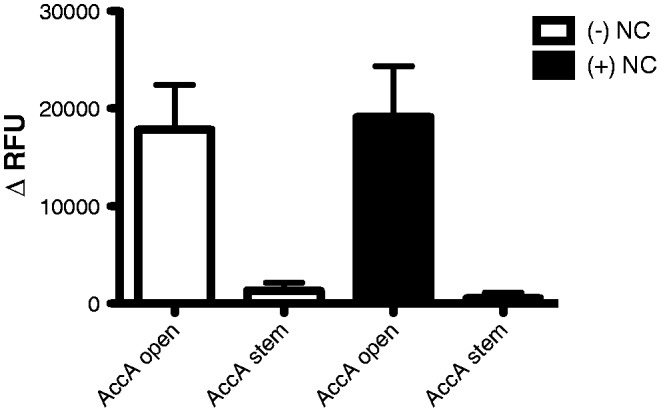
No effect of HIV-1 NC protein on altering the efficiency of A3G deamination. The AccA set 2 open and stem oligonucleotides were incubated in the presence or absence of HIV-1 NC protein (concentration of 5 nt per NC protein). The change in relative fluorescence units (ΔRFU) was calculated for each experiment by subtracting the RFU from the control 293 cell lysates (baseline negative control) from the 293 cell lysates that stably express A3G. Error bars represent the standard deviation from three independent experiments.
It has been previously demonstrated that UDG excises uracil residues more efficiently from ssDNA than dsDNA (28), and that the excision of uracil from loops is inefficient. In order to confirm that the FRET signal differences observed with the target cytosine base for cytosine deamination by A3G is in a non-paired region, or in a stem, bulge or DNA loop is actually due to A3G activity and not to UDG, we synthesized oligonucleotides that contain uracil in these different locations. Figure 4 shows that UDG is readily able to excise the uracil residue in each of these positions, indicating that the differences that we have observed are due to A3G activity and not due to UDG.
Figure 4.

UDG activity is undiminished on ssDNA secondary structures. The effect of uracil location in oligonucleotides was investigated. Four different oligonucleotides were used in which the target cytosine was replaced with a uracil that was located in a non-paired, stem, bulge or DNA loop region. The relative fluorescent units (RFU) from a uracil in the open, stem, three base loop and bulge location in the presence of UDG is shown. The average and standard deviation from three independent experiments is shown.
Experimental confirmation of mFold ssDNA structural predictions
The oligonucleotides that were used in this study were selected in part based upon their having a single structural prediction in the mFold program (24). Specific parameters have been designed into the mFold program for ssDNA folding that were based upon NMR data generated with ssDNA sequences (29–34). In order to experimentally validate that these predictions for the oligonucleotides used in this study, a small subset were analysed that possessed DNA secondary structures that created restriction enzyme sites. Confirmation of the presence of these restriction sites would provide one line of experimental evidence in support of the structure predicted by the mFold program for oligonucleotide test. Oligo GccG set 1 stem, when folded, creates a Msp I restriction site that is not present in the non-folded version of the oligonucleotide. Figure 5a resulted in a strong FRET signal when oligonucleotide GccG set 1 stem was incubated in the presence of Msp I, but not when the non-folding version of the oligonucleotide (i.e., GccG set 1 open) was incubated with Msp I. This data suggest that the predicted structure for the GccG set 1 stem is correct. We conducted a similar analysis with GccG set 2 stem and GccG set 2 open, where the predicted folded structure for GccG set 2 stem resulted in the creation of an Aci I restriction site, which does not occur in GccG set 2 open (Figure 5b). Incubation of each oligonucleotide with Aci I lead to a strong FRET signal only with the GccG set 2 stem oligonucleotide, which also suggests that the structural predictions by mFold for the oligonucleotides used in this study are correct. Although this data support the proposed intramolecular structural predictions, it is formally possible that stable structures could also arise using the set 1 stem or set 2 stem oligonucleotides by the formation of intermolecular homodimers. For instance, the restriction enzyme analysis conducted above with Msp I and Aci I would not be able differentiate per se between a single intramolecular stem versus that of a intermolecular homodimer stem—though stem formation of the participating nucleotide bases would be confirmed.
Figure 5.

Experimental confirmation of ssDNA secondary structures. Two sets of oligonucleotides with restriction enzyme sites in the stem oligonucleotide were used ((A) GCCG Set 2 Stem and GCCG Set 2 Open, and (B) GCCG Set 1 Stem and GCCG Set 1 Open). The stem bases in the structured oligonucleotides create an Aci I restriction site (A) or a Msp I restriction site (B). The oligonucleotides GCCG Set 2 Open and GCCG Set 1 Open did not fold to form the restriction enzyme sites and remained intact. The average and standard deviation from three independent experiments are shown.
DISCUSSION
The goal of this study was to investigate the determinants for A3G hotspots. To do this, we used an experimental model system in which we used oligonucleotides that were dual-labeled with TAMRA and FAM fluorophores. Lysates from cells stably expressing A3G were incubated with these oligonucleotides, and A3G activity was detected by FRET. Oligonucleotides were designed to test (i) the role of nucleotide bases adjacent to the cytosine dinucleotide target site that is critical for cytosine deamination; and (ii) the role of ssDNA secondary structure, including DNA duplexes, loop sequences and bulges.
We observed that the ability of A3G to deaminate was found to be greatly dependent upon the nucleotide bases immediately adjacent to the cytosine dinucleotide. Specifically, A3G efficiently deaminates when cytosines, adenines or thymines are adjacent to the cytosine dinucleotide. A3G activity was low when guanine bases were on either side of the cytosine dinucleotide. Previous studies have indicated that A3G prefers a sequence context of 5′-CCCA-3′ or 5′-T/CCC-3′ (12,13). This corresponds well with the data in our study. In addition, a study has been reported in which 5′-TCCA-3′, 5′-ACCA-3′, and 5′-ACCG-3′ were found to be good substrates for A3G cytosine deaminase activity, whereas 5′-GCCA-3′ and 5′-ACCT-3′ were found to have no or minimal activity, respectively (35). Although these studies support parts of our current study, our observations represent a more extensive and complete study of the preferred bases, and therefore provides greater insight into being able to predict and identify A3G hotspots.
The results from our studies are supported by studies investigating A3G hotspots identified in HIV-1 sequences recovered from infected individuals. Coffin and colleagues found that of the available sites for A3G-mediated cytosine deamination, 40% of 5′-CCCC-3′ sequences, 21% of 5′-ACCA-3′ sequences, 11% of 5′-TCCT-3′ sequences and 0% of 5′-GCCG-3′ sequences were A3G cytosine deamination sites, which is a striking correlation to our data (2). Furthermore, 5′-TCCA-3′, 5′-TCCC-3′ and 5′-ACCT-3′ sequences were found be locations that the authors concluded that A3G-mediated cytosine deamination had occurred, while no cytosine deamination occurred at 5′-GCCT/A-3′ sequences. Finally, HIV-1 proviral sequencing data from our previous studies of A3G-mediated cytosine deamination of HIV-1 in cell culture found G-to-A mutations (in the positive strand) at either 5′-GGGG-3′, 5′-TGGT-3′ or 5′-TGGA-3′ sequences and few or no mutations at either 5′-AGGA-3′, 5′-AGGT-3′, 5′-TGGC-3′ or 5′-CGGC-3′ sequences (3,36). Taken together, these data indicate that the A3G hotspot sequence preferences in our cell-free study using oligonucleotides corresponds with strong predictive power to the A3G hotspot preferences observed from HIV-1 proviral DNA sequencing. In addition, A3G mutational hot spot sites are clearly far more complex than the widely cited 5′-CCCA-3′ or 5′-T/CCC-3′ A3G nucleotide sequence preference.
A recent study investigated the features of nucleotide bases that can help define the sequence preference of A3G (11). Exocyclic groups in pyrimidines that are located 1 or 2 nt 5′ of the cytosine targeted by A3G were found to dictate substrate recognition. The exocyclic groups were speculated to be important for stacking or for electrostatic interactions among adjacent bases. When these interactions are disrupted, it was conjectured that it could affect the ability of A3G to recognize the substrate. This hypothesis is supported by our data with the sequence 5′-T/CCCG/A/C-3′. However, we observed that the sequence 5′-ACCA-3′ can also be an efficient target for A3G-mediated cytosine deamination. It is of particular interest to these observations that local sequence context has been found to influence the scanning ability of A3G and that A3G has been proposed to ‘hover’ over 5′-ACCCA-3′ sequences longer than 5′-TCCCT-3′ sequences (37). This observation suggests that additional features of adenine bases may be important in the attraction of A3G to sites of cytosine deamination.
We have also demonstrated in this study that ssDNA secondary structure plays a vital role in the identification of A3G hotspots. In particular, the data presented here indicate that A3G either has no or low activity deamination activity for the cytosine dinucleotides located in ssDNA oligonucleotide stem structures or cytosine dinucleotides located in three base loops. The low activity observed on stems may be due to base unzipping from the stem at a low frequency, which would expose the CC dinucleotide. Furthermore, only cytosine dinucleotides flanked by cytosines or adenines were efficiently deaminated when located in ssDNA loops up to 8 bases in size or 8–10 bases respectively, whereas cytosine dinucleotides flanked by adenines resulted in only moderate level of A3G-mediated cytosine deamination in ssDNA oligonucleotide loops 5–7 bases. A related cytosine deaminase family member, AID, has also been reported to be inefficient in deaminating cytosines in ssDNA secondary structures involving stems and loops (38). Since A3G cannot deaminate dsDNA, we hypothesize that ssDNA stems mimic dsDNA in an efficient enough of a manner in order to avoid cytosine deamination. Furthermore, A3G has been previously demonstrated to be processive along ssDNA substrates by sliding and jumping (39,40). When A3G encounters partially dsDNA, the sliding ability was lost but the jumping ability was retained (39). Therefore, it is tempting to speculate that A3G could ‘jump’ over stems and loops—and this could help explain at least part of the reduced level of cytosine deamination in those regions. However, the ability of A3G to ‘jump’ over stems and loops does not account for the differences observed with 5′-CCCC-3′ and 5′-ACCA-3′ sequences in ssDNA loops. While the tortional bend of nucleotides in ssDNA loops may not allow for the proper contacts between A3G and the nucleotide bases, this may be more readily overcome with cytosines rather than adenines. Further studies are needed in order to determine the specific factors behind these observations. It will be important and beneficial to compare the sequence and secondary structural preferences of other APOBEC3 family members to our findings on APOBEC3G. The addition of HIV-1 NC protein was found in our experiments to have no effect on A3G activity when the CC dinucleotide was either in the AccA set 2 open or stem oligonucleotide. These observations suggest that ssDNA secondary structure (e.g., stem structures) could act as an accessibility barrier for A3G, even in the presence of physiologically relevant concentrations of HIV-1 NC. It is presently unclear how generally applicable these observations are to other CC dinucleotide position locations.
In summary, the observations made in this study provide the first demonstration that A3G cytosine deamination hotspots are defined by both the sequence context of the cytosine dinucleotide target as well as the ssDNA secondary structure. These observations provide useful information for predicting the locations of cytosine deamination by A3G. Such predictions are important for investigations directed at investigating the origins of mutations that are associated with HIV-1 genetic variation (14). Given the high HIV-1 mutation rate (41), and the high rate of G-to-A transition mutations (42), there is intense interest in the origins of mutations that arise during HIV-1 replication. Knowledge on the origins of mutations during HIV-1 replication is important for developing a better understanding of HIV-1 genetic variation and evolution as well as for efforts to purposely elevate HIV-1 mutation to induce lethal mutagenesis (43,44).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figure 1.
FUNDING
National Institutes of Health (NIH) [R01 GM56615 to L.M.M., T32 DE007288 and T90 DE022732 to C.M.H.]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Malim MH. APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009;364:675–687. doi: 10.1098/rstb.2008.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jern P, Russell RA, Pathak VK, Coffin JM. Likely role of APOBEC3G-mediated G-to-A mutations in HIV-1 evolution and drug resistance. PLoS Pathog. 2009;5:e1000367. doi: 10.1371/journal.ppat.1000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sadler HA, Stenglein MD, Harris RS, Mansky LM. APOBEC3G contributes to HIV-1 variation through sublethal mutagenesis. J. Virol. 2010;84:7396–7404. doi: 10.1128/JVI.00056-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hache G, Mansky LM, Harris RS. Human APOBEC3 proteins, retrovirus restriction, and HIV drug resistance. AIDS Rev. 2006;8:148–157. [PubMed] [Google Scholar]
- 5.Goila-Gaur R, Strebel K. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology. 2008;5:51. doi: 10.1186/1742-4690-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 2004;4:868–877. doi: 10.1038/nri1489. [DOI] [PubMed] [Google Scholar]
- 7.Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003;424:94–98. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 9.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113:803–809. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 10.Kohli RM, Maul RW, Guminski AF, McClure RL, Gajula KS, Saribasak H, McMahon MA, Siliciano RF, Gearhart PJ, Stivers JT. Local sequence targeting in the AID/APOBEC family differentially impacts retroviral restriction and antibody diversification. J. Biol. Chem. 2010;285:40956–40964. doi: 10.1074/jbc.M110.177402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rausch JW, Chelico L, Goodman MF, Le Grice SF. Dissecting APOBEC3G substrate specificity by nucleoside analog interference. J. Biol. Chem. 2009;284:7047–7058. doi: 10.1074/jbc.M807258200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 2004;14:1392–1396. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 13.Yu Q, Konig R, Pillai S, Chiles K, Kearney M, Palmer S, Richman D, Coffin JM, Landau NR. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 2004;11:435–442. doi: 10.1038/nsmb758. [DOI] [PubMed] [Google Scholar]
- 14.Kijak GH, Janini M, Tovanabutra S, Sanders-Buell EE, Birx DL, Robb ML, Michael NL, McCutchan FE. HyperPack: a software package for the study of levels, contexts, and patterns of APOBEC-mediated hypermutation in HIV. AIDS Res. Hum. Retrov. 2007;23:554–557. doi: 10.1089/aid.2006.0279. [DOI] [PubMed] [Google Scholar]
- 15.Thielen BK, Klein KC, Walker LW, Rieck M, Buckner JH, Tomblingson GW, Lingappa JR. T cells contain an RNase-insensitive inhibitor of APOBEC3G deaminase activity. PLoS Pathog. 2007;3:1320–1334. doi: 10.1371/journal.ppat.0030135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iwatani Y, Takeuchi H, Strebel K, Levin JG. Biochemical activities of highly purified, catalytically active human APOBEC3G: correlation with antiviral effect. J. Virol. 2006;80:5992–6002. doi: 10.1128/JVI.02680-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Darlix JL, Godet J, Ivanyi-Nagy R, Fosse P, Mauffret O, Mely Y. Flexible nature and specific functions of the HIV-1 nucleocapsid protein. J. Mol. Biol. 2011;410:565–581. doi: 10.1016/j.jmb.2011.03.037. [DOI] [PubMed] [Google Scholar]
- 18.Levin JG, Mitra M, Mascarenhas A, Musier-Forsyth K. Role of HIV-1 nucleocapsid protein in HIV-1 reverse transcription. RNA Biol. 2010;7:754–774. doi: 10.4161/rna.7.6.14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muriaux D, Darlix JL. Properties and functions of the nucleocapsid protein in virus assembly. RNA Biol. 2010;7:744–753. doi: 10.4161/rna.7.6.14065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mirambeau G, Lyonnais S, Gorelick RJ. Features, processing states, and heterologous protein interactions in the modulation of the retroviral nucleocapsid protein function. RNA Biol. 2010;7:724–734. doi: 10.4161/rna.7.6.13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Godet J, Mely Y. Biophysical studies of the nucleic acid chaperone properties of the HIV-1 nucleocapsid protein. RNA Biol. 2010;7:687–699. doi: 10.4161/rna.7.6.13616. [DOI] [PubMed] [Google Scholar]
- 22.Thomas JA, Gorelick RJ. Nucleocapsid protein function in early infection processes. Virus Res. 2008;134:39–63. doi: 10.1016/j.virusres.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW, Jr, Swanstrom R, Burch CL, Weeks KM. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009;460:711–716. doi: 10.1038/nature08237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stenglein MD, Burns MB, Li M, Lengyel J, Harris RS. APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat. Struct. Mol. Biol. 2010;17:222–229. doi: 10.1038/nsmb.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu W, Henderson LE, Copeland TD, Gorelick RJ, Bosche WJ, Rein A, Levin JG. Human immunodeficiency virus type 1 nucleocapsid protein reduces reverse transcriptase pausing at a secondary structure near the murine leukemia virus polypurine tract. J. Virol. 1996;70:7132–7142. doi: 10.1128/jvi.70.10.7132-7142.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henderson LE, Bowers MA, Sowder RC, II, Serabyn SA, Johnson DG, Bess JW, Jr, Arthur LO, Bryant DK, Fenselau C. Gag proteins of the highly replicative MN strain of human immunodeficiency virus type 1: posttranslational modifications, proteolytic processings, and complete amino acid sequences. J. Virol. 1992;66:1856–1865. doi: 10.1128/jvi.66.4.1856-1865.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar NV, Varshney U. Inefficient excision of uracil from loop regions of DNA oligomers by E. coli uracil DNA glycosylase. Nucleic Acids Res. 1994;22:3737–3741. doi: 10.1093/nar/22.18.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peyret N, Seneviratne PA, Allawi HT, SantaLucia J., Jr Nearest-neighbor thermodynamics and NMR of DNA sequences with internal A.A, C.C, G.G, and T.T mismatches. Biochemistry. 1999;38:3468–3477. doi: 10.1021/bi9825091. [DOI] [PubMed] [Google Scholar]
- 30.SantaLucia J., Jr A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl Acad. Sci. USA. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allawi HT, SantaLucia J., Jr Thermodynamics and NMR of internal G.T mismatches in DNA. Biochemistry. 1997;36:10581–10594. doi: 10.1021/bi962590c. [DOI] [PubMed] [Google Scholar]
- 32.Allawi HT, SantaLucia J., Jr Nearest neighbor thermodynamic parameters for internal G.A mismatches in DNA. Biochemistry. 1998;37:2170–2179. doi: 10.1021/bi9724873. [DOI] [PubMed] [Google Scholar]
- 33.Allawi HT, SantaLucia J., Jr Thermodynamics of internal C.T mismatches in DNA. Nucleic Acids Res. 1998;26:2694–2701. doi: 10.1093/nar/26.11.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allawi HT, SantaLucia J., Jr Nearest-neighbor thermodynamics of internal A.C mismatches in DNA: sequence dependence and pH effects. Biochemistry. 1998;37:9435–9444. doi: 10.1021/bi9803729. [DOI] [PubMed] [Google Scholar]
- 35.Beale RC, Petersen-Mahrt SK, Watt IN, Harris RS, Rada C, Neuberger MS. Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. J. Mol. Biol. 2004;337:585–596. doi: 10.1016/j.jmb.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 36.Dapp MJ, Holtz CM, Mansky LM. Concomitant lethal mutagenesis of human immunodeficiency virus type 1. J. Mol. Biol. 2012;419:158–170. doi: 10.1016/j.jmb.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Senavirathne G, Jaszczur M, Auerbach PA, Upton TG, Chelico L, Goodman MF, Rueda D. Single-stranded DNA scanning and deamination by APOBEC3G at single molecule resolution. J. Biol. Chem. 2012;287:15826–15835. doi: 10.1074/jbc.M112.342790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larijani M, Martin A. Single-stranded DNA structure and positional context of the target cytidine determine the enzymatic efficiency of AID. Mol. Cell Biol. 2007;27:8038–8048. doi: 10.1128/MCB.01046-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chelico L, Pham P, Calabrese P, Goodman MF. APOBEC3G DNA deaminase acts processively 3' –> 5' on single-stranded DNA. Nat. Struct. Mol. Biol. 2006;13:392–399. doi: 10.1038/nsmb1086. [DOI] [PubMed] [Google Scholar]
- 40.Chelico L, Sacho EJ, Erie DA, Goodman MF. A model for oligomeric regulation of APOBEC3G cytosine deaminase-dependent restriction of HIV. J. Biol. Chem. 2008;283:13780–13791. doi: 10.1074/jbc.M801004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mansky LM, Temin HM. Lower in vivo mutation rate of human immunodeficiency virus type 1 than predicted from the fidelity of purified reverse transcriptase. J. Virol. 1995;69:5087–5094. doi: 10.1128/jvi.69.8.5087-5094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Kuyl AC, Berkhout B. The biased nucleotide composition of the HIV genome: a constant factor in a highly variable virus. Retrovirology. 2012;9:92. doi: 10.1186/1742-4690-9-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clouser CL, Patterson SE, Mansky LM. Exploiting drug repositioning for discovery of a novel HIV combination therapy. J. Virol. 2010;84:9301–9309. doi: 10.1128/JVI.01006-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dapp MJ, Patterson SE, Mansky LM. Back to the future: revisiting HIV-1 lethal mutagenesis. Trends Microbiol. 2013;21:56–62. doi: 10.1016/j.tim.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.