

Abstract
Holprosencephaly (HPE) is the most common disorder of the developing forebrain in humans, and is characterized by varying degrees of abnormal union of the cerebral hemispheres. These defects are typically co-associated with midline craniofacial anomalies. The combination of forebrain and craniofacial defects that comprise HPE can present along a broad and variable phenotypic spectrum. Both the SHH and NODAL signaling pathways play important roles in the pathogenesis of this disorder. Disruption of these pathways by chromosomal rearrangements, mutations in pathway-related genes and/or biochemical alterations are proposed to contribute to HPE in a large number of patients. Additional factors that are not yet fully delineated are also very likely to be involved in the pathogenesis and phenotypic heterogeneity of the disorder. Genetic loss of GAS1, a cell membrane receptor and positive regulator of SHH, has been demonstrated to contribute to the HPE phenotypic spectrum in animal models. We have evaluated the coding and flanking sequence of GAS1 in 394 patients who have clinical findings within the HPE phenotypic spectrum, and now report five novel missense sequence variants among five unrelated HPE probands. Finally, we tested the effect of these variants (as well as previously reported GAS1 variants) on the ability of GAS1 to bind to SHH. Here, we demonstrate that sequence variants in GAS1 can impair its physical interaction with SHH, suggesting a decrease in the SHH downstream signaling cascade as a pathogenic mechanism of disease.
Introduction
Holoprosencephaly (HPE) is the most common disorder of the developing forebrain in humans and affects 1:250 pregnancies and 1:10,000 live births (Matsunaga and Shiota 1977). This etiologically complex disorder is characterized by a broad phenotypic spectrum including abnormal union of the cerebral hemispheres. The forebrain defects are classified into common HPE subtypes, including the alobar, semilobar, lobar, and middle interhemispheric variant forms. Associated (but highly variable) craniofacial anomalies include cyclopia/synophthalmia, hypotelorism, flat nasal bridge, single nostril, congenital nasal pyriform aperture stenosis, cleft lip with or without palate and a single central maxillary incisor (Demyer et al. 1964; Hahn and Barnes 2010; Solomon et al. 2010b).
Alterations in the components of the SHH and NODAL pathways have an important role in the pathogenesis of HPE (Roessler and Muenke 2010). Chromosomal rearrangements disrupting genes encoding proteins related to these and other key signaling pathways are identified in ~50% of patients, and another 25% of HPE occurs in the context of other multi-malformation syndromes (e.g. Smith-Lemli-Opitz, and others). Among patients with non-syndromic, non-chromosomal HPE, 25% have mutations in genes in the aforementioned pathways; these genes include SHH (Roessler et al. 1996), SIX3 (Wallis et al. 1999; Domené et al. 2008; Lacbawan et al. 2009), ZIC2 (Brown et al. 1998; Solomon et al. 2010a), TGIF (Gripp et al. 2000), GLI2 (Roessler et al. 2003; Roessler et al. 2005), NODAL (Roessler et al. 2009c), PTCH (Ming et al. 2002), and others. Additional environmental risk factors that potentially increase the risk of HPE include prenatal exposure agents such as retinoic acid, alcohol, gestational diabetes, and low maternal cholesterol (Muenke and Beachy 2000; Haas et al. 2007; Pineda-Alvarez et al. 2010). Private familial mutations are the most frequently observed type of genetic changes. These mutations often segregate within pedigrees (Muenke lab, unpublished data); however, incomplete penetrance and variable expressivity are extremely common, particularly in those with mutations in SHH and SIX3 (Solomon et al. 2009, 2010b).
Although substantial progress has recently been made in identifying many of the principal genetic and environmental factors that contribute to the development of HPE, the cause in a significant proportion of affected individuals remains unclear, making the delineation of additional contributory factors essential (Pineda-Alvarez et al. 2010). Furthermore, clarification of just how gene–gene and gene–environmental factors contribute to the incidence and severity of HPE spectrum conditions remains a significant challenge.
During development of the brain and face in animals, SHH acts as a morphogen, wherein a tightly regulated gradient is required to generate an adequate signal throughout a target field. Many factors are involved in the regulation of such a gradient, such as the bi-lipid modification of the Hedgehog (HH) ligands, the proposed macro-molecular multimers influencing free ligand diffusion, the intercellular matrix within which the ligand particles diffuse, and the induction of HH binding proteins such as PTCH1 and HIP that limit the range of action on target cells (Roessler and Muenke 2003; Muenke and Beachy 2000; Ming et al. 2002). While many components of this pathway have been implicated in human HPE causation, the full extent of sequence variations in the extended Hedgehog pathway is yet to be determined.
Growth arrest-specific 1 gene (GAS1) encodes a glycosylphosphatidyl inositol (GPI)-anchored membrane receptor protein (Stebel et al. 2000), which acts as co-receptor and positive modulator of at least two hedgehog (HH) proteins (SHH and IHH). In mice, Gas1 null mutations (through targeted disruption) produces a phenotype consistent with the microform HPE spectrum, including brain, craniofacial, and limb anomalies that are more severe with co-morbid genetic reductions of Shh gene dosage or deletion of another hedgehog co-receptor, Cdo (Martinelli and Fan 2007a; Seppala et al. 2007; Allen et al. 2007).
SHH physically interacts with GAS1 through several specific residues. At least one amino acid substitution (p.Asn115Lys) in those residues has been directly implicated in human HPE and causes a decrease in the affinity between SHH and GAS1, highlighting a role in human HPE (Nanni et al. 1999; Roessler et al. 2009a) related to the way this interaction affects downstream signaling (Martinelli and Fan 2009).
In order to generate an adequate signaling response, GAS1 acts in synergy with PTCH1; cells expressing both GAS1 and PTCH have an increased capacity to bind SHH compared to the situation in which either is expressed alone (Seppala et al. 2007; Martinelli and Fan 2007a, b). The fly ortholog of Cdo, ihog, demonstrates multiple complex interactions with ptc that influence hh pathway activity. In vertebrates the orthologs CDO and BOC are also considered to be cell membrane co-receptors and positive regulators of SHH, and also interact with PTCH and GAS1 to stabilize the binding affinity with SHH, resulting in subsequent activation of the downstream pathway (Beachy et al. 2010; Martinelli and Fan 2007a). The requirement for this type of interaction is exemplified by the occurrence of both severe HPE and typically accompanying craniofacial defects in Gas1−/−; Cdo−/− double null mice, while the null condition for either gene alone produces relatively milder midline craniofacial anomalies (Allen et al. 2007).
GAS1 is also important in initiating and maintaining the SHH morphogenic gradient. In early development, SHH and GAS1 co-localize in the floor plate of the neural plate/tube. However, SHH expression decreases GAS1 expression in the most ventral aspect of the neural tube, and conversely increases the expression of GAS1 in more dorsal locations where the SHH concentration is lower. This supports the important role of GAS1 in maintaining SHH long-range signaling (Kang et al. 2007; Martinelli and Fan 2007a). Taken together, we conclude that GAS1 constitutes an excellent pathway-derived candidate gene that may contribute to the phenotypic spectrum of human HPE.
A recent study screened a Brazilian cohort of 54 unrelated patients with findings in the HPE phenotypic spectrum for mutations in GAS1 (Ribeiro et al. 2010). This study described four novel sequence variants. Two of these patients did not present with chromosomal anomalies or detectable mutations in the genes most commonly implicated in HPE (specifically SHH, SIX3, ZIC2, TGIF, and GLI2). Interestingly, the remaining two individuals presented with potentially pathogenic mutations in SHH in addition to the newly described GAS1 sequence variants. The significance of this observation will be discussed.
In addition to extending our sequencing analysis of GAS1 (NM_002048.2) to a larger cohort of unrelated patients with HPE than previously reported, we now also present detailed molecular and clinical findings of all known sequence variants in this replication study, including experimental functional data on the ligand (SHH) to receptor (GAS1) binding affinity acquired from cultured cells.
Materials and methods
Patient selection and DNA sequencing of GAS1
All subjects in this study provided written informed consent for participation in HPE-related research and clinical protocols in accordance with our NHGRI IRB guidelines.
Sequencing analysis was performed at two different phases during this study. The initial sample set was composed of 310 unrelated probands with HPE. Later, given the positive results of the ligand–receptor affinity assays (see “Results”), an additional set of 84 patients with HPE (more recently recruited) and a corresponding panel of 96 Human Random Controls (HRC) (Sigma-Aldrich, MO, USA) were also analyzed with the goal of identifying additional variants, as well as interrogating whether any of the novel variants identified in HPE cohorts were also present in controls.
GAS1 (NM_002048.2) reference sequence annotation was obtained from publicly available databases provided by NCBI (http://www.ncbi.nlm.nih.gov/) and the UCSC Genome Browser (http://www.genome.ucsc.edu/). Synthetic oligonucleotide primers were designed and optimized to cover this gene’s coding sequence (1 kb) and immediate flanking sequences (amplicon 1: Forward primer 5′ GTG GGC AGG ACT TGG ACA AAC 3′, and reverse primer: 5′ CTG CTC AAC GAC TGC GTG TGC 3′; amplicon 2: forward primer: 5′ CTG AGC CGC TAC CTG ACC TAC 3′, and reverse primer: 5′ GTG GCT TGG GAC AGA TAG AAG G 3′). PCR amplification was performed using 25 ng of genomic DNA template with the FastStart® Polymerase PCR Kit (Roche Applied Sciences, IN, USA) on a 25 μl total reaction volume, under the following conditions: 1 × (2.5 μl) of amplification buffer (10 × containing 20 mM of MgCl2), 0.20 mM (0.5 μl) of dNTP mix (10 mM), 1 × (5 μl) of GC-rich solution (5 ×), 0.30 mM of each oligonucleotide primer, and 1 U (0.2 μl) of FastStart® Polymerase (5 U/μl). Subsequently, PCR products were purified using QIAquick® 96 PCR purification kit (Qiagen, MD, USA).
Sequencing reactions were performed using the BigDye Terminator v3.1 chemistry, and capillary electrophoresis was performed in an ABI 3730xl genetic analyzer (Applied Biosystems, CA, USA) as recommended by the manufacturer. Chromatograms were aligned to the reference sequence (NM_002048.2) and analyzed using Sequencher version 4.9 (GeneCodes Corp, MI, USA).
Bioinformatics analysis
Nucleotide and protein multi-species alignments were performed using EvoPrinter HD provided by the National Institute of Neurological Disorders and Stroke (NINDS) (http://evoprinter.ninds.nih.gov/), and Constraint-based Multiple Alignment Tool (COBALT) provided by NCBI (http://www.ncbi.nlm.nih.gov/tools/cobalt/) in order to estimate the conservation throughout higher vertebrate species of the nucleotide and protein residues where variants have occurred. Conserved functional domains of GAS1 were determined using UniProt (http://expasy.org/sprot/) and literature review (Stebel et al. 2000; Cabrera et al. 2006; Lee and Fan 2001). SignalP 3.0 Server (http://www.cbs.dtu.dk/services/SignalP/) was used to evaluate the likely effect of the variant occurring within GAS1’s signaling peptide. Sequence variations were named using standard nomenclature rules http://www.hgvs.org/mutnomen/) and confirmed by on-line Name Checker using Mutalyzer (http://www.mutalyzer.nl/2.0).
SHH-N-AP surface binding assay
Missense sequence variants in GAS1 found in the first set of patients (310 patients), and relevant variants described in previous studies from the literature (Ribeiro et al. 2010) were included in this arm of the study (Tables 1, 2). Interestingly, one variant detected in our study (c.928C > T, p.Pro310Ser), could not be successfully propagated in E. coli, probably due to cellular toxicity; consequently, it could not be tested in the receptor–ligand binding assay. The observed toxicity is intriguing and could reflect an interaction with related GPI-linked enzymes in bacterial cells. Synonymous or non-coding variations were not tested in functional studies. The full length GAS1 cDNA was subjected to site-directed mutagenesis by a contractor (Transponics) using commercial reagents. Sequence verification of both coding strands documented construct integrity.
Table 1.
Experimentally determined variation of the human GAS1 gene
| GAS1 variant
|
RAF in patients (Na = 896) |
RAF in control (Nb = 192) |
Loss of SHH-GAS1 binding affinity | Reference | |
|---|---|---|---|---|---|
| Position | Change | ||||
| c.16C > G | p.Leu6Val | 0.11% (1) | – | ND | This study |
| c.96C > G | p.Gly32Gly | 0.11% (1) | – | ND | This study |
| c.133C > G | p.Arg45Gly | 0.11% (1) | – | ~20% | This study |
| c.264C > Tc | p.Ala88Ala | 3.57% (32) | 0.52% (1) | ND | This study |
| c.482G > A | p.Gly161Asp | – | 0.52% (1) | ND | This study |
| c.599C > G | p.Thr200Arg | 0.11% (1) | – | ~95% | Ribeiro et al. (2010) |
| c.660C > G | p.Asn220Lys | 0.11% (1) | – | 20–25% | This study |
| c.736G > T | p.Ala246Ser | 0.11% (1) | – | 0% | Ribeiro et al. (2010) |
| c.741G > A | p.Glu247Glu | 0.11% (1) | – | ND | This study |
| c.775G > A | p.Gly259Argd | 0.11% (1) | – | ~10%d | Ribeiro et al. (2010) |
| c.808G > T | p.Asp270Tyr | 0.11% (1) | – | 5–10% | Ribeiro et al. (2010) |
| c.825C > A | p.Thr275Thr | 0.11% (1) | – | ND | This study |
| c.831C > G | p.Gly277Gly | 0.11% (1) | – | ND | Ribeiro et al. (2010) |
| c.840T > G | p.Gly280Gly | 0.11% (1) | – | ND | Ribeiro et al. (2010) |
| c.863A > G | p.Asp288Gly | 0.11% (1) | – | 15–20% | Ribeiro et al. (2010) |
| c.882G > A | p.Pro294Pro | 0.11% (1) | – | ND | Ribeiro et al. (2010) |
| c.927G > C | p.Leu309Leu | – | 0.52% (1) | ND | This study |
| c.928C > T | p.Pro310Ser | 0.11% (1) | – | NDe | This study |
| c.958G > T | p.Gly320Cys | 0.11% (1) | – | 0% | This study |
| c.960C > G | p.Gly320Gly | 0.11% (1) | – | ND | Ribeiro et al. (2010) |
| c.1038*46T > C | NA | – | 0.52% (1) | ND | This study |
RAF Rare allele frequency
Total number of chromosomes analyzed to determine the RAF including the patients in this study (394) and the patients of the Brazilian study (Ribeiro et al. 2010) (54)
Total number of control chromosomes analyzed to determine the RAF
This was reported in the Brazilian study (Ribeiro et al. 2010) as c.260C > T
c.775G > A was reported in the Brazilian study (Ribeiro et al. 2010) as p.Gly259Gln (5–10% loss-of-affinity in the SHH-N-AP surface biding assay—Fig. 2a), but the described nucleotide variation corresponds to p.Gly259Arg
Variant did not propagate in E. coli, therefore, it was not tested in the binding assay
Table 2.
Clinical characteristics of affected individuals with missense variants in GAS1
| Individual | GAS1 variant
|
Inheritance | Other HPE-associated genes | Relevant clinical data | Reference | |
|---|---|---|---|---|---|---|
| Position | Change | |||||
| 10042 | c.16C > G | p.Leu6Val | Paternal | SHH, SIX3, ZIC2 and TGIF are normal | Lobar HPE, hypotelorism, single nostril, cleft lip/palate. Father is phenotypically unaffected, mother described as having clear “microforms” of HPE | This study |
| 9490 | c.133C > G | p.Arg45Gly | Paternal | de novo ZIC2: c.994_1005dup, p. Cys335_Pro338dup; SHH, SIX3, TGIF are normal | Semilobar HPE, developmental delay, agenesis of the corpus callosum, microcephaly and spasticity | This study |
| Patient 1 | c.599C > G | p.Thr200Arg | NA | SHH, SIX3, ZIC2 and TGIF are normal | Semilobar HPE, developmental delay, hypoplastic midface, upslanting palpebral fissures, proptosis, depressed nasal bridge, absent collumella, and bilateral cleft lip and palate | Ribeiro et al. (2010) |
| 1024 | c.660C > G | p.Asn220Lys | Paternal | ZIC2: c.709_711dup; p.His239dup (polymorphism); SHH, SIX3 and TGIF are normal | Hypoplastic corpus callosum, diabetes insipidus, craniosynostosis of metopic suture, pyriform apeture stenosis, midline cleft lip with complete premaxillary agenesis | This study |
| Patient 7 | c.736G > T | p.Ala246Ser | NA | No data available | No data available | Ribeiro et al. (2010) |
| Patient 2 | c.775G > A | p.Gly259Arg | NA | Normal karyotype, SHH, SIX3, TGIF, ZIC2 and GLI2 are normal | Hypotelorism, with proptosis, hypoplastic midface, depressed nasal bridge, absent collumella with notching of the tip, bilateral cleft lip and palate | Ribeiro et al. (2010) |
| Patient 3 | c.808G > T | p.Asp270Tyr | NA | Paternally inherited SHH: c.653T > C, p.Leu218Pro; SIX3, ZIC2, TGIF and GLI2 are normal | Microcephaly, hypotelorism, hypoplastic midface, depressed nasal bridge and nasal bones, right cleft lip and palate, no brain structural anomalies | Ribeiro et al. (2010) |
| Patient 4 | c.863A > G | p.Asp288Gly | de novo | de novo SHH: c.1088G > A, p.Cys363Tyr | Microcephaly, hypotelorism, midface hypoplasia, hypoplastic nasal bones, depressed nasal bridge, midline cleft lip and palate | Ribeiro et al. (2010) |
| 6386 | c.928C > T | p.Pro310Ser | Paternal | SHH, SIX3, ZIC2 and TGIF are normal | Semilobar HPE, absent pituitary and olfactory bulbs, microcephaly, hypotelorism, midface hypoplasia, depressed nasal bridge and hypoplastic septum, midline cleft lip with intact palate, micropenis | This study |
| 4953 | c.958G > T | p.Gly320Cys | Mother is normal; father is unavailable | ZIC2: c.709_711dup; p.His239dup (polymorphism); SHH, SIX3 and TGIF: are normal | HPE, no further clinical data available | This study |
SHH-N-alkaline phosphatase (SHH-N-AP) conditioned media was produced in HEK293T cells by transient transfection using pcDNA3-SHH-N-AP (Lee et al. 2001). Concentration was determined by AP activity (1 pmol of SHH-N-AP = 22.8 OD 405/h at 37°C) using p-nitrophenyl-phosphate as a substrate in standard assay condition (Lee et al. 2001). A binding titration curve was obtained by using COS cells transfected with 1 μg/well of pcDNA3- human GAS1 and 3 μl of Fugen-HD (Roche) in 6-well dishes. 48 h later, 1 ml of twofold serially diluted SHH-N-AP conditioned media was applied to each well. Cell surface binding and washing conditions were described in (Lee et al. 2001). For the relative binding assay, a sub-maximal (near saturation) concentration was used for all point mutants of human GAS 1 (in pcDNA3) and wild type in parallel and the bound SHH-N-AP activities were determined. Kds for wildtype (WT) and p.Thr200Arg were determined using serial concentrations of SHH-N-AP for binding, followed by Scatchard plot analysis (Lee et al. 2001).
Western-blot analyses
One microgram of pcDNA3 vectors carrying wild type and point mutants of human GAS1 cDNA were transfected into COS cells (in 6-well dishes); 0.25 μg pcDNA3-gpi-YFP (Martinelli and Fan 2007a) was co-transfected as control; Empty vector was used as a control for the specificity of antibody detection of GAS1 and YFP. Forty-eight hours after transfection, cells were lysed in 300 μl of RIPA buffer. Cleared lysates were supplemented with 100 μl of 4 × SDS sample buffer, and 10 μl of each lysate were subjected to 12.5% SDS–PAGE, followed by Western transfer to PVDF membrane. Western blot analyses were performed using a goat anti-human GAS1 (1:1,000; R&D Systems) and rabbit anti-GFP (1:2,000; Torre Pines), followed by appropriate HRP-conjugated secondary antibodies (Bio-Rad) and the ECL kit (GE).
Results
DNA sequence analysis
Sequence analysis of 394 unrelated probands referred for HPE spectrum clinical findings identified 5 novel missense sequence variants (not reported in dbSNP v.132—http://www.ncbi.nlm.nih.gov/projects/SNP/) in 5 unrelated individuals (c.16C > G, p.Leu6Val; c.133C > G, p.Arg45Gly; c.660C > G, p.Asn220Lys; c.928C > T, p.Pro310Ser; and c958G > T, p.Gly320Cys). In addition, three silent variants were identified, which were exclusive to this study’s patient cohort (c.96C > G, p.Gly32Gly; c.741G > A, p.Glu247Glu; and c.825C > A, p.Thr275Thr), as well as one common polymorphism (c.264C > T, p.Ala88Ala) that occurred in all patients cohorts, including those reported by Ribeiro et al. (2010) in the Brazilian study, and in our control group. Analysis of 192 chromosomes from the Human Random Control collection (Sigma-Aldrich, MO, USA) revealed the presence of three additional novel rare variants, 1 missense (c.482G > A, p.Gly161Asp), 1 synonymous (c.927G > C, p.Leu309Leu) and 1 non-coding variant (c.1038*46T > C) present exclusively in this latter group (summarized in Fig. 1; Table 1). These data represent a significant sampling of the genetic variation present in the GAS1 gene, and suggests that non-synonymous alterations are fairly common among HPE probands and their direct blood relatives.
Fig. 1.

Structural motifs of GAS1 and the position of missense variations. The signal peptide (tan) and the COOH–terminus removed by the process of GPI-anchor addition (grey) are sites of genetic variation (red). The patient-associated mutations from this study are shown in red, while those of the Brazilian study are shown in blue. The non-synonymous p.Gly161Asp detected among controls (green) is observed in the non-conserved region between two conserved motifs (yellow)
SHH-N-AP surface binding assay
All clonable mutations in GAS1 detected in the present study were combined with those of the Brazilian study (Ribeiro et al. 2010) to evaluate their degree of SHH-N-AP (ligand) and GAS1 (receptor) interaction. Two point mutations (c.928C > T, p.Pro310Ser) that could not be propagated in E. coli, and the missense sequence variant that occurred in the random human control individual (p.Gly161Asp) were not included the experiments.
Of the nine mutations tested with this assay (Fig. 2a, b), seven had measurable reductions of binding capacity between SHH-N-AP and the mutant receptor. One mutation in particular (c.599C > G, p.Thr200Arg) demonstrated an approximately 95% loss of total binding with SHH compared to wild-type constructs. Scatchard analysis for this c.599C > G GAS1 construct demonstrates its failure to bind SHH with measurable affinity despite comparable protein expression as determined by Western blot (Fig. 2a, b). The second mutation that is significantly affected encodes c.660C > G, p.Asn220Lys. This variant reduces GAS1’s binding activity to SHH by approximately 20–25% as determined by the AP (SHH-N-AP) reporter bound on the transfected cells. Interestingly, these mutations are located in the second cysteine-rich domain, which is thought to perform the receptor binding function, per se.
Fig. 2.
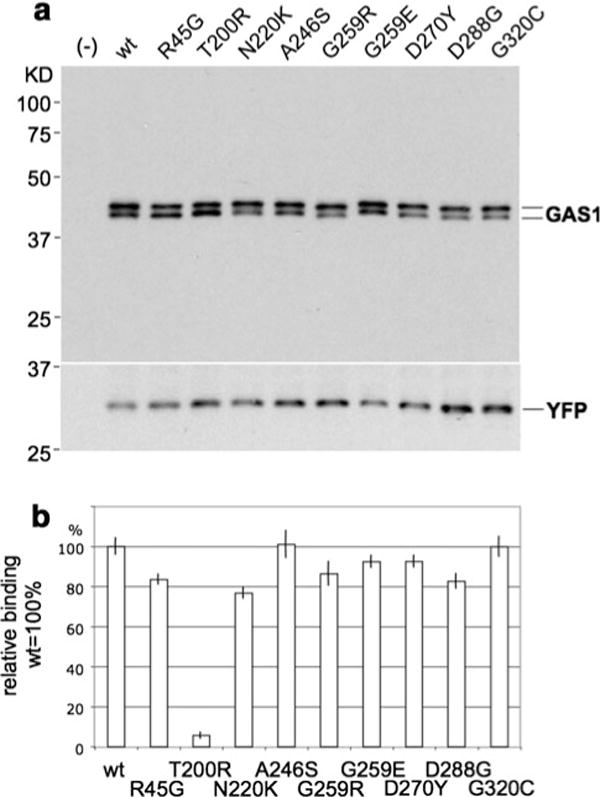
a The upper panel, depicting a Western blot, illustrates that transfected pcDNA3 constructs with the wild-type version and all point mutations of GAS1 presented a uniform level of expression of GAS1. This excludes differences of expression levels as the causeofsignificant variation of binding affinity in the SHH-N-alkaline phosphatase (SHH-N–AP) surface binding assay. b The lower panel shows relative percentage surface-binding of SHH-N-AP to GAS1. Constructs containing the wild-type version of the protein and each point mutation in GAS1 were tested. While the variant p.Thr200Arg fails to bind with SHH, several other variants show measurably hypomorphic reduced affinity (p.Arg45Gly, p.Asn220Lys, p.Gly259Arg, p.Gly259Glu, p.Asn270Tyr, and p.Asn288Gly) compared to the wild-type while others (p.Ala246Ser and p.Gly320Asp) demonstrate essentially the same affinity as the wild-type (100%). Note that a fully processed, GPI-linked protein translated from the p.Gly320Asp missense construct would lack this residue in the membrane form
The remaining missense mutations had measurable binding reductions ranging between 20 and 5%. These variants are located outside of cysteine-rich domains, but still present in highly conserved regions based on multiple species alignments. Two sequence variants did not affect the ability of GAS1 to bind SHH; the explanation regarding one of these variants (c.958G > T, p.Gly320Cys) likely relates to the fact that it is located in the GPI-domain (hydrophobic carboxyl end of the protein) that becomes replaced by the GPI-anchor. The second such variant (c.736G > T, p.Ala246Ser) is located three residues to the carboxyl end of the second cysteine-rich domain (Fig. 1). Furthermore, the nature of the amino acid change suggests that it is likely a benign variant (Ribeiro et al. 2010).
Discussion
In this present study, we provide functional data regarding all types of alterations (benign, hypomorphic and loss-of-function alleles) in GAS1. When these data are combined with the mutation data of other genes in the Hedgehog pathway an interesting pattern begins to emerge. While extreme dysfunction of GAS1 is relatively infrequent (with the p.Thr200Arg variant as the best example in this study), we find an unexpectedly frequent occurrence of hypomorphic alleles with measurably reduced SHH-AP binding activity. This is precisely what was previously predicted from gene–gene interaction studies in animal models: studies that imply that Gas1 variants can function as modifiers of other Hedgehog-related gene products, including HH itself. It is also intriguing that when two mutations are identified in the same proband (a situation that is clearly uncommon overall) one of the mutations is predicted to be strong or nearly null, while the companion variation is hypomorphic. We propose that the former are the primary drivers of the malformation and that the hypomorphic variants account for much of the clinical phenotypic variability.
In order to adequately interpret the effect of specific variations of GAS1 on the activation of the SHH signaling pathway, the former’s protein structure and basic biology must be understood. As shown in Fig. 1, the protein structure of GAS1 consists of two conserved hydrophobic domains, a signaling peptide and a propeptide/GPI-signaling domain at the amino and carboxyl ends of the protein (amino acids 1–39 and 318–345, respectively), two cysteine-rich domains analogous to the Glial cell-derived neurotrophic factor (GDNF), which composes the portion of the receptor that interacts with its ligands, and a GPI-anchor placed in residue serine 318. The two hydrophobic domains are cleaved off the mature protein in the endoplasmatic reticulum during processing (Cabrera et al. 2006; Lee and Fan 2001; Stebel et al. 2000). Presumably, the conserved residues of the remaining polypeptide are critical for the three-dimensional structure of the receptor, as well as its presentation (anchoring) on the cell membrane.
Two missense variants observed in both hydrophobic regions are interpreted as likely benign (Figs. 1, 2). The variant c.16C > G, p.Leu6Val, occurs in the signaling peptide, which was not predicted by SignalP 3.0 to be affected by this amino acidic change; additionally, it was noted to be inherited from an unaffected parent (Table 2). The second variant, c.958G > T, p.Gly320Cys, occurs at the end of the gene in an evolutionarily conserved residue in vertebrates. However, this variant did not have an effect on the observed binding affinity between GAS1 and SHH (Fig. 2b) since it is effectively eliminated once the GPI-anchor is attached.
Another benign variant was elucidated, c.736G > T, p.Ala246Ser. Multi-species alignments show that this amino acid is poorly conserved among vertebrates, and in addition, the SHH-N-AP surface-binding assay (Fig. 2b) showed no loss-of-binding. Furthermore, Ribeiro et al. (2010) classified this rare variant as benign based on in-silico prediction software, such as, Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/; Adzhubei et al. 2010).
Five GAS1’s variants were interpreted as hypomorphic, with decreased SHH binding-capacity, ranging from 5 to 25% reduction in SHH-binding capacity (Fig. 2b). Interestingly, three of these variants co-occurred in unrelated patients with previously identified mutations in ZIC2 and SHH (see Table 2). A de novo ZIC2 mutation, c.994_1005dup, occurred in a patient with semilobar HPE, and co-segregated with the paternally inherited p.Arg45Gly variant in GAS1. The novel ZIC2 mutation, which alters the C2H2 motif of the third zinc-finger key element for this transcription factor, therefore represents a likely pathogenic variant (Roessler et al. 2009b). However, the hypomorphic change in GAS1 possibly contributes to the phenotypic characteristics of this patient, as it potentially further reduces the effective SHH signaling for the ventral midline structures. Other variants of GAS1 (p.Asp270Tyr and p.Asp288Gly) that co-occured with SHH mutations were identified in two patients of the cohort described by our Brazilian colleagues (Ribeiro et al. 2010). Both SHH variants occurred in the COOH-terminus. Although the C-terminus of SHH is not part of the mature ligand, it has a fundamental role for N-terminus (ligand) processing and bi-lipidation. Mutations in this domain of the SHH protein have been proven to be hypomorphic (Roessler et al. 2009a; Tokhunts et al. 2010). The co-segregating variants in GAS1 found in this study (Fig. 2b) may contribute to further detriment the already altered SHH gradient in the ventral forebrain. Additional hypomorphic variants in GAS1 (p.Asn220Lys and p.Gly259Lys) occurred in patients with microforms of HPE and without co-segregation of any other known pathogenic mutation in HPE-associated genes. With these results, we feel that we have documented instances where hypomorphic mutations in known HPE-associated genes interact with others of related function and thus contribute to the variability of the phenotypic spectrum of this disorder (Solomon et al. 2010b). This represents the first objective (i.e., functionally demonstrated) example of the so-called “multiple hit hypothesis” for the genesis of HPE (Ming and Muenke 2002) (Fig. 3).
Fig. 3.
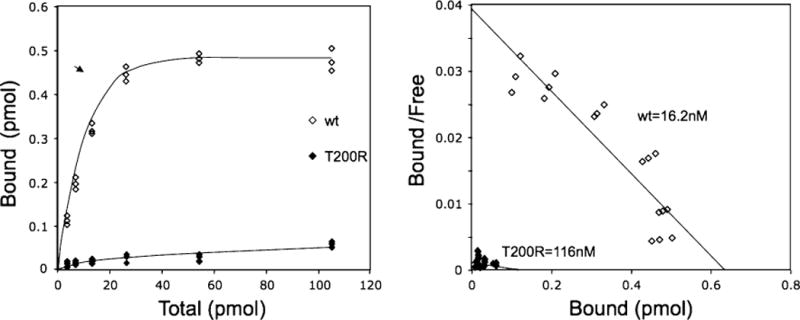
Scatchard plot demonstrating the inability of SHH-N alkaline phosphatase (SHH-AP) to bind to the p.Thr200Arg GAS1 construct compared to the wild-type version, which shows a normal titration curve that plateaus with increasing dose of SHH-N AP
Only one variant (p.Thr200Arg) presented with near-null binding affinity between GAS1 and SHH, and occurred in a patient with the semilobar form of HPE accompanied by moderately severe craniofacial anomalies (see Table 2 for details) (Ribeiro et al. 2010). With this GAS1 mutation being near null, the severity of the effect is less than might be expected (Solomon et al. 2010b). We suggest that other SHH co-receptors such as CDO and BOC may compensate to some degree for the GAS1 loss.
In summary, we have demonstrated that various mutations in GAS1 result in variable reductions in the binding affinity for its ligand, SHH. These mutations may independently cause phenotypic anomalies along the HPE phenotypic spectrum or act to modify the effect of bona fide mutations in other HPE-associated genes. The developmental anomalies that occur likely are the result of a direct adverse effect or a deepening of a defect in both the SHH morphogenic gradient and its downstream signaling cascade. This may be the basis for phenotypic heterogeneity observed in families segregating mutations in SHH and SIX3 (Solomon et al. 2010b; Solomon et al. 2009). Although sequence variations that occurred in the control group were not functionally tested in this study, evidence derived from preliminary data and other studies unrelated to this disorder suggest that hypomorphic modifying variants occur in the general population at higher frequency than more detrimental variants. We hypothesize that the allelic frequency of these hypomorphic variants maybe more common than more severe mutations, as the former require other variants to produce a detectable phenotypic effect. Our results further suggest that in future genomic research, more emphasis should be placed upon studying the effects of relatively minor genomic variations that may cause subtle anomalies only revealed by careful phenotypic characterization.
Overall, our interpretation of the results is nevertheless preliminary and, therefore, limited since the SHH-N-AP surface-binding assay only measures the physical interaction between SHH and GAS1, leaving the signaling cascade not fully assessed. Moreover, GAS1, CDO, BOC and PTCH, which are all co-receptors of SHH, synergize in order to produce a single effect (Zheng et al. 2010; Beachy et al. 2010), and therefore, the interactions between them have not yet been evaluated.
In conclusion, we have demonstrated that pathogenic variation of GAS1 occurs in more than 1% of patients with non-syndromic and non-chromosomal HPE (6 of 448 patients in this, combined with the Brazilian study). Following the current recommendations for molecular evaluation of patients with abnormalities that are within the HPE phenotypic spectrum (Pineda-Alvarez et al. 2010), we now propose that GAS1 should be screened for mutations as part of the routine molecular diagnostic approach for affected probands.
Acknowledgments
We thank the families who participated in these research studies, and the National Institute of Neurological Disorders and Stroke’s (NINDS) DNA Sequencing Facility for their technical support with DNA sequencing. This work was supported in part by the Division of Intramural Research (DIR) of the National Human Genome Research Institute (MM) and RO1 DK084963 (C-M F).
Contributor Information
Daniel E. Pineda-Alvarez, Medical Genetics Branch, National Human Genome Research Institute (NHGRI), National Institutes of Health, 35 Convent Drive, Building 35, Room 1B403, Bethesda, ML 20892-3717, USA
Erich Roessler, Medical Genetics Branch, National Human Genome Research Institute (NHGRI), National Institutes of Health, 35 Convent Drive, Building 35, Room 1B403, Bethesda, ML 20892-3717, USA.
Ping Hu, Medical Genetics Branch, National Human Genome Research Institute (NHGRI), National Institutes of Health, 35 Convent Drive, Building 35, Room 1B403, Bethesda, ML 20892-3717, USA.
Kshitij Srivastava, Medical Genetics Branch, National Human Genome Research Institute (NHGRI), National Institutes of Health, 35 Convent Drive, Building 35, Room 1B403, Bethesda, ML 20892-3717, USA.
Benjamin D. Solomon, Medical Genetics Branch, National Human Genome Research Institute (NHGRI), National Institutes of Health, 35 Convent Drive, Building 35, Room 1B403, Bethesda, ML 20892-3717, USA
C. Evan Siple, Department of Embryology, Carnegie Institution for Science, Baltimore, ML 21218, USA.
Chen-Ming Fan, Department of Embryology, Carnegie Institution for Science, Baltimore, ML 21218, USA.
Maximilian Muenke, Medical Genetics Branch, National Human Genome Research Institute (NHGRI), National Institutes of Health, 35 Convent Drive, Building 35, Room 1B403, Bethesda, ML 20892-3717, USA, mamuenke@mail.nih.gov.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BL, Tenzen T, McMahon AP. The Hedgehog-binding proteins Gas1 and Cdo cooperate to positively regulate Shh signaling during mouse development. Genes Dev. 2007;21(10):1244–1257. doi: 10.1101/gad.1543607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beachy PA, Hymowitz SG, Lazarus RA, Leahy DJ, Siebold C. Interactions between Hedgehog proteins and their binding partners come into view. Genes Dev. 2010;24(18):2001–2012. doi: 10.1101/gad.1951710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SA, Warburton D, Brown LY, Yu CY, Roeder ER, Stengel-Rutkowski S, Hennekam RC, Muenke M. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat Genet. 1998;20(2):180–183. doi: 10.1038/2484. [DOI] [PubMed] [Google Scholar]
- Cabrera JR, Sanchez-Pulido L, Rojas AM, Valencia A, Manes S, Naranjo JR, Mellstrom B. Gas1 is related to the glial cell-derived neurotrophic factor family receptors alpha and regulates Ret signaling. J Biol Chem. 2006;281(20):14330–14339. doi: 10.1074/jbc.M509572200. [DOI] [PubMed] [Google Scholar]
- Demyer W, Zeman W, Palmer CG. The Face Predicts the Brain: Diagnostic Significance of Median Facial Anomalies for Holoprosencephaly (Arhinencephaly) Pediatrics. 1964;34:256–263. [PubMed] [Google Scholar]
- Domené S, Roessler E, El-Jaick KB, Snir M, Brown JL, Velez JI, Bale S, Lacbawan F, Muenke M, Feldman B. Mutations in the human SIX3 gene in holoprosencephaly are loss of function. Hum Mol Genet. 2008;17(24):3919–3928. doi: 10.1093/hmg/ddn294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripp KW, Wotton D, Edwards MC, Roessler E, Ades L, Meinecke P, Richieri-Costa A, Zackai EH, Massague J, Muenke M, Elledge SJ. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat Genet. 2000;25(2):205–208. doi: 10.1038/76074. [DOI] [PubMed] [Google Scholar]
- Haas D, Morgenthaler J, Lacbawan F, Long B, Runz H, Garbade SF, Zschocke J, Kelley RI, Okun JG, Hoffmann GF, Muenke M. Abnormal sterol metabolism in holoprosencephaly: studies in cultured lymphoblasts. J Med Genet. 2007;44(5):298–305. doi: 10.1136/jmg.2006.047258. doi:jmg.2006.047258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn JS, Barnes PD. Neuroimaging advances in holoprosencephaly: Refining the spectrum of the midline malformation. Am J Med Genet C Semin Med Genet. 2010;154C(1):120–132. doi: 10.1002/ajmg.c.30238. [DOI] [PubMed] [Google Scholar]
- Kang JS, Zhang W, Krauss RS. Hedgehog signaling: cooking with Gas1. Sci STKE. 2007;2007(403):pe50. doi: 10.1126/stke.4032007pe50. [DOI] [PubMed] [Google Scholar]
- Lacbawan F, Solomon BD, Roessler E, El-Jaick K, Domene S, Velez JI, Zhou N, Hadley D, Balog JZ, Long R, Fryer A, Smith W, Omar S, McLean SD, Clarkson K, Lichty A, Clegg NJ, Delgado MR, Levey E, Stashinko E, Potocki L, Vanallen MI, Clayton-Smith J, Donnai D, Bianchi DW, Juliusson PB, Njolstad PR, Brunner HG, Carey JC, Hehr U, Musebeck J, Wieacker PF, Postra A, Hennekam RC, van den Boogaard MJ, van Haeringen A, Paulussen A, Herbergs J, Schrander-Stumpel CT, Janecke AR, Chitayat D, Hahn J, McDonald-McGinn DM, Zackai EH, Dobyns WB, Muenke M. Clinical spectrum of SIX3-associated mutations in holoprosencephaly: correlation between genotype, phenotype and function. J Med Genet. 2009;46(6):389–398. doi: 10.1136/jmg.2008.063818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CS, Fan CM. Embryonic expression patterns of the mouse and chick Gas1 genes. Mech Dev. 2001;101:1–2. 293–297. doi: 10.1016/s0925-4773(01)00283-0. S092 5477301002830[pii] [DOI] [PubMed] [Google Scholar]
- Lee CS, Buttitta L, Fan CM. Evidence that the WNT-inducible growth arrest-specific gene 1 encodes an antagonist of sonic hedgehog signaling in the somite. Proc Natl Acad Sci USA. 2001;98(20):11347–11352. doi: 10.1073/pnas. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli DC, Fan CM. Gas1 extends the range of Hedgehog action by facilitating its signaling. Genes Dev. 2007a;21(10):1231–1243. doi: 10.1101/gad.1546307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli DC, Fan CM. The role of Gas1 in embryonic development and its implications for human disease. Cell Cycle. 2007b;6(21):2650–2655. doi: 10.4161/cc.6.21.4877. 4877[pii] [DOI] [PubMed] [Google Scholar]
- Martinelli DC, Fan CM. A sonic hedgehog missense mutation associated with holoprosencephaly causes defective binding to GAS1. J Biol Chem. 2009;284(29):19169–19172. doi: 10.1074/jbc.C109.011957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga E, Shiota K. Holoprosencephaly in human embryos: epidemiologic studies of 150 cases. Teratology. 1977;16(3):261–272. doi: 10.1002/tera.1420160304. [DOI] [PubMed] [Google Scholar]
- Ming JE, Muenke M. Multiple hits during early embryonic development: digenic diseases and holoprosencephaly. Am J Hum Genet. 2002;71(5):1017–1032. doi: 10.1086/344412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming JE, Kaupas ME, Roessler E, Brunner HG, Golabi M, Tekin M, Stratton RF, Sujansky E, Bale SJ, Muenke M. Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Hum Genet. 2002;110(4):297–301. doi: 10.1007/s00439-002-0695-5. [DOI] [PubMed] [Google Scholar]
- Muenke M, Beachy PA. Genetics of ventral forebrain development and holoprosencephaly. Curr Opin Genet Dev. 2000;10(3):262–269. doi: 10.1016/s0959-437x(00)00084-8. S0959-437X(00)00084-8[pii] [DOI] [PubMed] [Google Scholar]
- Nanni L, Ming JE, Bocian M, Steinhaus K, Bianchi DW, Die-Smulders C, Giannotti A, Imaizumi K, Jones KL, Campo MD, Martin RA, Meinecke P, Pierpont ME, Robin NH, Young ID, Roessler E, Muenke M. The mutational spectrum of the sonic hedgehog gene in holoprosencephaly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly. Hum Mol Genet. 1999;8(13):2479–2488. doi: 10.1093/hmg/8.13.2479. ddc285[pii] [DOI] [PubMed] [Google Scholar]
- Pineda-Alvarez DE, Dubourg C, David V, Roessler E, Muenke M. Current recommendations for the molecular evaluation of newly diagnosed holoprosencephaly patients. Am J Med Genet C Semin Med Genet. 2010;154C(1):93–101. doi: 10.1002/ajmg.c.30253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro LA, Quiezi RG, Nascimento A, Bertolacini CP, Richieri-Costa A. Holoprosencephaly and holoprosencephaly-like phenotype and GAS1 DNA sequence changes: Report of four Brazilian patients. Am J Med Genet A. 2010;152A(7):1688–1694. doi: 10.1002/ajmg.a.33466. [DOI] [PubMed] [Google Scholar]
- Roessler E, Muenke M. How a Hedgehog might see holoprosencephaly. Hum Mol Genet. 2003;12(Spec No 1):R15–R25. doi: 10.1093/hmg/ddg058. [DOI] [PubMed] [Google Scholar]
- Roessler E, Muenke M. The molecular genetics of holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010;154C(1):52–61. doi: 10.1002/ajmg.c.30236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, Tsui LC, Muenke M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14(3):357–360. doi: 10.1038/ng1196-357. [DOI] [PubMed] [Google Scholar]
- Roessler E, Du YZ, Mullor JL, Casas E, Allen WP, Gillessen-Kaesbach G, Roeder ER, Ming JE, Ruiz i Altaba A, Muenke M. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc Natl Acad Sci USA. 2003;100(23):13424–13429. doi: 10.1073/pnas. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E, Ermilov AN, Grange DK, Wang A, Grachtchouk M, Dlugosz AA, Muenke M. A previously unidentified amino-terminal domain regulates transcriptional activity of wild-type and disease-associated human GLI2. Hum Mol Genet. 2005;14(15):2181–2188. doi: 10.1093/hmg/ddi222. [DOI] [PubMed] [Google Scholar]
- Roessler E, El-Jaick KB, Dubourg C, Velez JI, Solomon BD, Pineda-Alvarez DE, Lacbawan F, Zhou N, Ouspenskaia M, Paulussen A, Smeets HJ, Hehr U, Bendavid C, Bale S, Odent S, David V, Muenke M. The mutational spectrum of holoprosencephaly-associated changes within the SHH gene in humans predicts loss-of-function through either key structural alterations of the ligand or its altered synthesis. Hum Mutat. 2009a;30(10):E921–E935. doi: 10.1002/humu.21090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E, Lacbawan F, Dubourg C, Paulussen A, Herbergs J, Hehr U, Bendavid C, Zhou N, Ouspenskaia M, Bale S, Odent S, David V, Muenke M. The full spectrum of holoprosencephaly-associated mutations within the ZIC2 gene in humans predicts loss-of-function as the predominant disease mechanism. Hum Mutat. 2009b;30(4):E541–E554. doi: 10.1002/humu.20982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E, Pei W, Ouspenskaia MV, Karkera JD, Velez JI, Banerjee-Basu S, Gibney G, Lupo PJ, Mitchell LE, Towbin JA, Bowers P, Belmont JW, Goldmuntz E, Baxevanis AD, Feldman B, Muenke M. Cumulative ligand activity of NODAL mutations and modifiers are linked to human heart defects and holoprosencephaly. Mol Genet Metab. 2009c;98(1–2):225–234. doi: 10.1016/j.ymgme.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seppala M, Depew MJ, Martinelli DC, Fan CM, Sharpe PT, Cobourne MT. Gas1 is a modifier for holoprosencephaly and genetically interacts with sonic hedgehog. J Clin Invest. 2007;117(6):1575–1584. doi: 10.1172/JCI32032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon BD, Lacbawan F, Jain M, Domene S, Roessler E, Moore C, Dobyns WB, Muenke M. A novel SIX3 mutation segregates with holoprosencephaly in a large family. Am J Med Genet A. 2009;149A(5):919–925. doi: 10.1002/ajmg.a.32813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon BD, Lacbawan F, Mercier S, Clegg NJ, Delgado MR, Rosenbaum K, Dubourg C, David V, Olney AH, Wehner LE, Hehr U, Bale S, Paulussen A, Smeets HJ, Hardisty E, Tylki-Szymanska A, Pronicka E, Clemens M, McPherson E, Hennekam RC, Hahn J, Stashinko E, Levey E, Wieczorek D, Roeder E, Schell-Apacik CC, Booth CW, Thomas RL, Kenwrick S, Cummings DA, Bous SM, Keaton A, Balog JZ, Hadley D, Zhou N, Long R, Velez JI, Pineda-Alvarez DE, Odent S, Roessler E, Muenke M. Mutations in ZIC2 in human holoprosencephaly: description of a Novel ZIC2 specific phenotype and comprehensive analysis of 157 individuals. J Med Genet. 2010a;47(8):513–524. doi: 10.1136/jmg.2009.073049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon BD, Mercier S, Velez JI, Pineda-Alvarez DE, Wyllie A, Zhou N, Dubourg C, David V, Odent S, Roessler E, Muenke M. Analysis of genotype-phenotype correlations in human holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010b;154C(1):133–141. doi: 10.1002/ajmg.c.30240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebel M, Vatta P, Ruaro ME, Del Sal G, Parton RG, Schneider C. The growth suppressing gas1 product is a GPI-linked protein. FEBS Lett. 2000;481(2):152–158. doi: 10.1016/s0014-5793(00)02004-4. S0014-5793(00)02004-4[pii] [DOI] [PubMed] [Google Scholar]
- Tokhunts R, Singh S, Chu T, D’Angelo G, Baubet V, Goetz JA, Huang Z, Yuan Z, Ascano M, Zavros Y, Therond PP, Kunes S, Dahmane N, Robbins DJ. The full-length unprocessed hedgehog protein is an active signaling molecule. J Biol Chem. 2010;285(4):2562–2568. doi: 10.1074/jbc.M109.078626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis DE, Roessler E, Hehr U, Nanni L, Wiltshire T, Richieri-Costa A, Gillessen-Kaesbach G, Zackai EH, Rommens J, Muenke M. Mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nat Genet. 1999;22(2):196–198. doi: 10.1038/9718. [DOI] [PubMed] [Google Scholar]
- Zheng X, Mann RK, Sever N, Beachy PA. Genetic and biochemical definition of the Hedgehog receptor. Genes Dev. 2010;24(1):57–71. doi: 10.1101/gad.1870310. [DOI] [PMC free article] [PubMed] [Google Scholar]