


Abstract
DNA double-strand breaks (DSB) are considered as the most deleterious DNA lesions, and their repair is further complicated by increasing damage complexity. However, the molecular effects of clustered lesions are yet not fully understood. As the locally restricted phosphorylation of H2AX to form γH2AX is a key step in facilitating efficient DSB repair, we investigated this process after localized induction of clustered damage by ionizing radiation. We show that in addition to foci at damaged sites, H2AX is also phosphorylated in undamaged chromatin over the whole-cell nucleus in human and rodent cells, but this is not related to apoptosis. This pan-nuclear γH2AX is mediated by the kinases ataxia telangiectasia mutated and DNA-dependent protein kinase (DNA–PK) that also phosphorylate H2AX at DSBs. The pan-nuclear response is dependent on the amount of DNA damage and is transient even under conditions of impaired DSB repair. Using fluorescence recovery after photobleaching (FRAP), we found that MDC1, but not 53BP1, binds to the nuclear-wide γH2AX. Consequently, the accumulation of MDC1 at DSBs is reduced. Altogether, we show that a transient dose-dependent activation of the kinases occurring on complex DNA lesions leads to their nuclear-wide distribution and H2AX phosphorylation, yet without eliciting a full pan-nuclear DNA damage response.
INTRODUCTION
Genotoxic agents, including ionizing radiation (IR), induce a variety of DNA lesions, with DNA double-strand breaks (DSB) being the most deleterious type of damage, if not properly repaired. A key process in the repair of DSBs is the phosphorylation of the histone variant H2AX on serine 139 that occurs in a megabasepair chromatin region surrounding the breaks, forming visible γH2AX immunofluorescent foci (1). The phosphatidylinositol 3-kinase-related kinases (PIKK) ataxia telangiectasia mutated (ATM), DNA-dependent protein kinase (DNA–PK) and ataxia telangiectasia and Rad3-related (ATR) are the main kinases that phosphorylate H2AX (2–4). H2AX is critical for the efficient accumulation of DNA repair factors at the break site, and H2AX deficient mice show increased radiosensitivity (5–7), with the specific phosphorylation at serine 139 promoting cellular survival after IR (8). Several studies have shown that the formation of γH2AX is central in the protein recruitment and signalling cascade of the DNA damage response (9). However, not only the formation of distinct γH2AX foci but also the induction of pan-nuclear γH2AX signals has been reported as a cellular reaction to various stressors other than IR. After transfection of human cells with small DNA fragments an intense pan-nuclear γH2AX signal has been described (10). After ultraviolet (UV) irradiation not only the S-phase-dependent induction of γH2AX was reported but also a weaker nuclear-wide γH2AX in G1 cells that is dependent on nucleotide excision repair (NER) (11). Moreover, changes in chromatin structure via hypotonic treatment led to the formation of pan-nuclear γH2AX (12). Also adeno-associated virus infection can induce a pan-nuclear γH2AX response (13,14). The formation of a ring-shaped γH2AX and a subsequent nuclear-wide γH2AX signal was also observed in apoptotic cells (15). Induction by different stressors suggests the presence of different regulation mechanisms. The consequences of pan-nuclear γH2AX also seem to be variable: pan-nuclear γH2AX has no effect on cell survival after UV irradiation (8) or hypotonic treatment (12), but the hyperactivation of DNA–PK by transfection of small DNA molecules compromises DNA repair (10,16).
Heavy ion irradiation is increasingly being used in cancer therapy because of its physical advantages and increased relative biological effectiveness (RBE) compared with photon irradiation (17). Heavy charged particles in cosmic rays are a major contributor to health risks in long-term manned space exploration (18). The increased effectiveness of ion irradiation is often attributed to the induction of less repairable clustered DNA lesions with multiple damage sites in close proximity (19). After ion irradiation at low energy, the ionization events produced are localized in a submicrometer volume around the ion track (18), and formation of γH2AX foci is clearly observed in the damaged nuclear region traversed by the ion track (20). In this study, we demonstrate that the localized sub-nuclear induction of complex DNA damage elicits the pan-nuclear activation of ATM and DNA–PK that are additionally able to phosphorylate nuclear-wide H2AX in undamaged chromatin.
MATERIALS AND METHODS
Cell culture and irradiation
Normal human primary skin fibroblasts (AG01522, passage 10–16) and xeroderma pigmentosum, complementation group A (XPA)-deficient human fibroblasts (21) (GM00710B) were obtained from Coriell Cell Repository and grown in EMEM with 15% fetal calf serum (FCS). AT cells (AT1BR) express truncated ATM (22) (obtained from ECACC) and were cultivated in Ham’s F10 and 15% FCS. U2-OS cells stably transfected with 53BP1-GFP or MDC1-GFP were kindly provided by C. Lukas and grown in Dulbecco’s modified Eagle’s medium (DMEM) and 10% FCS. Mouse embryonic fibroblasts (MEF) cells expressing truncated DNA–PKcs (23) were kindly provided by D. Chen and grown in α-medium with nucleosides and 15% FCS. The X-ray repair cross complementing 4 (XRCC4) −/− MEF cell line (24) (kindly provided by F. Alt) was cultivated in DMEM and 15% FCS. Hamster ovary cells (AA8) were obtained from ATCC and grown in RPMI and 10% FCS. Cells were cultured as described previously (20). For irradiation, cells were grown on 24- × 24-mm glass cover slips. Irradiation was performed at the accelerator facility of the GSI Helmholtz Center for Heavy Ion Research using ions <10 MeV/nucleon (20). Low angle irradiation was conducted as described previously (25) with a fluence of 3 × 106 particles/cm2 corresponding to two to three ion traversals per nucleus and a dose proportional to the fluence and LET (linear energy transfer). Control samples were mock irradiated. Targeted single-ion cell irradiation was used for the quantitative measurement of immunofluorescence signals. Cells grown on polypropylene foils were stained for cell nuclei recognition with 100 nM Hoechst 33342 1 h before irradiation at the GSI heavy ion microprobe as described previously (26). For irradiation, different heavy ions with increasing ionization densities were used dependent on the availability at the accelerator. On the basis of their ionization densities they can be divided into three groups of lighter ions (carbon, nitrogen and neon), intermediate ions (titanium, chromium, iron and nickel) and very heavy ions (ruthenium, xenon, gold, lead and uranium). Values for the LET of the different ion species: 12C 290 keV/µm, 14N 400 keV/µm, 20Ne 460 keV/µm, 50Ti 2180 keV/µm, 54Cr 3000 keV/µm, 56Fe 3050 keV/µm, 64Ni 3800 keV/µm, 96Ru 7060 keV/µm, 136Xe 9300 keV/µm, 197Au 12800 keV/µm, 208Pb 13400 keV/µm and 238U 15000 keV/µm. Dosimetry relied on secondary electron transmission counting calibrated by CR39 nuclear track detectors or at the microprobe on the detection of ion-induced secondary electron cloud emission from the vacuum window. For irradiation at the microprobe, the growth medium was replaced with HEPES-buffered medium (20 mM, pH 7.2); post-irradiation incubation was done in normal medium with inhibitors as indicated. To reduce the area of the DNA damage focus in the intensity quantitation, all ions were targeted to one spot within the cell nucleus, unless stated otherwise. Mock irradiated cells at the microprobe correspond to non-irradiated cells on the same dish. X-ray irradiation was conducted as described previously (27).
Drug treatment
DNA–PKcs inhibitor IC86621 and ATM inhibitor KU55933 (both Calbiochem) were dissolved in dimethyl sulphoxide (DMSO) and used at 200 or 10 µM, respectively. Caffeine (Sigma) was used at 20 mM in growth medium. Cells were incubated in the presence of the inhibitor 1 h before and after irradiation for the time indicated.
Antibodies and immunological techniques
Antibodies against γH2AX (mouse monoclonal, JBW301, Millipore, 1:500), 53BP1 (rabbit, ab36823, Abcam, 1:2000), tubulin (mouse, clone B-5-1-2, Sigma, 1:50000), MDC1 (sheep, 1:400, kindly provided by M. Lavin), phosphorylated ATM (S1981) (mouse, Rockland, 1:200) and DNA–PKcs (S2056) (rabbit, Abcam, 1:100) were used. For immunoblots, horseradish peroxidase-conjugated anti-mouse or -rabbit secondary antibodies (Amersham Biosciences) were diluted 1:10000. Secondary Alexa 488- and Alexa 568-conjugated goat anti-rabbit, anti-mouse and anti-sheep antibodies were used at 1:400 (Invitrogen) in immunofluorescence of cells fixed in 2% formaldehyde and permeabilized as described previously (25). For extraction of soluble proteins, cells were incubated with cytoskeleton and cytoskeleton stripping buffer and fixed for 30 min with modified Streck Tissue Fixative as described previously (20). DNA was counterstained with 1 µM ToPro-3 or 1.5 µM propidium iodide (PI) containing 100 µg/ml of ribonuclease.
Microscopy and measurement of pan-nuclear immunofluorescence intensity
Confocal laser scanning micrographs were obtained using a Leica confocal system as described previously (25). Representative images of the different experiments are shown in the figures. The γH2AX intensity was measured on maximum projections based on Image-Pro Plus 6.0 (Media Cybernetics Inc.) for every pixel within the nuclear area (DNA signal). The mean intensity was determined for each nucleus and for all analysed nuclei. To minimize the contribution of the single damage focus produced by microirradiation, γH2AX foci were chosen to be imaged with saturated intensity and excluded from calculation of the nuclear mean. The γH2AX intensity was normalized to the laser intensity. To assess the variability between identical samples, the percentage standard deviation was calculated for groups of up to 15 identical samples and ranged from 2 to 28% with a mean of 10%.
Fluorescence recovery after photobleaching measurement
Cells on glass cover slips (40 mm diameter) were mounted in a Focht chamber. A bleaching spot was generated using a 473-nm diode laser coupled to a Leica DMLA microscope (Leica Microsystems GmbH, Wetzlar, Germany). Further details and the evaluation of the intensity measurements are described in Tobias et al. (28).
Subcellular protein fractionation
Cells were lysed in cytoskeleton buffer (29) containing 1% Triton X-100 and additives [1× protease inhibitor ethylenediaminetetraacetic acid-free (Roche), 10 mM NaF, 500 µM AEBSF and 1 mM DTT) and incubated for 25 min on ice. The cytoplasmic fraction was separated by centrifugation, nuclei were washed and resuspended with buffer C (30) containing additives as described earlier in the text. The nuclei suspension was lysed by passing through a 27G syringe needle, shortly ultrasound treated (Bioruptor UCD-200) and incubated for 100 min on ice. The nucleoplasmic fraction was separated by centrifugation, and the chromatin pellet was incubated at 95°C in 100 mM Tris–HCl, pH 6.8, 2% sodium dodecyl sulphate and 20% glycerol to obtain the chromatin fraction.
RESULTS
Pan-nuclear formation of γH2AX in undamaged chromatin after localized densely IR
After localized ion irradiation, we reproducibly detected in addition to γH2AX foci a less intense γH2AX signal distributed over the whole nucleus in regions where no dose was deposited (Figure 1A). The fraction of cells showing pan-nuclear γH2AX was 95–99% for heavy ion-irradiated human fibroblasts but <2% in the mock-irradiated cells (Supplementary Figure S1A).
Figure 1.
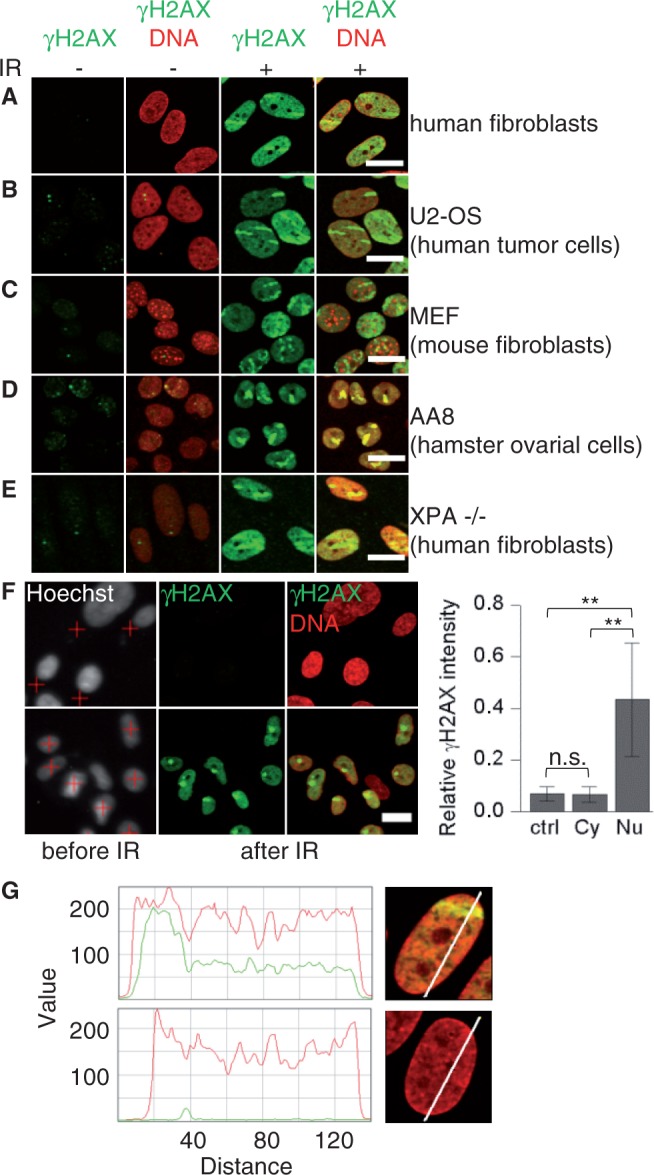
Pan-nuclear H2AX phosphorylation after localized IR-induced DNA damage to the nucleus. Different types of cell lines were fixed 1 h after ion irradiation and immunostained for γH2AX (green) and DNA (ToPro-3, red). Cells were irradiated (IR) at low angle with gold (A–C) or xenon ions (E), or with 40 carbon ions in 1 spot at the microprobe (D). (F) Nuclei of confluent human fibroblasts were detected by Hoechst staining and marked for irradiation (red crosses) to target either the cytoplasm (top) or the nucleus (bottom). Cells were irradiated with one gold ion. The mean pan-nuclear γH2AX signal was measured in unirradiated cells (ctrl) and after irradiation of the cytoplasm (Cy) or the nucleus (Nu) (right) (number of analyzed cells: 327, 193 and 53). Scale bar: 20 µm. (G) Exemplary intensity profiles of the γH2AX (green) and DNA (red) signal (relative value) are shown for a human fibroblast hit by a gold ion at low angle (top) and an unirradiated nucleus (bottom) fixed at 1 h. Error bars denote SD (**P < 0.01, n.s. = not significant, using two-sided U-test).
Nuclear-wide γH2AX occurred after ion irradiation in various different human and rodent cell lines, including human fibroblasts, the tumour cell line U2-OS, MEF and hamster ovary cells (AA8) (Figure 1A–D). As expected, after ion irradiation, an H2AX-deficient cell line did not show γH2AX neither pan-nuclear nor in foci, whereas the DNA damage sites could be visualized via phosphorylated meiotic recombination 11 protein (MRE11, S676/S678) (Supplementary Figure S1B).
We excluded an association of the particle-induced pan-nuclear γH2AX to apoptosis (15) because we did not detect annexin V-positive cells or morphological signs of apoptosis like chromatin condensation or apoptotic bodies up to 24 h post-irradiation of human fibroblasts with gold ions (Supplementary Figure S1C) that clearly induced pan-nuclear γH2AX (Figure 1A).
A UV-induced pan-nuclear H2AX phosphorylation dependent on proficient NER has been previously described (11). Therefore, we irradiated cells deficient for XPA, a factor involved in early steps of NER (31). A clear nuclear-wide γH2AX signal was readily detected after exposure to ions (Figure 1E), suggesting different mechanisms than for UV response.
For a precise quantification of pan-nuclear γH2AX, we took advantage of the heavy ion microprobe allowing the targeted irradiation of cells with a defined number of ions in a submicrometer area (26). All cells hit by ions (Supplementary Figure S1D), but none of the neighbouring unirradiated cells (Supplementary Figure S1D, arrows), showed a nuclear-wide γH2AX in addition to the focus created by the particle traversal. After ion irradiation, an ∼5- to 10-fold increase of nuclear-wide γH2AX was observed compared with unirradiated cells (Figure 1F) with standard deviations of ∼30–50%. No pan-nuclear-positive cells were observed when the particles hit the cytoplasm only, and no response in unirradiated cells was observed (Figure 1F). In addition to the γH2AX peak at the damage foci, an even but clearly enhanced γH2AX distribution compared with the unirradiated nucleus is detected in undamaged chromatin based on line profiles (Figure 1G). Moreover, we observed that pan-nuclear γH2AX is induced in G1 and G2 phase of the cell cycle (Supplementary Figure S1E).
Dose-dependent induction of pan-nuclear γH2AX by localized clustered DNA damage
To analyse whether different amounts of DNA damage generated by varying radiation doses influence the pan-nuclear γH2AX response, we irradiated confluent human fibroblasts in different irradiation fields within the same sample with an increasing number of ions per cell nucleus. Irradiation with increasing numbers of light ions (up to 40 carbon ions) per nucleus showed that the pan-nuclear response increased with number of traversals and, therefore, with dose (Figure 2). Already after irradiation with 1 carbon ion, corresponding to a mean nuclear dose of only 0.2 Gy, the induction of nuclear-wide γH2AX was measurable, although not yet microscopically apparent (Supplementary Figure S2A). In the lower dose range, the signal increase was linear (Figure 2), and it tends to saturate at higher doses. However, with heavier ions, we observed no clear saturation of the response (Supplementary Figure S2B). The pan-nuclear γH2AX signal was always less intense than γH2AX at damage foci (Supplementary Figure S2A), even after the highest tested dose of ∼70 Gy (obtained with eight very heavy ions). The distribution of damage sites in the nucleus, i.e. aiming four carbon ions either to one spot, resulting in a discrete submicrometer damage area, or to a square pattern of four spots with 3-µm distance between traversals had no marked effect on the nuclear-wide response (Figure 2). This was confirmed after irradiation with 10 or 15 ions of intermediate ionization density targeted to one spot or distributed in a five spot cross pattern (Supplementary Figure S2C). These results make it possible to use different ion types to induce the pan-nuclear response taking into account the delivered dose. A pan-nuclear response was not detected after irradiation with 1 Gy X-rays, the line profiles showing very low γH2AX levels in regions devoid of DSBs compared with γH2AX at the DSBs (Supplementary Figure S2D). However, an analysis after higher doses using conventional X-ray irradiation is hampered by the random distribution of DSBs in the nucleus. In summary, these data show that the pan-nuclear formation of γH2AX after ion irradiation is clearly dependent on the dose, and thus the amount of induced DNA damage.
Figure 2.

Pan-nuclear γH2AX increases with increasing dose. Confluent human fibroblasts were irradiated at the microprobe with increasing numbers of carbon or nitrogen ions (corresponding to a mean nuclear dose of ∼0.2 or 0.3 Gy/traversal, respectively) targeted to one spot or with four carbon ions targeted in a square pattern of four spots in 3-µm distance (carbon ions distributed). One hour after irradiation, the mean pan-nuclear γH2AX signal of every cell was quantified. The values were normalized to the intensity of the unirradiated control. The dashed line shows the exponential fitting curve for the complete data set. The inset shows the data for up to four carbon traversals in more detail. Error bars denote SD (**P < 0.01, n.s. = not significant, using two-sided U-test, doses were calculated based on a nuclear cross-section of 240 µm2 for the used cells, number of analyzed cells: 45–284 with an average of 144).
Formation of nuclear-wide γH2AX is transient
We analysed the pan-nuclear response up to 24 h after irradiation with different ions inducing increasing damage clustering. For all ion types and numbers of traversals, the kinetics of γH2AX intensity were similar (Figure 3A). A maximum occurred approximately at 1 h after irradiation, followed by a decline over the following 5–7 h and a weak but measurable pan-nuclear signal remaining 24 h after irradiation. This is in contrast to DSB repair kinetics, which have been shown to be significantly delayed by densely ionizing radiation (19,25,32), suggesting that disappearance of the pan-nuclear response is not linked to DSB repair. A decline of pan-nuclear γH2AX was also observed within the first 6 h in XRCC4 −/− MEF cells (Figure 3B), which are deficient in non-homologous end-joining (NHEJ), arguing against a dependency of the disappearance of the pan-nuclear signal on functional NHEJ repair. This result was confirmed for XRCC4 −/− cells using a different ion type, and similar results were also obtained for NHEJ-deficient Ku80 −/− cells (Supplementary Figure S3). Simultaneous depletion of the nucleases MRE11, C-terminal-binding protein interacting protein (CtIP) and exonuclease 1 (EXO1), involved in resection and homologous recombination (HR), did not suppress the decrease of pan-nuclear γH2AX (Supplementary Figure S3), indicating that processing of DNA ends by these nucleases is not necessary for the recovery from nuclear-wide chromatin phosphorylation.
Figure 3.

Transient nuclear-wide γH2AX after DNA damage induction of varying complexity and in NEHJ-deficient cells. (A) Confluent human fibroblasts were irradiated at the microprobe with 40 carbon (∼7.6 Gy), 14 nitrogen (∼3.4 Gy), 2 gold (∼17 Gy) or indicated numbers of lead ions (∼8.9 Gy/ion). The dashed line shows the exponential fitting curve for the measurement after carbon ion irradiation. For comparison of different experiments, the values were normalized to the intensity at 1 h. (B) MEF WT or XRCC4 −/− cells were irradiated using broad beam titanium ions. Cells were fixed at indicated time points and pan-nuclear γH2AX quantified. Error bars denote SD (number of analysed cells: 72–1344 with an average of 267).
ATM and DNA–PK mediate the pan-nuclear γH2AX response
We next investigated the role of PIKK family kinases in the pan-nuclear γH2AX response using specific inhibitors. The pan-nuclear H2AX phosphorylation after ion irradiation was clearly reduced but not completely inhibited in cells treated with only ATM or DNA–PKcs (DNA–PK catalytic subunit) inhibitor or caffeine. Simultaneous treatment with the specific inhibitors of both ATM and DNA–PKcs almost completely abolished the pan-nuclear H2AX phosphorylation (Figure 4A), whereas DMSO alone had no effect (Supplementary Figure S4A). The almost total suppression of pan-nuclear γH2AX by inhibition of both ATM and DNA–PKcs was confirmed using irradiation with a different ion (Supplementary Figure S4B).
Figure 4.

DNA–PK and ATM-dependent nuclear-wide γH2AX and dose-dependent pan-nuclear pATM (S1981) and pDNA–PKcs (S2056). (A) Confluent human fibroblasts were treated with DMSO, ATM inhibitor (Ai), DNA–PKcs inhibitor (Di) or caffeine and irradiated with three xenon ions (∼18.3 Gy). (B) WT or ATM-deficient human fibroblasts (AT), treated with DMSO or DNA–PKcs inhibitor (Di), and (C) WT MEF, DNA–PK −/− MEF, treated with DMSO or ATM inhibitor (Ai), were irradiated with five nickel ions (∼12.5 Gy). Cells were fixed 1 h after irradiation, and pan-nuclear γH2AX was quantified. (D) Confluent human fibroblasts were irradiated with different numbers of gold ions (∼8.5 Gy/ion), and pan-nuclear pDNA–PKcs (S2056) or pATM (S1981) was quantified 1 h after irradiation. For comparison of pATM and pDNA–PKcs signal intensities, the values were normalized to the intensity in unirradiated cells. (E) Confluent human fibroblasts were treated with DMSO or both ATM and DNA–PKcs inhibitors (Ai + Di) 1 h before until 1 h after irradiation and reseeded 24 h after irradiation with different doses of X-rays or neon ions for the survival assay (N = 3). Solid lines show the linear quadratic (X-rays) or linear (neon) fit for each data set. Error bars denote SD (**P < 0.01, using two-sided U-test, number of analysed cells for immunofluorescence quantification: 84–1228 with an average of 268).
The role of ATM and DNA–PK in nuclear-wide H2AX phosphorylation was confirmed irradiating ATM-deficient primary human fibroblasts that showed a decreased pan-nuclear γH2AX signal compared with WT fibroblasts. The pan-nuclear signal was further reduced to near control level after treatment with DNA–PKcs inhibitor (Figure 4B). Conversely, the ion irradiation of DNA–PKcs-deficient MEF cells induced a pan-nuclear γH2AX signal that was reduced compared with WT cells and largely suppressed after additional ATM inhibition (Figure 4C). Similar results were obtained using a different ion irradiation (Supplementary Figure S4C and D).
The aforementioned results imply the availability of nuclear-wide active ATM and DNA–PKcs. Using immunofluorescence microscopy, we examined the cellular distribution of the activated kinases after ion irradiation. Both phosphorylated DNA–PKcs (S2056) (Figure 5A) and ATM (S1981) (Figure 5B) formed pan-nuclear signals in a dose-dependent manner (Figure 4D), and the pan-nuclear pATM was suppressed by specific ATM inhibition (Supplementary Figure S4E). This is in line with the nuclear-wide formation of γH2AX mediated by ATM and DNA–PK.
Figure 5.
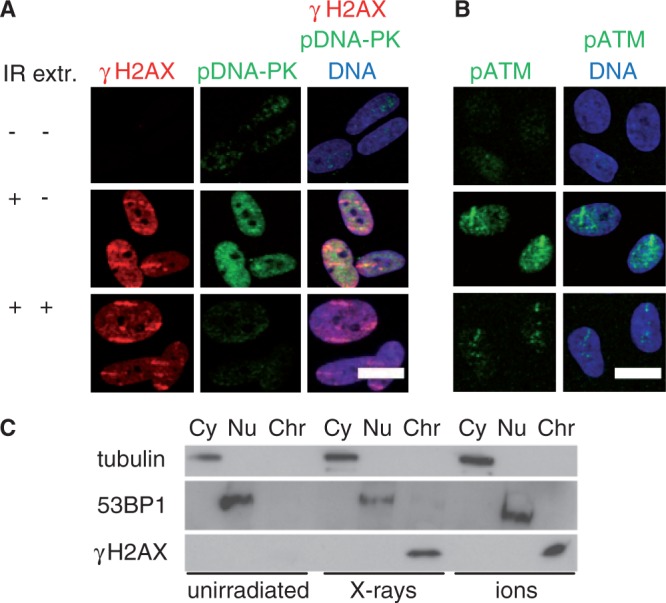
Pan-nuclear γH2AX is chromatin bound, and pan-nuclear pDNA–PKcs and pATM are extractable. Confluent human fibroblasts were irradiated (IR) with (A) uranium or (B) xenon ions, fixed after 1 h directly or after extraction of unbound proteins (extr.) and stained for γH2AX, pDNA–PKcs (S2056), pATM (S1981) and DNA. Scale bar: 20 µm. (C) Cells were harvested 1 h after irradiation with gold ions (3 × 106 particles/cm2) or 30 Gy X-rays, and the localization of γH2AX in the cytoplasmic (Cy), nucleoplasmic (Nu) and chromatin-bound (Chr) protein fractions was analysed using western blot. The 53BP1 and tubulin were detected as controls for the cytoplasma and nucleoplasma fraction, respectively.
Surprisingly, in these experiments, the formation of γH2AX at damage foci induced by very heavy ions was not suppressed when both ATM and DNA–PKcs were inhibited (Figure 6A). One possible explanation is that residual amounts of activated kinases were still present even under inhibiting conditions as detected by western blot analysis after ion irradiation (Supplementary Figure S4F). Interestingly, we found that after irradiation with single light ions, which produce a lower local damage density compared with heavier ones, the inhibition of both ATM and DNA–PK was able to completely suppress γH2AX foci at the damage sites. However, when 10 of these light ions were targeted in one spot, the γH2AX focus could be again detected (Supplementary Figure S4G). Thus, the effect of residual kinase activity induced by high amounts of localized DSBs can fully explain γH2AX foci detection under inhibiting conditions. In line with this notion, an additional downregulation of ATR by RNA interference had no effect on the formation of γH2AX foci (Supplementary Figure S4G).
Figure 6.
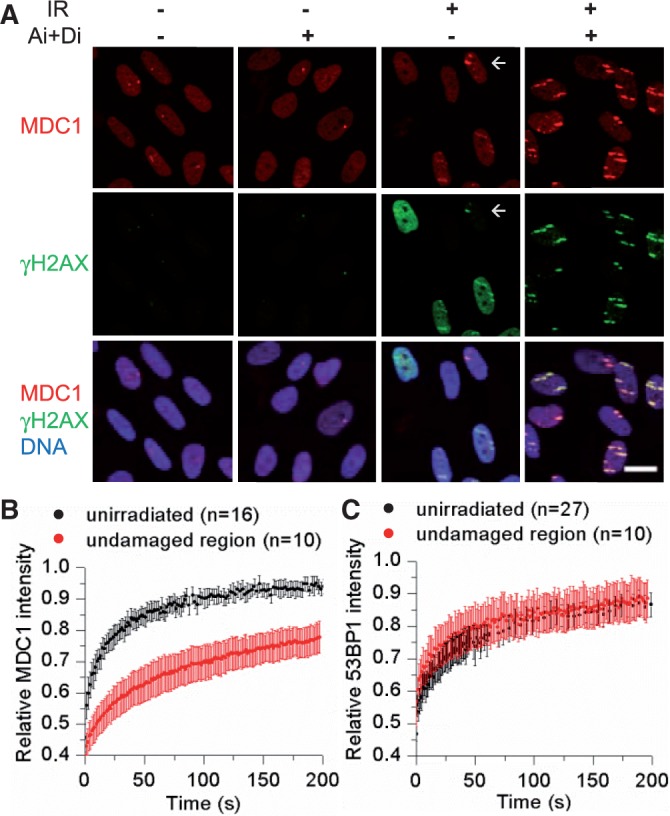
Influence of pan-nuclear γH2AX on MDC1 but not 53BP1 binding. (A) Confluent human fibroblasts were treated with DMSO or ATM and DNA–PKcs inhibitor (Ai + Di) and irradiated with gold ions at low angle. One hour after irradiation, MDC1, γH2AX and DNA were detected. Scale bar: 20 µm (arrow: cell with peripheral ion hit, resulting in low pan-nuclear γH2AX and efficient MDC1 recruitment). U2-OS cells expressing (B) MDC1-GFP or (C) 53BP1-GFP were irradiated with ruthenium ions at low angle, and the mobility of GFP-tagged proteins was measured by FRAP analysis in undamaged regions of irradiated cells or in unirradiated cells. Error bars denote 95% confidence interval.
We next asked whether the pan-nuclear γH2AX response was relevant for the biological effectiveness of ion irradiation, i.e. whether this effect could be partly responsible for the increased effectiveness of heavy ions compared with X- or γ-rays (17,18). Simultaneous treatment of confluent human fibroblasts with specific ATM and DNA–PKcs inhibitor 1 h before until 1 h after exposure to X-rays did not cause clonogenic toxicity (Figure 4E), indicating that the transient inhibition short time after irradiation was reversible and compensated later on. For exposure to densely ionizing irradiation, the inhibition of the pan-nuclear response by this treatment did not significantly influence cell survival (Figure 4E), suggesting that the nuclear-wide γH2AX does not contribute to the effectiveness of ion irradiation.
Pan-nuclear γH2AX is tightly associated to chromatin
Nuclear-wide γH2AX was induced by ion irradiation and the immunofluorescent signal detected after extraction of detergent-soluble protein. The signal was not changed compared with direct fixation (Figure 5A) showing that γH2AX is associated to chromatin also in the undamaged regions. This result was confirmed by subcellular protein fractionation using stringent conditions leaving only tightly bound proteins in the chromatin fraction. A fluence of about seven ion traversals per cell nucleus was used to induce efficient pan-nuclear H2AX phosphorylation, still leaving a large undamaged nuclear area. One hour after irradiation with ions or X-rays, γH2AX was located exclusively in the chromatin fraction (Figure 5C), indicating that also the ion-induced pan-nuclear γH2AX is bound to chromatin, whereas tubulin and p53-binding protein (53BP1) were detected in the cytoplasmic and nucleoplasmic fraction, respectively. We examined by immunofluorescence microscopy if a gradient of γH2AX could be measured through the cell nucleus early after irradiation. The increased γH2AX signal at 15 min was uniform over the whole nucleus (Supplementary Figure S5A), arguing against a slow spreading of γH2AX from damage sites. On the other hand, the nuclear-wide signal of pDNA–PKcs (S2056) and pATM (S1981) vanished after extraction treatment, supporting a loosely bound nucleoplasmic localization (Figure 5A and B). In H2AX −/− MEF cells, pan-nuclear pATM was also detected, demonstrating that the pan-nuclear distribution of pATM occurs upstream of the nuclear-wide H2AX phosphorylation (Supplementary Figure S5B).
Nuclear-wide binding of MDC1 but not 53BP1 is induced after localized irradiation
In addition to H2AX, a strong pan-nuclear phosphorylation of other kinase target proteins has been reported after transfection of small DNA molecules (10) and after virus infection (13,14). After ion irradiation, phosphorylated Nijmegen breakage syndrome 1 (NBS1, S343), a phosphorylation target of ATM (33), and phosphorylated replication protein A (RPA, S4/S8), which is phosphorylated by DNA–PK at these sites (34), formed only a faint pan-nuclear signal (Supplementary Figure S6A). Moreover, in the presence of microinjected DNA molecule-induced pan-nuclear γH2AX, secondary γH2AX foci did not form on irradiation (10). In contrast, a second ion irradiation clearly induced the formation of γH2AX foci (Supplementary Figure S6B), despite pan-nuclear H2AX phosphorylation. This suggests that DNA damage signalling is not inhibited.
As mediator of DNA damage checkpoint 1 (MDC1) binds to γH2AX directly (35), we analysed the influence of pan-nuclear γH2AX on the nuclear distribution of MDC1. The suppression of pan-nuclear γH2AX by ATM and DNA–PKcs inhibition resulted in an increased MDC1 signal at damage sites compared with cells showing pan-nuclear γH2AX (Figure 6A). A possible explanation is nuclear-wide binding of MDC1, leaving less unbound MDC1 available for the recruitment to damage sites. In fact, more intense MDC1 foci were detected in cells showing only weakly induced pan-nuclear γH2AX because of peripheral ion traversals (Figure 6A, arrow), and the few spontaneous MDC1 foci found in unirradiated cells were of comparable intensity (Figure 6A). This indicates efficient MDC1 recruitment when pan-nuclear γH2AX is absent. We measured nuclear-wide MDC1 binding by FRAP, showing a reduced mobility of MDC1 in the undamaged region of the nucleus in ion-irradiated compared with unirradiated cells (Figure 6B). As expected, MDC1-deficient MEF cells also showed pan-nuclear γH2AX (Supplementary Figure S6C) because MDC1 binding occurs downstream of H2AX phosphorylation. The binding affinity of 53BP1, which is recruited in an MDC1-dependent manner to damage sites but requires further regulatory signalling steps (36,37), was not influenced in undamaged regions after ion irradiation (Figure 6C). In line with this observation, suppression of pan-nuclear γH2AX did not change 53BP1 recruitment to damage sites (Supplementary Figure S6D). We conclude that the pan-nuclear H2AX phosphorylation does not lead to a full DNA damage response in undamaged chromatin.
DISCUSSION
Using localized irradiation, we describe here a novel response to clustered DNA lesions involving the formation of pan-nuclear γH2AX mediated by the kinases ATM and DNA–PK. This nuclear-wide H2AX phosphorylation triggered by IR differs from previously described γH2AX responses. The effect is not related to apoptosis and, in contrast to the UV-induced nuclear-wide γH2AX (11,38), it is not dependent on NER. The ion-induced reaction requires direct nuclear damage induction and did not occur after cytoplasmic irradiation or in cells adjacent to the irradiated ones. It can thus not be attributed to bystander effects as described for γH2AX foci (39). A potential connection of pan-nuclear γH2AX noticed after high power laser microirradiation (40) to the here described IR-induced response awaits further clarification.
The nuclear-wide signal was measurable already after a single carbon ion traversal and increased with the IR dose. We demonstrate that the pan-nuclear γH2AX response is mediated by the activation of both ATM and DNA–PK. With only one of the kinases active, pan-nuclear γH2AX was still visible, albeit reduced. The pan-nuclear γH2AX is tightly chromatin-bound, and γH2AX spreading is uniform at short times after irradiation. This suggests that γH2AX in undamaged chromatin is generated via direct phosphorylation of H2AX by the activated kinases, most likely diffusing from DNA damage sites. In agreement with this hypothesis, the autophosphorylated forms of ATM (S1981) and DNA–PKcs (S2056) were also distributed over the whole nucleus after ion irradiation in a dose-dependent manner, but not tightly chromatin bound. For efficient DNA–PK activation under physiological conditions, the DNA–PKcs needs to bind DNA ends associated with the Ku70/Ku80 heterodimer [reviewed in (41)], and kinase activation is achieved by immobilization of the N-terminal end (42). This begs the question of how the activity of DNA–PK can be triggered at large distances from sites of DNA damage. A possible explanation is that the pan-nuclear DNA–PK activity is associated to small DNA fragments, which are often produced by densely IR (43). A possible in vivo activation by DNA fragments is supported by studies showing that DNA–PK can be strongly activated by transfection of DNA molecules (10), virus infection (14) or during apoptotic DNA fragmentation (44). However, for the DNA molecule-induced response, an all-or-none pattern has been described (10), whereas the ion-induced response is dose dependent.
An alternative explanation for the nuclear-wide DNA–PK activity is that during protein exchange, the kinase could dissociate in active form from the damage sites. The finding that DNA–PKcs can promote its own dissociation from DNA by autophosphorylation (45,46), which would require kinase activity at the time of dissociation, supports this notion. Furthermore, DNA–PKcs has been suggested to be especially involved in the repair of complex DNA lesions (47), but the binding efficiency of Ku is apparently reduced compared with simple lesions (48). These observations are in line with a potentially increased exchange of DNA–PK at clustered damage. Based on the observation that the N-terminal region of DNA–PKcs is required for kinase inactivation (49), it would be interesting to find out whether this process is connected to a loss of contact of the N-terminus after dissociation of the still active DNA–PKcs.
ATM has been shown to be activated in vitro in human cell extracts by small DNA fragments (50). ATM activation has also been associated with structural changes in chromatin (51), and pan-nuclear γH2AX induced by chromatin de-condensation can be suppressed by the PIKK inhibitor wortmannin (12). We found chromatin de-condensation-induced nuclear-wide γH2AX to depend on ATM but not DNA–PK (Supplementary Figure S7). This argues against an extensive chromatin alteration as the only basis of kinase activation after ion irradiation. Moreover, we have no evidence for ion-induced major rearrangements in chromatin structure (52,53), and only localized chromatin de-condensation was observed after aimed ion irradiation (54). A pan-nuclear activity of ATM has previously been reported, leading to the transient pan-nuclear phosphorylation of KRAB-associated protein 1 (KAP1) in response to local laser irradiation (55).
The pan-nuclear γH2AX response is transiently detected during a few hours after irradiation with maximum ∼1 h. These kinetics were not influenced by the complexity of the induced damage, whereas DSB rejoining has been shown to be delayed with increasing damage complexity (19,32). Moreover, the kinetics of pan-nuclear γH2AX loss were not dependent on XRCC4- or Ku80-mediated NHEJ, which is the major repair pathway in G1-phase cells (56). Processing of DNA ends by CtIP, MRE11 and EXO1, which is a key step in HR initiation, is also dispensable for the decline of pan-nuclear γH2AX. Overall, our results show that completion of DSB repair is not a prerequisite for the reduction of pan-nuclear H2AX phosphorylation.
ATM and DNA–PK also phosphorylate other DNA damage response factors besides H2AX. After ion irradiation, only a faint pan-nuclear signal of phosphorylated NBS1 and RPA could be detected, in contrast to the clear pan-nuclear signals of phosphorylated RPA, NBS1 and other target proteins reported after virus infection (13,14) or transfection of small DNA molecules (10). In addition, after transfection of small DNA fragments, DNA repair is significantly compromised (10,16). Such repair impairment also for the ion-induced pan-nuclear response could be contributing to the increased RBE of densely IR, which constitutes one of the key advantages in the clinical application of heavy ions in tumour therapy (17). However, the formation of repair foci like 53BP1 or phosphorylated NBS1, RPA, ATM and MRE11 is not affected by the ion-induced pan-nuclear response. In addition, the γH2AX signal is always stronger at DNA DSBs than in undamaged chromatin, and γH2AX foci are induced by a second ion irradiation even in the presence of pan-nuclear γH2AX. These results indicate that the accumulation of repair factors at DNA lesions is not generally suppressed by the pan-nuclear reaction, and that the damage response is focused on chromatin areas directly surrounding DSBs. Only MDC1, which is recruited to damaged chromatin via direct interaction with γH2AX (35), was found to bind to pan-nuclear γH2AX leading to a decreased binding of MDC1 to damage sites. In addition, we could recently show that this nuclear-wide binding influences the exchange of MDC1 at damage sites (28). The 53BP1 binding, which is mediated by additional regulatory steps like ubiquitination (36,37), was not affected by the nuclear-wide response. Thus, the pan-nuclear γH2AX can influence downstream factors without inducing a full DNA damage response. In line with this notion, the temporary inhibition of ATM and DNA–PKcs, which suppresses pan-nuclear γH2AX, did not largely influence the survival of normal human fibroblasts after ion irradiation.
Generally, the level of phosphorylated H2AX is directly correlated to the yield of radiation-induced DNA DSBs. We show here that this correlation is hampered by pan-nuclear response after the induction of localized clustered DSBs. The transient formation of pan-nuclear γH2AX is caused by DNA–PK and ATM activity distant to sites of DNA damage. The pan-nuclear response triggers nuclear-wide MDC1 binding, reducing its recruitment to damage sites, but without a general inhibition of the DNA damage response. The nuclear-wide kinase activity may be elicited by IR-induced small DNA fragments or by diffusion of active kinases from DNA DSBs.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–7, Supplementary Materials and Methods and Supplementary References [57–61].
FUNDING
Bundesministerium für Bildung und Forschung [02NUK001A]. Funding for open access charge: GSI Helmholtz Center for Heavy Ion Research.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank G. Becker, A.L. Leifke and B. Kass for cell culturing and assistance, B. Merk, W. Becher, R. Khan and G. Lenz for technical irradiation support and Dr F.W. Alt (Harvard Medical School, Boston, MA, USA), Dr D. Chen (University of Texas Southwestern Medical Center, Dallas, TX, USA), Dr M. Lavin (Queensland Institute of Medical Research, Brisbane), Dr C. Lukas (Danish Cancer Society, Copenhagen) and Dr A. Nussenzweig (National Cancer Institute, Bethesda, MD, USA) for cell lines and antibodies.
REFERENCES
- 1.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001;276:47759–47762. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 3.Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Löbrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–2396. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- 4.Wang H, Wang M, Wang H, Böcker W, Iliakis G. Complex H2AX phosphorylation patterns by multiple kinases including ATM and DNA-PK in human cells exposed to ionizing radiation and treated with kinase inhibitors. J. Cell Physiol. 2005;202:492–502. doi: 10.1002/jcp.20141. [DOI] [PubMed] [Google Scholar]
- 5.Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell. Biol. 2003;5:675–679. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- 7.Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, Monroe BC, Ciccone DN, Yan C, Vlasakova K, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc. Natl Acad. Sci. USA. 2002;99:8173–8178. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Revet I, Feeney L, Bruguera S, Wilson W, Dong TK, Oh DH, Dankort D, Cleaver JE. Functional relevance of the histone gammaH2Ax in the response to DNA damaging agents. Proc. Natl Acad. Sci. USA. 2011;108:8663–8667. doi: 10.1073/pnas.1105866108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678–5694. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quanz M, Chassoux D, Berthault N, Agrario C, Sun J-S, Dutreix M. Hyperactivation of DNA-PK by double-strand break mimicking molecules disorganizes DNA damage response. PLoS One. 2009;4:e6298. doi: 10.1371/journal.pone.0006298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc. Natl Acad. Sci. USA. 2006;103:9891–9896. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baure J, Izadi A, Suarez V, Giedzinski E, Cleaver JE, Fike JR, Limoli CL. Histone H2AX phosphorylation in response to changes in chromatin structure induced by altered osmolarity. Mutagenesis. 2009;24:161–167. doi: 10.1093/mutage/gen064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fragkos M, Breuleux M, Clément N, Beard P. Recombinant adeno-associated viral vectors are deficient in provoking a DNA damage response. J. Virol. 2008;82:7379–7387. doi: 10.1128/JVI.00358-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwartz RA, Carson CT, Schuberth C, Weitzman MD. Adeno-associated virus replication induces a DNA damage response coordinated by DNA-dependent protein kinase. J. Virol. 2009;83:6269–6278. doi: 10.1128/JVI.00318-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Solier S, Pommier Y. The apoptotic ring: a novel entity with phosphorylated histones H2AX and H2B and activated DNA damage response kinases. Cell Cycle. 2009;8:1853–1859. doi: 10.4161/cc.8.12.8865. [DOI] [PubMed] [Google Scholar]
- 16.Quanz M, Berthault N, Roulin C, Roy M, Herbette A, Agrario C, Alberti C, Josserand V, Coll J-L, Sastre-Garau X, et al. Small-molecule drugs mimicking DNA damage: a new strategy for sensitizing tumors to radiotherapy. Clin. Cancer Res. 2009;15:1308–1316. doi: 10.1158/1078-0432.CCR-08-2108. [DOI] [PubMed] [Google Scholar]
- 17.Durante M, Loeffler JS. Charged particles in radiation oncology. Nat. Rev. Clin. Oncol. 2010;7:37–43. doi: 10.1038/nrclinonc.2009.183. [DOI] [PubMed] [Google Scholar]
- 18.Durante M, Cucinotta FA. Heavy ion carcinogenesis and human space exploration. Nat. Rev., Cancer. 2008;8:465–472. doi: 10.1038/nrc2391. [DOI] [PubMed] [Google Scholar]
- 19.Asaithamby A, Hu B, Chen DJ. Unrepaired clustered DNA lesions induce chromosome breakage in human cells. Proc. Natl Acad. Sci. USA. 2011;108:8293–8298. doi: 10.1073/pnas.1016045108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jakob B, Scholz M, Taucher-Scholz G. Biological imaging of heavy charged-particle tracks. Radiat. Res. 2003;159:676–684. doi: 10.1667/0033-7587(2003)159[0676:biohct]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 21.Satokata I, Tanaka K, Miura N, Narita M, Mimaki T, Satoh Y, Kondo S, Okada Y. Three nonsense mutations responsible for group A xeroderma pigmentosum. Mutat Res. 1992;273:193–202. doi: 10.1016/0921-8777(92)90080-m. [DOI] [PubMed] [Google Scholar]
- 22.Stankovic T, Kidd AM, Sutcliffe A, McGuire GM, Robinson P, Weber P, Bedenham T, Bradwell AR, Easton DF, Lennox GG, et al. ATM mutations and phenotypes in ataxia-telangiectasia families in the British Isles: expression of mutant ATM and the risk of leukemia, lymphoma, and breast cancer. Am. J. Hum. Genet. 1998;62:334–345. doi: 10.1086/301706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kurimasa A, Ouyang H, Dong LJ, Wang S, Li X, Cordon-Cardo C, Chen DJ, Li GC. Catalytic subunit of DNA-dependent protein kinase: impact on lymphocyte development and tumorigenesis. Proc. Natl Acad. Sci. USA. 1999;96:1403–1408. doi: 10.1073/pnas.96.4.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, Sekiguchi JM, Rathbun GA, Swat W, Wang J, et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998;95:891–902. doi: 10.1016/s0092-8674(00)81714-6. [DOI] [PubMed] [Google Scholar]
- 25.Jakob B, Splinter J, Taucher-Scholz G. Positional stability of damaged chromatin domains along radiation tracks in mammalian cells. Radiat. Res. 2009;171:405–418. doi: 10.1667/RR1520.1. [DOI] [PubMed] [Google Scholar]
- 26.Heiss M, Fischer BE, Jakob B, Fournier C, Becker G, Taucher-Scholz G. Targeted irradiation of Mammalian cells using a heavy-ion microprobe. Radiat. Res. 2006;165:231–239. doi: 10.1667/rr3495.1. [DOI] [PubMed] [Google Scholar]
- 27.Wiese C, Rudolph JH, Jakob B, Fink D, Tobias F, Blattner C, Taucher-Scholz G. PCNA-dependent accumulation of CDKN1A into nuclear foci after ionizing irradiation. DNA Repair (Amst) 2012;11:511–521. doi: 10.1016/j.dnarep.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 28.Tobias F, Löb D, Lengert N, Durante M, Drossel B, Taucher-Scholz G, Jakob B. Spatiotemporal dynamics of early DNA damage response proteins on complex DNA lesions. PLoS One. 2013;8:e57953. doi: 10.1371/journal.pone.0057953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin DI, Aggarwal P, Diehl JA. Phosphorylation of MCM3 on Ser-112 regulates its incorporation into the MCM2-7 complex. Proc. Natl Acad. Sci. USA. 2008;105:8079–8084. doi: 10.1073/pnas.0800077105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arva NC, Gopen TR, Talbott KE, Campbell LE, Chicas A, White DE, Bond GL, Levine AJ, Bargonetti J. A chromatin-associated and transcriptionally inactive p53-Mdm2 complex occurs in mdm2 SNP309 homozygous cells. J. Biol. Chem. 2005;280:26776–26787. doi: 10.1074/jbc.M505203200. [DOI] [PubMed] [Google Scholar]
- 31.Fornace AJ, Kohn KW, Kann HE. DNA single-strand breaks during repair of UV damage in human fibroblasts and abnormalities of repair in xeroderma pigmentosum. Proc. Natl Acad. Sci. USA. 1976;73:39–43. doi: 10.1073/pnas.73.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shibata A, Conrad S, Birraux J, Geuting V, Barton O, Ismail A, Kakarougkas A, Meek K, Taucher-Scholz G, Löbrich M, et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011;30:1079–1092. doi: 10.1038/emboj.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gatei M, Young D, Cerosaletti KM, Desai-Mehta A, Spring K, Kozlov S, Lavin MF, Gatti RA, Concannon P, Khanna K. ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nat. Genet. 2000;25:115–119. doi: 10.1038/75508. [DOI] [PubMed] [Google Scholar]
- 34.Zernik-Kobak M, Vasunia K, Connelly M, Anderson CW, Dixon K. Sites of UV-induced phosphorylation of the p34 subunit of replication protein A from HeLa cells. J. Biol. Chem. 1997;272:23896–23904. doi: 10.1074/jbc.272.38.23896. [DOI] [PubMed] [Google Scholar]
- 35.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 36.Wang B, Elledge SJ. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc. Natl Acad. Sci. USA. 2007;104:20759–20763. doi: 10.1073/pnas.0710061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435–446. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 38.de Feraudy S, Revet I, Bezrookove V, Feeney L, Cleaver JE. A minority of foci or pan-nuclear apoptotic staining of gammaH2AX in the S phase after UV damage contain DNA double-strand breaks. Proc. Natl Acad. Sci. USA. 2010;107:6870–6875. doi: 10.1073/pnas.1002175107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sokolov MV, Smilenov LB, Hall EJ, Panyutin IG, Bonner WM, Sedelnikova OA. Ionizing radiation induces DNA double-strand breaks in bystander primary human fibroblasts. Oncogene. 2005;24:7257–7265. doi: 10.1038/sj.onc.1208886. [DOI] [PubMed] [Google Scholar]
- 40.Ha K, Takeda Y, Dynan WS. Sequences in PSF/SFPQ mediate radioresistance and recruitment of PSF/SFPQ-containing complexes to DNA damage sites in human cells. DNA Repair (Amst) 2011;10:252–259. doi: 10.1016/j.dnarep.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meek K, Dang V, Lees-Miller SP. DNA-PK: the means to justify the ends? Adv. Immunol. 2008;99:33–58. doi: 10.1016/S0065-2776(08)00602-0. [DOI] [PubMed] [Google Scholar]
- 42.Meek K, Lees-Miller SP, Modesti M. N-terminal constraint activates the catalytic subunit of the DNA-dependent protein kinase in the absence of DNA or Ku. Nucleic Acids Res. 2012;40:2964–2973. doi: 10.1093/nar/gkr1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Psonka-Antonczyk K, Elsässer T, Gudowska-Nowak E, Taucher-Scholz G. Distribution of double-strand breaks induced by ionizing radiation at the level of single DNA molecules examined by atomic force microscopy. Radiat. Res. 2009;172:288–295. doi: 10.1667/RR1772.1. [DOI] [PubMed] [Google Scholar]
- 44.Mukherjee B, Kessinger C, Kobayashi J, Chen BP, Chen DJ, Chatterjee A, Burma S. DNA-PK phosphorylates histone H2AX during apoptotic DNA fragmentation in mammalian cells. DNA Repair (Amst) 2006;5:575–590. doi: 10.1016/j.dnarep.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 45.Uematsu N, Weterings E, ichi Yano K, Morotomi-Yano K, Jakob B, Taucher-Scholz G, Mari PO, van Gent DC, Chen BP, Chen DJ. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J. Cell Biol. 2007;177:219–229. doi: 10.1083/jcb.200608077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hammel M, Yu Y, Mahaney BL, Cai B, Ye R, Phipps BM, Rambo RP, Hura GL, Pelikan M, So S, et al. Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J. Biol. Chem. 2010;285:1414–1423. doi: 10.1074/jbc.M109.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reynolds P, Anderson JA, Harper JV, Hill MA, Botchway SW, Parker AW, O’Neill P. The dynamics of Ku70/80 and DNA-PKcs at DSBs induced by ionizing radiation is dependent on the complexity of damage. Nucleic Acids Res. 2012;40:10821–10831. doi: 10.1093/nar/gks879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang H, Zhang X, Wang P, Yu X, Essers J, Chen D, Kanaar R, Takeda S, Wang Y. Characteristics of DNA-binding proteins determine the biological sensitivity to high-linear energy transfer radiation. Nucleic Acids Res. 2010;38:3245–3251. doi: 10.1093/nar/gkq069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davis AJ, Lee KJ, Chen DJ. The N-terminal region of the DNA-dependent protein kinase catalytic subunit is required for its DNA double-stranded break-mediated activation. J. Biol. Chem. 2013;288:7037–7046. doi: 10.1074/jbc.M112.434498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shiotani B, Zou L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol. Cell. 2009;33:547–558. doi: 10.1016/j.molcel.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 52.Jakob B, Rudolph JH, Gueven N, Lavin MF, Taucher-Scholz G. Live cell imaging of heavy-ion-induced radiation responses by beamline microscopy. Radiat. Res. 2005;163:681–690. doi: 10.1667/rr3374. [DOI] [PubMed] [Google Scholar]
- 53.Jakob B, Splinter J, Durante M, Taucher-Scholz G. Live cell microscopy analysis of radiation-induced DNA double-strand break motion. Proc. Natl Acad. Sci. USA. 2009;106:3172–3177. doi: 10.1073/pnas.0810987106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jakob B, Splinter J, Conrad S, Voss KO, Zink D, Durante M, Löbrich M, Taucher-Scholz G. DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res. 2011;39:6489–6499. doi: 10.1093/nar/gkr230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 2006;8:870–876. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 56.Rothkamm K, Krüger I, Thompson LH, Löbrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003;23:5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, Hanitsch L, Celeste A, Muramatsu M, Pilch DR, et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Becherel OJ, Jakob B, Cherry AL, Gueven N, Fusser M, Kijas AW, Peng C, Katyal S, McKinnon PJ, Chen J, et al. CK2 phosphorylation-dependent interaction between aprataxin and MDC1 in the DNA damage response. Nucleic Acids Res. 2010;38:1489–1503. doi: 10.1093/nar/gkp1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boehme KA, Kulikov R, Blattner C. p53 stabilization in response to DNA damage requires Akt/PKB and DNA-PK. Proc. Natl Acad. Sci. USA. 2008;105:7785–7790. doi: 10.1073/pnas.0703423105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yuan J, Chen J. MRE11-RAD50-NBS1 complex dictates DNA repair independent of H2AX. J. Biol. Chem. 2010;285:1097–1104. doi: 10.1074/jbc.M109.078436. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.