

Abstract
The canonical exogenous trigger of RNA interference (RNAi) in mammals is small interfering RNA (siRNA). One promising application of RNAi is siRNA-based therapeutics, and therefore the optimization of siRNA efficacy is an important consideration. To reduce unfavorable properties of canonical 21mer siRNAs, structural and chemical variations to canonical siRNA have been reported. Several of these siRNA variants demonstrate increased potency in downstream readout-based assays, but the molecular mechanism underlying the increased potency is not clear. Here, we tested the performance of canonical siRNAs and several sequence-matched variants in parallel in gene silencing, RNA-induced silencing complex (RISC) assembly, stability and Argonaute (Ago) loading assays. The commonly used 19mer with two deoxythymidine overhangs (19merTT) variant performed similarly to canonical 21mer siRNA. A shorter 16mer variant (16merTT) did not perform comparably in our assays. Dicer substrate interfering RNA (dsiRNA) demonstrated better gene silencing by the guide strand (target complementary strand), better RISC assembly, persistence of the guide strand and relatively more loading of the guide strand into Ago. Hence, we demonstrate the advantageous properties of dsiRNAs at upstream, intermediate and downstream molecular steps of the RNAi pathway.
INTRODUCTION
The canonical trigger of the RNA interference (RNAi) mechanism for mammalian gene-silencing studies is small interfering RNA (siRNA). The canonical siRNA structure and chemistry consists of two 21 nucleotide (nt) strands, hybridized such that each 3′-end contains a 2 nt overhang (1). These canonical siRNAs, however, have disadvantages, including RNase susceptibility, incorrect strand selection and innate immune system immunogenicity, which can ultimately lead to poor gene-silencing potency and off-target effects. To combat these disadvantages, several structural and chemical variations of the canonical siRNA design have been reported [reviewed in (2,3)]. From a therapeutic standpoint, the prospect of using RNAi-based therapeutics as an entirely new drug platform is steadily developing (2–4). Optimizing the potency and efficacy of siRNAs is a critical step in the development of RNAi-based therapeutics.
As an example of a variant form of a canonical siRNA, we previously reported that a structurally asymmetric (25/27mer) and chemically modified longer duplex RNA termed a Dicer substrate interfering RNA (dsiRNA), demonstrated better gene silencing compared with its sequence-matched canonical siRNA (5,6). Any canonical siRNA can be transformed into the dsiRNA format. The dsiRNA format imparts functional polarity to promote selection of the guide strand (i.e. the strand complementary to the target mRNA) and discourages selection of the passenger strand (6) while remaining under the double-stranded RNA (dsRNA) length threshold that would otherwise trigger an interferon response (5). Another group has observed the improved potency of dsiRNAs over sequence-matched siRNAs as well (7,8). It has also been reported that short hairpin RNA (shRNA), which is a substrate for Dicer, is more potent than its sequence-matched siRNA (9). Notably, in contrast to longer siRNA variants, shorter siRNA variants with increased potency and functional polarity also have been reported (10–13).
Gene-silencing assays (i.e. a downstream read-out) were valuable in demonstrating that some siRNA variants, including dsiRNAs, are more efficacious than their sequence-matched canonical siRNAs, but these assays fail to provide information about the molecular mechanism underlying the improved potency. Here, we use identical target sequences to further characterize several sequence-matched siRNA variants and pinpoint the molecular basis of improved gene silencing.
MATERIALS AND METHODS
siRNA variants
All siRNA variants were ordered as RNase-free HPLC-purified single-stranded RNA (ssRNA) oligos (Integrated DNA Technologies). RNA oligos were resuspended in IDT annealing buffer (RNase-free 100 mM potassium acetate, 30 mM HEPES, pH 7.5). The sequences of siRNAs used are shown in Figure 1. Negative control siRNAs against enhanced green fluorescence protein (siGFP) had the following guide strand sequence: siGFP site S1C, 5′ CAGAUGAACUUCAGGGUCAGC; or siGFP site S1E, 5′ GAUGAACUUCAGGGUCAGCUU. Complementary strands of siRNA variants were annealed by mixing equimolar amounts, heating to 95°C for 3 min and removing the heating block from the heating source to allow slow cooling to room temperature. siRNA variants were stored at −20°C. The siRNAs were diluted serially with DEPC-treated water to achieve concentrations to be used for transfection.
Figure 1.

Design of sequence-matched siRNA variants used in this study. (a) H5 series of variants. (b) H6 series of variants. DNA nucleotides are represented as underlined ‘d_’. The predicted in vitro Dicer cleavage sites are indicated by the open arrowheads. All variants are sequence-matched with respect to the 5′-end of the 21mer bottom strand. The color scheme used here will be consistent throughout the remainder of this report.
Plasmids and cell lines
Plasmids encoding myc_Dicer, FLAGHA_Ago1 and FLAGHA_Ago2 have been described (14,15). The plasmid encoding myc_TRBP has been described (16). The psiCHECK-hnRNPH –S (sensor for the bottom strands) and –AS (sensor for the top strands) were used as previously described (6,17). For experiments with exogenously provided top strand target, the psiCHECK-hnRNPH-AS was used. HCT116 and HEK293 cell lines were purchased from American Tissue Culture Collection. For 50% inhibitory concentration (IC50) determination experiments, HCT116 colon carcinoma and HEK293 cells were used. For all other transient transfection assays, HEK293 cells were used. For RNA immunoprecipitation (RIP) experiments, HEK293 cells that stably express FLAGHA_Ago1 or FLAGHA_Ago2 were used. Stable cell lines were generated via drug selection as per Landthaler et al. (14). After selection, stable cell lines were maintained in drug-free media.
Co-transfections and IC50 determination
Co-transfections for IC50 determination were performed using the Dual Luciferase Assay kit (Promega) as previously described (17). Briefly, cells were seeded in 48- or 96-well plates. The following day, the cells were co-transfected with the indicated concentration of siRNA variant and the indicated top or bottom strand psiCHECK-hnRNPH sensor plasmid. Non-specific pcDNA3.1 was used to equalize the total nucleic acid mass content per well. All silencing results were normalized to an equimolar concentration of canonical 21mer siGFP site 1C or site 1E as previously reported (17). Graphpad Prism was used to calculate the IC50 values. For all data points, transfections were performed n ≥ 2 in duplicate, represented as the mean ± standard error of the mean (SEM).
Electrophoretic mobility shift assays
Native HEK293 cell lysate was recovered using Cytobuster reagent (EMD Biosciences). Lysates were cleared via centrifugation, and only the soluble supernatant was used for subsequent experiments. Protein concentrations were determined by Bradford Assay.
The 32P radiolabeling protocol was performed essentially as previously described (17). Briefly, the top strands of the siRNA variants were 5′ 32P labeled using T4 PNK and G25 column purified (GE Healthcare). Equimolar amounts of 32P-labeled and complementary cold bottom strands were annealed using the annealing procedure described above.
Fifteen micrograms of HEK293 endogenous lysate (or lysate from 24 h post-transfected cells transiently overexpressing epitope-tagged RNAi proteins) were incubated with 1 nM of each of the siRNA variants for 45 min at room temperature. Samples were run on a 5% native polyacrylamide gel for 1.5–2 h at 190 V. The gel was dried for 1 h at 80°C. Gel images were acquired via phosphorimaging (Typhoon, GE) and exposed to film for quantitative and qualitative analysis, respectively. The percentage bound is calculated as the density of the shifted band(s) divided by the density of the corresponding input (lysate-free) lane. The signal from the ‘well bottom’ is not included in the calculation.
Small RNA deep sequencing
HEK293 cells were transfected with 500 pM of each of the siRNA variants in triplicate (designated as ‘xxxx_1, xxxx_2, xxxx_3’) using Lipofectamine RNAiMAX (Invitrogen) in six-well plates. Non-transfected cells (‘non’) and cells transfected with RNAiMAX alone (‘mock’) served as negative controls. Twenty-four hours after transfection, total RNA was collected with RNA-STAT 60 (Amsbio) following the manufacturer’s protocol. RNA quality was verified spectrophotometrically (Nanodrop). One microgram total RNA was submitted to the City of Hope DNA Sequencing/Solexa Core for small RNA deep sequencing (HiSeq, Illumina). Barcoded adaptor ligation, amplification and small RNA isolation was performed using the manufacturer’s protocol (Illumina). The manufacturer’s protocol isolates small RNAs between 18–30 nt in size.
Small RNA deep sequencing analysis
For quantification of exogenous siRNA variants, all analysis was done using R statistical language and Bioconductor packages. The sequences were trimmed to remove the 3′ Illumina adapter first. To identify the siRNA products and the unique trimmed sequences with their number of occurrence, we counted the total number of reads in each sample scaled to 50 million. These unique sequences were then matched to the bottom and top strand of the siRNA variants separately. The products were identified with the following criteria: (i) >1000 scaled counts; (ii) at least 16 perfect contiguous matches to the reference bottom or top strand sequence; (iii) no more than two untemplated 5′ additions; and (iv) no more than six untemplated 3′ additions. When displaying the strand count data, we format the values as RNA counts per million. For quantification of microRNAs (miRNAs), sequence data analysis and statistical comparisons were carried out using Bioconductor packages and an in-house developed analysis pipeline using R statistical environment (18). After mapping the deep sequencing data onto the human genome hg18 and counting the reads for the mature miRNAs in the miRBase v19, raw miRNA expression data were ported to ‘edgeR’ bioconductor package for TMM (trimmed mean of M values) normalization.
Immunoblotting
Unless otherwise noted, all antibodies were diluted 1:3000 in PBS-T. Expression of epitope-tagged Dicer, TAR RNA binding protein (TRBP) and Argonaute 2 (Ago2) were verified via immunoblotting using antibodies to FLAG (Sigma or Cell Signaling) or myc (Santa Cruz Biotechnology). Endogenous proteins were probed with the following antibodies: hnRNP H 1:200 in 5% milk PBS-T (Santa Cruz Biotechnology), HSP70 (Santa Cruz Biotechnology), alpha-actin (Santa Cruz Biotechnology), TRBP 1:500 (Abcam), Dicer 1:500 (Santa Cruz Biotechnology), GAPDH (Cell Signaling).
Small RIP
HEK293 cells stably expressing FLAGHA_Ago2 (HEK293FLAGHA_Ago2) or FLAGHA_Ago1 (HEK293FLAGHA_Ago1) were transfected with 10 nM of each siRNA variant using Lipofectamine RNAiMAX (Invitrogen). After 24 h, cell lysates were collected using lysis and immunoprecipitation (IP) wash buffer [50 mM Tris–HCl, pH 8, 137 mM NaCl, 1% v/v Triton X-100, 1× Proteinase Inhibitor Cocktail (Roche), 0.1 U RNasIN (Promega)]. Lysates were cleared via centrifugation, and only the soluble supernatant was used for subsequent experiments. IP was performed with 2 mg of cell lysate and 20 μl anti-FLAG M2 affinity gel (Sigma). Immunoblotting, using rabbit anti-FLAG antibody (Cell Signaling), was performed to confirm expression of FLAGHA_Ago2 or FLAGHA_Ago1 in both the input (30 μg) and immunoprecipitated (∼3 μl of pellet) samples. The immunoprecipitated nucleic acid was dissociated from the affinity matrix via boiling for 5 min in 2× RNA loading dye. Northern blotting (8% PAGE, 8 M urea) of the immunoprecipitated RNAs was performed and respective strands were detected with the following 32P-labeled probes: probe against H5 and H6 bottom strand (5′ TGAATCAGAAGATGAAGTC); probe against H5 and H6 top strand (5′ GACTTCATCTTCTGATTCA); probe against mature miR-19b 3′ arm (5′ TCAGTTTTGCATGGATTTGCACA); probe against mature miR-21 5′ arm (5′ TCAACATCAGTCTGATAAGCTA). The membranes were stripped and reprobed for each small RNA. Eighty fmol of single strand bottom or top strand, both 21 nt and 25/27 nt, were loaded as positive control size markers. For input RNA lanes, 12 or 25 μg (∼40% v/v) of input RNA was used. Densitometry was performed by phosphorimaging.
Co-transfection of targets when investigating the role of target presence
For studies exploring the effects of endogenous heterogeneous nuclear ribonucleoprotein H (hnRNPH) levels in dual luciferase assay conditions, the same transfection protocol used for dual luciferase assay was proportionally scaled up to a 12-well format at the indicated siRNA concentrations. For studies exploring the amount of target mRNA in RIP assays in HEK293FLAGHA_Ago2 cells, co-transfections of 10 nM of the siRNA variants with 2.4 μg of the top-strand sensor psiCHECK plasmid or psiCHECK plasmid lacking the target sequence (negative control) were performed in 10 cm plates. The samples were assayed using the RIP procedure mentioned above.
RESULTS
Sequences used and nomenclature
We abbreviate a canonical siRNA as a ‘21mer’. The canonical site H5 (Figure 1a, blue and red strands) and site H6 (Figure 1b, green and orange strands) 21mers were designed previously by a single nucleotide siRNA walk along the luciferase reporter plasmid psiCHECK-hnRNPH (17). Several sequence-matched variations on the canonical siRNA were synthesized for this study (Figure 1). For example, the 19mer with two deoxythymidine overhang (19merTT) variant (1) is available commercially and is used in many RNAi studies in mammalian systems. The rationally designed dsiRNA variant, featured by longer length, asymmetric termini and placement of DNA nucleotides at the 3′-end of the undesired strand, integrates these three additional design features to improve silencing of the intended strand (as drawn, the bottom strand). The 16mer with two deoxythymidine overhang (16merTT) variant, one of a class of variants shorter than the canonical siRNA (10,12), was reported to be more potent than its sequence-matched 21mer (11) yet challenges our understanding of the minimal length requirements for an effective RNAi trigger. Collectively, we refer to these different constructs as ‘siRNA variants’. In Figure 1, as drawn, the bottom strands of the siRNA variants are perfectly complementary to a segment of the coding region of the hnRNPH mRNA sequence. The top strands of the siRNA variants are not perfectly complementary to any known human gene. We will use the ‘bottom’ and ‘top’ strand nomenclature consistently for all our assays.
Improved potency and bottom strand selection by dsiRNA
The siRNA variants were subjected to a dual luciferase gene-silencing assay in parallel. Our assay uses different luciferase reporter plasmids to quantify the utilization of the top or bottom strand and to allow a relative comparison of the silencing ability of each strand. The R- (designed to preferentially load the bottom strand and discourage top strand loading compared with the parent canonical 21mer) or L-form (designed to preferentially load the top strand and discourage bottom strand loading compared with the parent canonical 21mer) dsiRNAs exhibit the expected strand biasing properties (6) compared with their sequence-matched 21mers (Supplementary Figure S1). With this strand polarity verified, for simplicity for the remainder of the study, ‘dsiRNA’ indicates the R-form dsiRNA, which biases strand selection toward the bottom strand.
The IC50 was determined for the siRNA variants (Figure 2, insets) to compare their gene-silencing potency. No differences in gene-silencing trends were observed using HCT116 (Figure 2) or HEK293 cells (Supplementary Figure S2), indicating no cell-type specific differences in siRNA performance.
Figure 2.

Gene-silencing activity of siRNA variants. Gene silencing was assessed by dual luciferase assay using plasmids that report the RNAi activity of the bottom (blue and green) or top (red and orange) strands for the H5 (a) and H6 (b) set of variants. Assay performed in HCT116 cells. All data points were normalized to an equimolar amount of non-targeting siRNA. n ≥ 2, in duplicate, mean ± SEM. IC50 values for each strand of each siRNA variant are included as insets.
The H5 21mer demonstrated ∼2.5-fold better silencing by the top strand compared with the bottom strand (Figure 2a), consistent with its predicted 5′-end thermodynamics (17) and strand-selection properties (19,20). The H5 19merTT bottom and top strands were slightly less potent in gene silencing but mirrored the trend of the H5 21mer (Figure 2a). In contrast, the IC50 for the H5 dsiRNA bottom strand was ∼2-fold lower compared with the H5 21mer bottom strand, and the IC50 of the H5 dsiRNA top strand was ∼3-fold higher compared with the H5 21mer top strand. The strands of the 16merTT, in either the H5 or H6 (below) variants, were 10–10 000× less potent than the other sequence-matched variants.
The H6 set of siRNA variants demonstrates the strand biasing properties of the dsiRNA variant in a different context. For the H6 series (Figure 2b), the IC50 values for the 21mer bottom and top strands are similar, also consistent with computational predictions. Again, gene silencing by the bottom and top strands of the H6 19merTT are slightly less potent, yet mirror the gene-silencing trend of the H6 21mer. The IC50 for H6 dsiRNA bottom strand, however, is ∼2-fold lower than the H6 21mer bottom strand, and the H6 dsiRNA top strand was ∼2-fold higher than the H6 21mer top strand. These results independently confirm the previous finding that the intended strand (i.e. the bottom strands) of dsiRNAs are more potent in silencing compared with the bottom strands of sequence-matched canonical siRNAs.
The siRNA variants used in this study can target endogenous levels of hnRNPH protein. Therefore we were curious whether, in the co-transfection conditions used for dual luciferase assays, endogenous hnRNPH is affected. In our co-transfection conditions, the amount of target transcript generates a quantifiable level of Renilla luciferase luminescence. At 1 nM transfections of the H6 21mer and H6 dsiRNA co-transfected with the sensor plasmids for either the bottom or top strand, we observed no decrease in expression of hnRNPH protein (Supplementary Figure S3). As the bulk of the siRNA concentrations tested are ≤1 nM, we do not believe that the amount of target transcript in these conditions influences the IC50 value calculations of our studies.
Taken together, the H5 and H6 series of 21mer siRNAs represent two different contexts. On one hand, the H5 21mer (and H5 19merTT) represents a scenario where the potency of the bottom strand is poorer than its top strand. On the other hand, the H6 21mer (and H6 19merTT) represents a scenario where the bottom strand is equally potent as the top strand. Regardless of the context, however, dsiRNAs displayed better gene-silencing potency of the bottom strands and relatively worse silencing of the top strands compared with their sequence-matched 21mers and 19merTT.
dsiRNAs form high-molecular-weight complexes more robustly than the other variants in vitro
We wanted to explore the molecular basis for the results of the reporter-based gene-silencing assays. Therefore, we conducted native gel electrophoretic mobility shift assays (EMSAs) to monitor RNA-induced silencing complex (RISC) assembly. Formation of the RISC is an essential step in RNAi-mediated gene silencing, and proceeds from a pre-RISC (sometimes referred to as a RISC loading complex, RLC) to a mature RISC. Top strands of the siRNA variants were 32P radiolabeled, annealed to equimolar amounts of bottom strand and incubated with cell lysate from HEK293 cells. Addition of lysates to the labeled siRNAs resulted in a band shift in native acrylamide gel electrophoresis. The progression of shifted complexes [pre-RISC (lower) to RISC (higher)] is labeled as in Liu et al. (21). The nomenclature for these pre-RISC and RISC complexes is similar to another naming convention (Complex V and Complex II, respectively) used by the Tomari group (22). For both the H5 and H6 siRNA variants, the dsiRNAs showed 2- to 3-fold better formation of shifted complexes compared with the H5 21mer and 19merTT (Figure 3a and b). The 16merTT failed to form any appreciable shifted complexes.
Figure 3.

Robust high-molecular-weight complex formation by dsiRNAs in RISC assembly assays. Immunoblotting confirmed the transient expression of epitope-tagged RNAi proteins (top). ‘non’ = cells without respective plasmid transfection. EMSA assay performed with the H5 (a) or H6 (b) sets of variants. Asterisk (*) indicates position of the 5′ 32P label. Labeled RNAs in the absence of cell lysate (‘−’) serve as negative controls. Amount of band shift was calculated as the sum of shifted bands divided by the respective negative control lane. Images and densitometry are representative of two biological replicates.
As an additional step to identify which proteins form these higher-molecular-weight complexes, we transiently overexpressed epitope-tagged Dicer, TRBP or Ago2 in HEK293 cells and repeated the EMSA analysis using these lysates. These three proteins are the core constituents of a functional minimal RISC in vitro (23). Cells treated with negative control empty vector showed identical band shift patterns as endogenous HEK293 cell extract (data not shown). The overexpression of the various key RNAi proteins was verified by immunoblotting (Figure 3, top). With all lysates, the H5 and H6 dsiRNAs continued to show better formation of pre and mature RISC compared with the other siRNA variants (Figure 3). Lysates containing overexpressed TRBP showed the most pronounced band shift, particularly for the pre-RISC complex, consistent with the role of TRBP in small RNA loading before mature RISC formation. Compared with endogenous HEK293 cell lysate, overexpression of Dicer led to a moderate increase in binding of the dsiRNA species compared with the other variants. The band shift patterns using lysates from HEK293 cells overexpressing Ago2 was similar to the pattern using endogenous HEK293 cell lysate, again with dsiRNAs showing more robust band shifts than any of the other variants. Consistently, the 16merTT variants did not show appreciable band shifts in any of the EMSAs. These results demonstrate that dsiRNAs form pre and mature RISC more robustly than the other variants in vitro, suggesting that improved initial loading of siRNA variants at an upstream step in the RNAi pathway contributes to their improved downstream potency.
dsiRNA bottom strands persist more than top strands
During siRNA strand selection, small RNA strands that are not loaded into RISC complexes are degraded, while loaded strands are stabilized. To correlate the abundance of the bottom and top strands of the siRNA variants to gene-silencing function in a cell-based assay, the siRNA variants were transfected at 500 pM into HEK293 cells in triplicate and RNA was collected for small RNA deep sequencing. This concentration corresponds to the log phase of the dual luciferase gene-silencing assay and is greater than the IC50 values of the strands of the siRNA variants. To verify the integrity and reproducibility amongst replicates of the small RNA sample preparation, a similar number of raw counts for small RNAs (primarily miRNAs) were observed for all replicates (Supplementary Table S1). Nine of the top ten highest endogenously expressed miRNAs were constant among samples (Supplementary Table S2), confirming that the transfection of siRNA variants at this concentration does not dramatically perturb miRNA levels.
We counted the number of respective bottom and top strands for the 21mers and dsiRNAs, displayed as RNA strand counts per 1 million small RNA reads. In both H5 and H6 sets, the number of dsiRNA bottom strands, as well as the bottom:top strand ratio, is higher than their sequence-matched 21mer (Table 1). Our data are consistent with previous reports, demonstrating that more bottom strand counts and a higher bottom:top strand ratio correlates with stronger bottom strand silencing (24). Likely due to the technical limitations of the adaptor ligation step for small RNA deep sequencing sample preparation, strands with terminal 3′ deoxythymidine nucleotides had low counts and were not deemed to be reliable for analysis. These data support our model that dsiRNAs bias strand selection toward the bottom strand compared with sequence-matched 21mers, leading to persistence of the bottom strand and therefore improved bottom strand gene silencing at equimolar concentrations.
Table 1.
Strand counts and bottom:top strand ratio of siRNA variants, assessed by small RNA deep sequencing, displayed as counts per million
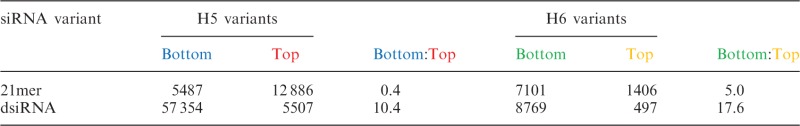 |
Values are the average from n = 3 transfections.
Enhanced gene silencing by dsiRNAs is coupled to Dicer processing, not Dicer products
Previous in vitro Dicer processing studies predict that dsiRNAs are processed primarily into 21mers (6). In contrast, when transfecting our dsiRNAs and counting each strand by deep sequencing, the predominant strand counts (Table 2) of the H5 dsiRNAs would assemble into a 20 + 2mer, not a 21mer, and the predominant strand counts of the H6 dsiRNA would assemble into a 19 + 3mer. Although these species of complementary strands may not be in duplex form intracellularly, we nonetheless cannot rule out the possibility that the H5 20 + 2mer and H6 19 + 3mer structure, and not the Dicer-dependent dsiRNA format per se, provides the gene-silencing improvement. To test this, we chemically synthesized the H5 20 + 2mer and H6 19 + 3mer (Figure 4a and b) and transfected these in parallel with their parent 21mer and dsiRNAs in a dual luciferase assay. If the H5 20 + 2mer and H6 19 + 3mer, and not the parent dsiRNA, were responsible for improved gene silencing, we would expect equivalent levels of gene silencing (more specifically, equivalent ratios of bottom:top strand silencing). This was not the case (Figure 4c and d), indicating that the dsiRNA format, and not its Dicer processed product, is responsible for the more efficacious gene silencing.
Table 2.
Length and percentage of top three most abundant strand counts for each strand, averaged from triplicate transfections
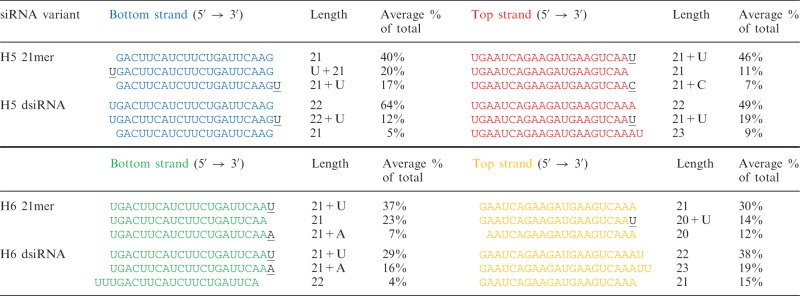 |
Underlined letters represent unambiguous untemplated additions.
Figure 4.

Synthetic dsiRNA products not similar to dsiRNA in gene silencing. (a and b) Schematic of H5 20 + 2mer and H6 19 + 3mer siRNA variants, compared with their sequence-matched 21mer and dsiRNA format. Boxed nucleotides represent nucleotides not predicted by in vitro Dicer processing. (c and d) Dual luciferase gene-silencing assay. n = 3, in duplicate, mean ± SEM. P-value shown from unpaired Student’s t-test.
Further, previous in vitro dicing experiments (6) would have predicted that H5 dsiRNA would be processed by Dicer into a species reminiscent of the H5 21mer with 5′ terminal guanine on the bottom strand (which is thermodynamically unfavorable for strand selection and gene silencing). The H5 20 + 2mer bottom strand species shown in our deep sequencing results, however, has a 5′ terminal uridine on the bottom strand (which is relatively more favorable for strand selection and gene silencing). Therefore, again, it is still unclear whether the improved gene-silencing properties of our dsiRNAs are due to Dicer processing per se or whether the Dicer products coincidentally have favorable end thermodynamics. To test this, we conducted gene-silencing experiments with a canonical siRNA and sequence-matched dsiRNA shifted one nucleotide upstream from site 5, targeting site 4 (H4 21mer and H4 dsiRNA, respectively) (17) (Supplementary Figure S4a). If Dicer processing generates a 22mer from H4 dsiRNA, the bottom strand would contain a 5′ terminal guanine and would not be predicted to be favorable for strand selection compared with the H4 21mer bottom or top strand. Consistent with our model, however, the H4 dsiRNA again demonstrated better gene silencing by its bottom strand and worse silencing by its top strand compared with its sequence-matched H4 21mer (Supplementary Figure S4b). These results continue to show that Dicer processing, necessitated by the 25/27mer length of dsiRNAs, improves gene silencing by the bottom strand.
More dsiRNA bottom strands are loaded into Ago proteins
Ago2 is the core effector protein of the RISC and executes the ‘slicer’ step of siRNA-mediated RNAi. One strand of an siRNA must be loaded into Ago2 to activate the RISC and identify and cleave target mRNA. To test whether the improved efficacy of the dsiRNA may be due to better bottom strand incorporation into the RISC, a RIP assay was performed. HEK293 cells stably expressing FLAG-Ago2 (HEK293FLAGHA_Ago2 cells) were transfected with 10 nM of each of the siRNA variants. Untransfected cells and cells transfected with siGFP were used as negative controls. FLAGHA_Ago2 was immunoprecipitated and Ago2-bound RNAs were isolated and purified for a northern blot assay. Successful and specific Ago2 IP was verified by immunoblotting (Figure 5a and b). In the northern blots, the top strand of H5 21mer and 19merTT was detected at similar levels, but the bottom strand was not detectable (Figure 5c). These results are consistent with the gene-silencing data. For the H5 dsiRNA, however, a stronger relative abundance of processed bottom strand to top strand was detected compared with other variants (Figure 5c). Northern blots performed with the H6 variants also mirrored the gene-silencing trends; the processed H6 bottom and top strand are detected at nearly equal amounts for the 21mer and 19merTT variants, but a stronger relative abundance of processed bottom strand to top strand for the dsiRNA was detected (Figure 5d). Both Dicer processed (∼21–22 nt) and unprocessed (∼25–27 nt) dsiRNA strands were immunoprecipitated (Figure 5c and d, white arrowheads), but we only consider the Dicer processed product when assessing the amount of loaded bottom or top strand. The 16merTT strands did not show appreciable Ago2 loading with the exception of the H6 16merTT top strand, the only 16merTT strand that had a sub-100 pM IC50 value.
Figure 5.

More bottom strands load into Ago2 for dsiRNAs compared with canonical siRNAs. Experiment performed in HEK293FLAGHA_Ago2 cells. Immunoblotting confirmed the IP procedure for the H5 (a) and H6 (b) set of variants, respectively. Lysate from wild-type HEK293 cells transiently transfected with pFLAG-Ago2 or pFLAG empty vector serve as a positive and negative control, respectively. Northern blots showing the levels of input and immunoprecipitated small RNAs for the H5 (c) and H6 (d) set of variants. Non-transfected cells or cells transfected with siGFP served as negative controls. White arrows indicate full-length (not Dicer processed) dsiRNA strands. Representative images from three independent biological replicates.
Four Ago proteins (Ago1-4) exist in mammals. For most adult tissues, Ago2 is typically expressed at the highest level, followed by Ago1. All Ago proteins randomly load endogenous miRNAs (25) and exogenous shRNAs (26) at levels proportional to each individual Ago protein expression level. Ago2 is the only mammalian family member that possesses catalytic ‘slicer’ activity when loaded with a small RNA that is perfectly complementary to a target mRNA. Agos 1/3/4 repress target mRNAs via a slicer-independent miRNA-like mechanism (27). We were curious about the strand loading of our variants in slicer-deficient Ago1. We repeated the RIP assay by immunoprecipitating Ago1 using HEK293FLAGHA_Ago1 cells. In the northern blots, the bottom strand signal is nearly undetectable for 21mer-, 19merTT- and 16merTT-treated cells, but dsiRNA-treated cells show a detectable bottom strand band. Hence, similar to Ago2, dsiRNAs show a higher relative amount of Ago1-loaded bottom strand compared with top strand when compared with other variants (Supplementary Figure S5a and b). These data further support our model of improved gene silencing by dsiRNAs compared with sequence-matched canonical siRNAs, demonstrated here at the level of superior Ago2 and Ago1 loading of the dsiRNA bottom strand relative to the top strand.
In mammals, it is unclear whether the amount of target mRNA makes a difference in siRNA potency. For miRNAs, target mRNA is shown to facilitate miRNA turnover by untemplated addition (tailing) or subtraction (trimming) of uridines and adenosines to the 3′ end of the miRNA (28). In several mammalian small RNA deep sequencing datasets, including our own (Table 2), tailing is observed (29–31), but the basis of the tailing in mammalian cells for exogenously delivered siRNA, and whether it is related to target amount, is not clear. To investigate the influence of the amount of target mRNA in our RIP assays, we repeated our RIP assays in cells co-transfected with the top strand sensor psiCHECK-hnRNPH vector. As stated before, the amount of top strand sensor plasmid generates a quantifiable amount of luciferase luminescence. We found that exogenously providing an mRNA target for the top strand did not influence Ago2 loading (Figure 6a and b), consistent with other findings (32).
Figure 6.

Loading of each strand of siRNA variants into Ago2 is not dependent on mRNA target presence. Co-transfections of the indicated psiCHECK strand sensor (scaling up from the amount used in Figure 2) and 10 nM of the indicated H5 (a) or H6 (b) siRNA variant were transfected into HEK293FLAGHA_Ago2 cells. Small RNA immunoprecipitation assay followed by Northern blotting was conducted as in Figure 5. Cells treated with siGFP served as negative controls. Additional size markers (lanes 1–4) are included to indicate the size of the probed small RNA species.
Ago2 has been reported to exist in a minimal pre-RISC complex with Dicer and TRBP (33). Dicer possesses one double-stranded RNA binding domain (dsRBD), and TRBP contains three dsRBDs. Our EMSA studies demonstrated that TRBP robustly binds small dsRNA ≥19 bp. To determine if contaminating TRBP or Dicer, not exclusively Ago2, is responsible for immunoprecipitating our small RNA variants in our IP buffer conditions, we immunoblotted against TRBP or Dicer in one of our FLAG-immunoprecipitated samples. Small yet detectable amounts of Dicer, but not TRBP, appeared in the immunoprecipitated samples (Supplementary Figure S6). Taking into account the strong Dicer protein signal in input lanes at 1/66× the mass of the immunoprecipitated samples, however, we believe any small RNA immunoprecipitated by Ago2-bound Dicer to be minor, and that Ago2 is the primary protein immunoprecipitating the small RNAs.
DISCUSSION
In each of our assays, probing various stages of the RNAi mechanism, the dsiRNAs consistently demonstrated better gene silencing of the bottom strand, better in vitro complex formation with proteins known to be important for RNAi, more accumulation of the bottom strand and better bottom strand loading into Ago proteins compared with sequence-matched canonical and other siRNA variants. These properties of dsiRNAs were demonstrated in the context of two different sets of canonical siRNA sequences. First, the dsiRNA format improved the potency of a canonical siRNA with a poor bottom strand (H5 21mer). Admittedly, an siRNA with poor strand selection thermodynamic properties would likely not be the basis for a therapeutic siRNA. Strikingly, we also observe that the dsiRNA format can further improve the potency of a reasonably strong canonical siRNA bottom strand (H6 21mer). Rather than iterative screens or sequence-specific modifications aimed at improving siRNA potency (34,35), we propose the dsiRNA format as a widely applicable design feature to improve siRNA potency. Combining the results from this study with previous reports, we have observed at least 9 of 11 dsiRNAs show better efficacy compared with their sequence-matched canonical 21mers (5,6,36,37). Several independent reports have used dsiRNAs for their studies in vitro and in vivo (7,8,38–42). These reports exemplify the suitability of dsiRNAs for therapeutic RNAi applications. In terms of therapeutic efficacy, the ability to use the same molar amount of a dsiRNA, achieve the same amount of knock-down as a canonical siRNA, and discourage use of the unintended strand are advantageous from the perspective of reducing RISC competition (43), using lower doses and therefore more cost-effective manufacturing for RNAi-based therapeutics, and reducing siRNA- and miRNA-like off-target effects by the unintended strand (44).
In contrast to previous reports focusing solely on downstream read-out assays, our study explored the mechanistic basis underlying canonical siRNA versus dsiRNA performance. Further, we provided information about strand persistence and the involvement and fate of the top strand. At saturating concentrations, the strand bias conferred by dsiRNAs is lost, serving as a potential explanation for some, but not all, of the data in a study claiming that dsiRNAs confer no improved silencing potency compared with canonical siRNAs (45). Another potential explanation for the apparent discrepancy in canonical siRNA versus dsiRNA gene-silencing potency is assay choice (dual luciferase assay versus branched DNA assay versus quantitative polymerase chain reaction).
Unless otherwise specified to the manufacturer, strands of siRNA oligos are typically chemically synthesized with a 5′ hydroxyl (5′ OH). Once transfected into cells, the 5′ OH is monophosphorylated (46) by endogenous Clp1 (47), a necessary step for RISC loading. Clp1 readily 5′ monophosphorylates RNA within the time frame of 10–30 min in vitro and is independent of single or double strandedness, overhang type, overhang chemistry or nucleotide identity (47). Using sequence-matched siRNAs synthesized with a 5′ OH or 5′ monophosphate, it has been demonstrated that siRNAs with asymmetric strand functionality are not affected by 5′ monophosphorylation in vitro (20), and that the 5′ monophosphorylation step is not a rate-limiting step for gene silencing in vivo (48). These reports argue against the idea that 5′ monophosphorylation may be affecting the asymmetric gene-silencing properties, conducted on the time frame of 24 h after transfection, by our siRNA variants.
The next generations of RNAi-based therapeutics will use chemically modified (e.g. 2′ O-methyl, 2′ fluoro) siRNAs to reduce innate immunogenicity and impart increased RNase resistance. Our study, using unmodified siRNA variants, did not explore the innate immunogenicity as a possible reason for increased potency of dsiRNAs because other studies demonstrated that chemical modifications to siRNAs and dsiRNAs reduced inflammatory cytokine induction without compromising gene silencing (36,45). Careful placement of chemical modifications (e.g. not in the Dicer-processing site) should not disrupt the favorable strand biasing and RISC-loading properties of dsiRNAs.
The 19merTTs were only slightly less potent in gene-silencing assays and were comparable in Ago2 loading compared with 21mers. Both Dicer and Ago2 contain Piwi-Argonaute-Zwille (PAZ) domains, which bind to 3′ RNA overhangs. The fact that deoxythymidine overhangs are poor substrates for PAZ domains in vitro (49) may explain the slightly reduced potency of 19merTTs. In vitro EMSA assays revealed, however, that the band shift patterns between sequence-matched 21mers and 19merTTs were not similar. For example, in the presence of Dicer-overexpressed HEK293 lysates, the H5 19merTT variant displays several smaller intermediate-molecular-weight complexes compared with the H5 21mer. Interestingly, the H6 19merTT variant does not form these intermediate-molecular-weight complexes. Sequence-specific parameters may determine how Dicer binds to certain 19merTTs.
The current literature highlights a debate whether the presence or binding affinity of Dicer or Dicer/TRBP with canonical siRNAs (i.e. Dicer products) plays a role in the potency of canonical siRNA (17,23,50); other groups propose that Dicer is dispensable for proper strand selection during RNAi silencing (22,51) and that loading into Ago2 is the best indicator of siRNA potency (52). Previous in vitro studies have shown that purified/recombinant Ago2 does not bind directly to 21mer siRNA (i.e. short dsRNA) but rather ssRNA or pre-miRNA (53,54), suggesting additional proteins are required to load the duplex form of siRNAs into the RISC. Numerous methods of assembling an active RISC in vitro have been described, with a variety of ss- or dsRNA substrates and various cell extracts or purified proteins (e.g. Dicer, TRBP, Ago2, HSP90, hC3PO) (22,23,52). Our EMSA data show that increased Ago2 protein in HEK293 lysates did not markedly increase formation of the high-molecular-weight complexes with a canonical siRNA in vitro. This finding suggests dsRNAs that resemble Dicer products, even with excess Ago2, are not directly loaded into Ago proteins, and further supports a ‘hand-off’ mechanism beginning earlier in the RNAi pathway (17). This hand-off mechanism has been reported for pre-miRNAs (i.e. a Dicer substrate) (55). Though uncertainty exists in the case of canonical siRNAs, we show here that dsiRNAs robustly bind Dicer and TRBP to form pre and/or mature RISC, load more of the bottom strand into Ago2 and indeed demonstrate improved efficacy of the bottom strand. Also, we see unprocessed dsiRNA in the Ago2 immunoprecipitated samples, suggesting that an additional benefit of dsiRNAs could be that their 25/27mer length meets the minimum length requirement of a dsRNA that can be directly loaded into Ago2, similar to direct Ago2 loading of pri-miRNAs (56), pre-miRNAs (54) or shRNAs in the absence of Dicer (57).
In sum, we report the parallel assessment of several sequence-matched siRNA variants, and provide mechanistic information at several stages of the RNAi pathway as to why equimolar amounts of dsiRNAs are more efficacious in gene silencing. Optimizing siRNA variants is essential for the development of RNAi-based therapeutics.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1 and 2 and Supplementary Figures 1–6.
ACKNOWLEDGEMENTS
We thank the following investigators who kindly provided plasmids for this study: Dr. Thomas Tuschl for the myc-Dicer and FLAG_HA_Ago1 and -Ago2 plasmids; Dr. Anne Gatignol for the myc_TRBP plasmid. We thank Harry Gao, Jinhui Wang and other members of the COH Solexa Core for small RNA deep sequencing sample preparation. We thank members of the Rossi lab for helpful scientific discussion. We thank the H.N. and Frances C. Berger Foundation Fellowship and the Helen and Morgan Chu Fellowship for their support of N.M.S.
FUNDING
United States National Institutes of Health [HL074704 to J.J.R.]. Funding for open access charge: National Institutes of Health [HL074704].
Conflict of interest statement: J.J.R. is a co-founder and scientific advisory board member of Dicerna Pharmaceuticals, a company employing therapeutic dsiRNAs. All other authors declare no conflict of interest.
REFERENCES
- 1.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 2.Snead NM, Rossi JJ. RNA interference trigger variants: getting the most out of RNA for RNA interference-based therapeutics. Nucleic Acid Ther. 2012;22:139–146. doi: 10.1089/nat.2012.0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Fougerolles A, Vornlocher HP, Maraganore J, Lieberman J. Interfering with disease: a progress report on siRNA-based therapeutics. Nat. Rev. Drug Discov. 2007;6:443–453. doi: 10.1038/nrd2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davidson BL, McCray PB., Jr Current prospects for RNA interference-based therapies. Nat. Rev. Genet. 2011;12:329–340. doi: 10.1038/nrg2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim DH, Behlke MA, Rose SD, Chang MS, Choi S, Rossi JJ. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat. Biotech. 2005;23:222–226. doi: 10.1038/nbt1051. [DOI] [PubMed] [Google Scholar]
- 6.Rose SD, Kim DH, Amarzguioui M, Heidel JD, Collingwood MA, Davis ME, Rossi JJ, Behlke MA. Functional polarity is introduced by Dicer processing of short substrate RNAs. Nucleic Acids Res. 2005;33:4140–4156. doi: 10.1093/nar/gki732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kubo T, Zhelev Z, Ohba H, Bakalova R. Modified 27-nt dsRNAs with dramatically enhanced stability in serum and long-term RNAi activity. Oligonucleotides. 2007;17:445–464. doi: 10.1089/oli.2007.0096. [DOI] [PubMed] [Google Scholar]
- 8.Kubo T, Zhelev Z, Ohba H, Bakalova R. Chemically modified symmetric and asymmetric duplex RNAs: an enhanced stability to nuclease degradation and gene silencing effect. Biochem. Biophys. Res. Commun. 2008;365:54–61. doi: 10.1016/j.bbrc.2007.10.116. [DOI] [PubMed] [Google Scholar]
- 9.Siolas D, Lerner C, Burchard J, Ge W, Linsley PS, Paddison PJ, Hannon GJ, Cleary MA. Synthetic shRNAs as potent RNAi triggers. Nat. Biotechnol. 2005;23:227–231. doi: 10.1038/nbt1052. [DOI] [PubMed] [Google Scholar]
- 10.Chang CI, Yoo JW, Hong SW, Lee SE, Kang HS, Sun X, Rogoff HA, Ban C, Kim S, Li CJ, et al. Asymmetric shorter-duplex siRNA structures trigger efficient gene silencing with reduced nonspecific effects. Mol. Ther. 2009;17:725–732. doi: 10.1038/mt.2008.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu CY, Rana TM. Potent RNAi by short RNA triggers. RNA. 2008;14:1714–1719. doi: 10.1261/rna.1161908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun X, Rogoff HA, Li CJ. Asymmetric RNA duplexes mediate RNA interference in mammalian cells. Nat. Biotechnol. 2008;26:1379–1382. doi: 10.1038/nbt.1512. [DOI] [PubMed] [Google Scholar]
- 13.Li Z, Kim SW, Lin Y, Moore PS, Chang Y, John B. Characterization of viral and human RNAs smaller than canonical MicroRNAs. J. Virol. 2009;83:12751–12758. doi: 10.1128/JVI.01325-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Landthaler M, Gaidatzis D, Rothballer A, Chen PY, Soll SJ, Dinic L, Ojo T, Hafner M, Zavolan M, Tuschl T. Molecular characterization of human Argonaute-containing ribonucleoprotein complexes and their bound target mRNAs. RNA. 2008;14:2580–2596. doi: 10.1261/rna.1351608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol. Cell. 2004;15:185–197. doi: 10.1016/j.molcel.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 16.Daher A, Laraki G, Singh M, Melendez-Pena CE, Bannwarth S, Peters AH, Meurs EF, Braun RE, Patel RC, Gatignol A. TRBP control of PACT-induced phosphorylation of protein kinase R is reversed by stress. Mol. Cell Biol. 2009;29:254–265. doi: 10.1128/MCB.01030-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sakurai K, Amarzguioui M, Kim DH, Alluin J, Heale B, Song MS, Gatignol A, Behlke MA, Rossi JJ. A role for human Dicer in pre-RISC loading of siRNAs. Nucleic Acids Res. 2011;39:1510–1525. doi: 10.1093/nar/gkq846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weng L, Wu X, Gao H, Mu B, Li X, Wang JH, Guo C, Jin JM, Chen Z, Covarrubias M, et al. MicroRNA profiling of clear cell renal cell carcinoma by whole-genome small RNA deep sequencing of paired frozen and formalin-fixed, paraffin-embedded tissue specimens. J. Pathol. 2010;222:41–51. doi: 10.1002/path.2736. [DOI] [PubMed] [Google Scholar]
- 19.Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 20.Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 21.Liu X, Jin DY, McManus MT, Mourelatos Z. Precursor microRNA-programmed silencing complex assembly pathways in mammals. Mol. Cell. 2012;46:507–517. doi: 10.1016/j.molcel.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoda M, Kawamata T, Paroo Z, Ye X, Iwasaki S, Liu Q, Tomari Y. ATP-dependent human RISC assembly pathways. Nat. Struct. Mol. Biol. 2010;17:17–23. doi: 10.1038/nsmb.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang HW, Noland C, Siridechadilok B, Taylor DW, Ma E, Felderer K, Doudna JA, Nogales E. Structural insights into RNA processing by the human RISC-loading complex. Nat. Struct. Mol. Biol. 2009;16:1148–1153. doi: 10.1038/nsmb.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou J, Song M-S, Jacobi AM, Behlke MA, Wu X, Rossi JJ. Deep sequencing analyses of DsiRNAs reveal the influence of 3′ terminal overhangs on dicing polarity, strand selectivity, and RNA editing of siRNAs. Mol. Ther. Nucleic Acids. 2012;1:e17. doi: 10.1038/mtna.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang D, Zhang Z, O'Loughlin E, Lee T, Houel S, O'Carroll D, Tarakhovsky A, Ahn NG, Yi R. Quantitative functions of Argonaute proteins in mammalian development. Genes Dev. 2012;26:693–704. doi: 10.1101/gad.182758.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu S, Jin L, Zhang F, Huang Y, Grimm D, Rossi JJ, Kay MA. Thermodynamic stability of small hairpin RNAs highly influences the loading process of different mammalian Argonautes. Proc. Natl Acad. Sci. USA. 2011;108:9208–9213. doi: 10.1073/pnas.1018023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Broderick JA, Salomon WE, Ryder SP, Aronin N, Zamore PD. Argonaute protein identity and pairing geometry determine cooperativity in mammalian RNA silencing. RNA. 2011;17:1858–1869. doi: 10.1261/rna.2778911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ameres SL, Horwich MD, Hung JH, Xu J, Ghildiyal M, Weng Z, Zamore PD. Target RNA-directed trimming and tailing of small silencing RNAs. Science. 2010;328:1534–1539. doi: 10.1126/science.1187058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baccarini A, Chauhan H, Gardner TJ, Jayaprakash AD, Sachidanandam R, Brown BD. Kinetic analysis reveals the fate of a microRNA following target regulation in mammalian cells. Curr. Biol. 2011;21:369–376. doi: 10.1016/j.cub.2011.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi YS, Patena W, Leavitt AD, McManus MT. Widespread RNA 3′-end oligouridylation in mammals. RNA. 2012;18:394–401. doi: 10.1261/rna.029306.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, Johnston WK, Russ C, Luo S, Babiarz JE, et al. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev. 2010;24:992–1009. doi: 10.1101/gad.1884710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dallas A, Ilves H, Ge Q, Kumar P, Shorenstein J, Kazakov SA, Cuellar TL, McManus MT, Behlke MA, Johnston BH. Right- and left-loop short shRNAs have distinct and unusual mechanisms of gene silencing. Nucleic Acids Res. 2012;40:9255–9271. doi: 10.1093/nar/gks662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacRae IJ, Ma E, Zhou M, Robinson CV, Doudna JA. In vitro reconstitution of the human RISC-loading complex. Proc. Natl Acad. Sci. USA. 2008;105:512–517. doi: 10.1073/pnas.0710869105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allerson CR, Sioufi N, Jarres R, Prakash TP, Naik N, Berdeja A, Wanders L, Griffey RH, Swayze EE, Bhat B. Fully 2′-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J. Med. Chem. 2005;48:901–904. doi: 10.1021/jm049167j. [DOI] [PubMed] [Google Scholar]
- 35.Bramsen JB, Pakula MM, Hansen TB, Bus C, Langkjaer N, Odadzic D, Smicius R, Wengel SL, Chattopadhyaya J, Engels JW, et al. A screen of chemical modifications identifies position-specific modification by UNA to most potently reduce siRNA off-target effects. Nucleic Acids Res. 2010;38:5761–5773. doi: 10.1093/nar/gkq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collingwood MA, Rose SD, Huang L, Hillier C, Amarzguioui M, Wiiger MT, Soifer HS, Rossi JJ, Behlke MA. Chemical modification patterns compatible with high potency dicer-substrate small interfering RNAs. Oligonucleotides. 2008;18:187–200. doi: 10.1089/oli.2008.0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Amarzguioui M, Lundberg P, Cantin E, Hagstrom J, Behlke MA, Rossi JJ. Rational design and in vitro and in vivo delivery of Dicer substrate siRNA. Nat. Protoc. 2006;1:508–517. doi: 10.1038/nprot.2006.72. [DOI] [PubMed] [Google Scholar]
- 38.Howard KA, Paludan SR, Behlke MA, Besenbacher F, Deleuran B, Kjems J. Chitosan/siRNA nanoparticle-mediated TNF-alpha knockdown in peritoneal macrophages for anti-inflammatory treatment in a murine arthritis model. Mol. Ther. 2009;17:162–168. doi: 10.1038/mt.2008.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bohle H, Lorenzen N, Schyth BD. Species specific inhibition of viral replication using dicer substrate siRNAs (DsiRNAs) targeting the viral nucleoprotein of the fish pathogenic rhabdovirus viral hemorrhagic septicemia virus (VHSV) Antiviral Res. 2011;90:187–194. doi: 10.1016/j.antiviral.2011.03.174. [DOI] [PubMed] [Google Scholar]
- 40.Darniot M, Schildgen V, Schildgen O, Sproat B, Kleines M, Ditt V, Pitoiset C, Pothier P, Manoha C. RNA interference in vitro and in vivo using DsiRNA targeting the nucleocapsid N mRNA of human metapneumovirus. Antiviral Res. 2012;93:364–373. doi: 10.1016/j.antiviral.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 41.Dore-Savard L, Roussy G, Dansereau MA, Collingwood MA, Lennox KA, Rose SD, Beaudet N, Behlke MA, Sarret P. Central delivery of Dicer-substrate siRNA: a direct application for pain research. Mol. Ther. 2008;16:1331–1339. doi: 10.1038/mt.2008.98. [DOI] [PubMed] [Google Scholar]
- 42.Lundberg P, Yang HJ, Jung SJ, Behlke MA, Rose SD, Cantin EM. Protection against TNFalpha-dependent liver toxicity by intraperitoneal liposome delivered DsiRNA targeting TNFalpha in vivo. J. Control. Release. 2011;160:194–199. doi: 10.1016/j.jconrel.2011.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Castanotto D, Sakurai K, Lingeman R, Li H, Shively L, Aagaard L, Soifer H, Gatignol A, Riggs A, Rossi JJ. Combinatorial delivery of small interfering RNAs reduces RNAi efficacy by selective incorporation into RISC. Nucleic Acids Res. 2007;35:5154–5164. doi: 10.1093/nar/gkm543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- 45.Foster DJ, Barros S, Duncan R, Shaikh S, Cantley W, Dell A, Bulgakova E, O′Shea J, Taneja N, Kuchimanchi S, et al. Comprehensive evaluation of canonical versus Dicer-substrate siRNA in vitro and in vivo. RNA. 2012;18:557–568. doi: 10.1261/rna.031120.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nykanen A, Haley B, Zamore PD. ATP requirements and small interfering RNA structure in the RNA interference pathway. Cell. 2001;107:309–321. doi: 10.1016/s0092-8674(01)00547-5. [DOI] [PubMed] [Google Scholar]
- 47.Weitzer S, Martinez J. The human RNA kinase hClp1 is active on 3′ transfer RNA exons and short interfering RNAs. Nature. 2007;447:222–226. doi: 10.1038/nature05777. [DOI] [PubMed] [Google Scholar]
- 48.Kenski DM, Willingham AT, Haringsma HJ, Li JJ, Flanagan WM. In vivo activity and duration of short interfering RNAs containing a synthetic 5′-phosphate. Nucleic Acid Ther. 2012;22:90–95. doi: 10.1089/nat.2011.0333. [DOI] [PubMed] [Google Scholar]
- 49.Ma JB, Ye K, Patel DJ. Structural basis for overhang-specific small interfering RNA recognition by the PAZ domain. Nature. 2004;429:318–322. doi: 10.1038/nature02519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Noland CL, Ma E, Doudna JA. siRNA repositioning for guide strand selection by human Dicer complexes. Mol. Cell. 2011;43:110–121. doi: 10.1016/j.molcel.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Betancur JG, Tomari Y. Dicer is dispensable for asymmetric RISC loading in mammals. RNA. 2012;18:24–30. doi: 10.1261/rna.029785.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ye X, Huang N, Liu Y, Paroo Z, Huerta C, Li P, Chen S, Liu Q, Zhang H. Structure of C3PO and mechanism of human RISC activation. Nat. Struct. Mol. Biol. 2011;18:650–657. doi: 10.1038/nsmb.2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lima WF, Wu H, Nichols JG, Sun H, Murray HM, Crooke ST. Binding and cleavage specificities of human Argonaute2. J. Biol. Chem. 2009;284:26017–26028. doi: 10.1074/jbc.M109.010835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tan GS, Garchow BG, Liu X, Yeung J, Morris JPt, Cuellar TL, McManus MT, Kiriakidou M. Expanded RNA-binding activities of mammalian Argonaute 2. Nucleic Acids Res. 2009;37:7533–7545. doi: 10.1093/nar/gkp812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu X, Jin DY, McManus MT, Mourelatos Z. Precursor microRNA-programmed silencing complex assembly pathways in mammals. Mol. Cell. 2012;46:507–517. doi: 10.1016/j.molcel.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trujillo RD, Yue SB, Tang Y, O′Gorman WE, Chen CZ. The potential functions of primary microRNAs in target recognition and repression. EMBO J. 2010;29:3272–3285. doi: 10.1038/emboj.2010.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang JS, Maurin T, Lai EC. Functional parameters of Dicer-independent microRNA biogenesis. RNA. 2012;18:945–957. doi: 10.1261/rna.032938.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.