

Abstract
Metastatic traits appear to be acquired by transformed cells with progenitor-like cancer-initiating properties, but there remains little mechanistic insight into this linkage. In this report, we show that the polarity protein Numbl, which is expressed normally in neuronal progenitors, becomes overexpressed and mislocalized in cancer cells from a variety of human tumors. Numbl overexpression relies on loss of the tumor suppressor microRNA-296-5p (miR-296), which actively represses translation of Numbl in normal cells. In turn, deregulated expression of Numbl mediates random tumor cell migration and invasion, blocking anoikis and promoting metastatic dissemination. In clinical specimens of non-small cell lung cancer, we found that Numbl overexpression correlated with a reduction in overall patient survival. Mechanistically, Numbl-mediated tumorigenesis involved suppression of a "stemness" transcriptional program driven by the stem cell programming transcription factor Klf4, thereby preserving a pool of progenitor-like cells in lung cancer. Our results reveal that Numbl-Klf4 signaling is critical to maintain multiple nodes of metastatic progression, including persistence of cancer-initiating cells, rationalizing its therapeutic exploitation to improve the treatment of advanced lung cancer
Keywords: Klf4, Numbl, cell invasion, metastasis, cancer-initiating cells, lung cancer
INTRODUCTION
The ability of tumor cells to colonize distant organs from a primary site, namely metastasis (1), heralds an incurable and almost invariably fatal disease course. It is now clear that this process involves broad remodeling of transcriptional programs, loss of tumor suppressor mechanisms, interplay with stromal components and changes in cellular properties that control cell migration and invasion (2).
One expanding gene regulatory network implicated in tumor cell motility includes microRNAs (miRs) (3). These molecules can function as oncogenes or tumor suppressors (4), but changes in miR expression in disparate tumor types, with concomitant downstream modulation of gene transcription, have been often linked to the acquisition of metastatic traits (5). In some malignancies, changes in gene expression controlled by oncogenic miRs may also influence the pool of cancer-initiating, progenitor-like cells (6, 7). Although still phenotypically ill-defined (8), these are considered rare cells with stem-like properties that may participate in the progression of at least certain tumors (9), for their resistance to drug-induced therapy (10), and propensity to metastatic dissemination (11).
In this context, decreased expression or complete loss of miR-296-5p (miR-296) has been observed in several types of human cancers (12–14), and often correlated with disease progression. Although elevated levels of miR-296 have been demonstrated in tumor-associated angiogenic endothelial cells, and contributed to growth factor signaling in these settings (15), other data have proposed a pivotal role of miR-296 in tumor suppression, mechanistically linked to modulation of HMGA1 oncogene expression (16), and deregulated function of the Scribble (17) cell polarity module (18).
In this study, we sought to further map the miR-296 tumor suppressor network for potential regulation of novel metastatic traits, specifically in lung cancer.
MATERIALS AND METHODS
Cell culture and miR-296 in vitro modulation
Human lung carcinoma A549, H23, H460, H1299, H1437, and H1792 or MDA-MB-231 breast cancer cells were purchased from the American Type Culture Collection (ATCC). Human embryonic kidney HEK293 cells were available in our laboratories. All cell lines were maintained in a 5% humidified incubator at 37°C, and kept in culture as recommended by the supplier. Immortalized human bronchial epithelial cells (HBEC3) were a generous gift from Dr. Marcelo Kazanietz (University of Pennsylvania, Perelman School of Medicine). HBEC3 cells were cultured in keratinocyte-SFM containing 50 µg/mL bovine pituitary extract and 5 ng/ml epidermal growth factor media until passage 7. All cell culture reagents were from Gibco-Invitrogen (Life Technologies, Carlsbad, CA, USA).
Immunoblotting and immunofluorescence
Aliquots of lung, breast cancer or HBEC3 cells were harvested 48 or 72 h after transfection and solubilized in 150 µl RIPA buffer supplemented with 1× complete protease and phosphatase inhibitors cocktails (Roche). Cell lysates (50 µg) were separated by electrophoresis on 10–12% SDS-polyacrylamide gels, transferred to PVDF membranes (Millipore), and probed with 1 µg/µl of antibodies against Numbl (Proteintech Group Inc., Chicago, IL), Scrib (Santa Cruz Biotechnology, Santa Cruz), Numb (Proteintech), β-catenin (Thermoscientific), c-Src (Santa Cruz), Tyr416-phosphorylated Src (p-Src, Biosource International), fibronectin (H-300, Santa Cruz), p21WAF1/Cip1 (Calbiochem, EMD Millipore Corporation, Billerica, MA), HA (Sigma-Adrich), laminin A (Santa Cruz), β-tubulin or β-actin (all from Sigma-Aldrich). Antibodies to Focal Adhesion Kinase (FAK), Tyr397-phosphorylated FAK (p-FAK), vimentin, Nanog, or Klf4 were from Cell Signaling. Reactive bands were visualized with ECL Plus reagents (GE Health Care).
For immunofluorescence experiments, lung cancer or HBEC3 cells were grown on cover-glasses, fixed in 4% paraformaldehyde for 15 min, permeabilized in ice-cold methanol, and incubated with an antibody to Numbl or Numb (both 10 µg/µl, Proteintech) for 16 h at 4°C, followed by a FITC-conjugated anti-rabbit secondary antibody (1:100, ThermoScientific) with or without an antibody to HA-tagged Klf4 (1:100, Sigma-Aldrich).
Slides were scored by light or fluorescent microscopy and photographed images were arranged with Adobe Photoshop CS5 for Windows. When confocal or two-photons microscopy analyses were performed, samples were imaged using a Leica TCS SP2 confocal or a Prairie Instruments Ultima 2 Photon microscopes, respectively.
Side population analysis
Transfected A549 cells were labeled with Hoechst 33342 (Cell Signaling Technology Inc, Danvers, MA), as described (19, 20). Briefly, cells were suspended at 1×106/ml in prewarmed DMEM-2% FCS and 10 mM HEPES buffer. Hoechst 33342 was added at a final concentration of 5 µg/ml in the presence or absence of reserpine (50 µM; Sigma-Aldrich). Cells were incubated for 2 h at 37°C with intermittent shaking, washed by centrifugation at 4°C with ice-cold HBSS-2% FCS and 10 mM HEPES (HBSS+), and suspended in ice-cold HBSS+ at a final concentration of 2×107/ml. PI (BD Bioscience) was added at a final concentration of 2 µg/ml to exclude dead cells. Before sorting, cells were filtered through a 40-µm cell strainer to obtain single cell suspension. All media reagents were from Gibco-Invitrogen (Life Technologies). Cell sorting and side population analyses were performed on a FACSAria using the FACSDiva (version 6.1.2, BD Bioscience) or FlowJo software (version 7.6.5, Tree Star Inc., Ashland, OR). The Hoechst 33342 dye was excited at 357 nm and its fluorescence was dual-wavelength analyzed (blue, 402–446 nm; red, 650–670 nm).
Motility and directional cell migration
Subconfluent (70%) cells were transfected with miR-296 mimics, target siRNAs or controls and cultured for 48 h. Wounds in the cell monolayers were created using a P200 micropipette tip, and the migration distance (units) was determined as reduction in the wound’s gap and quantified using NIH Image-J software, as described (17). For analysis of directional cell migration, monolayers of A549 cells were harvested 60 h after transfection, scratched and examined for Golgi orientation relative to the nucleus and the migration front after additional 6 h, by immunofluorescence (17, 21). Golgi staining was performed using Alexa Fluor555-conjugated anti-GM130 antibody (BD Bioscience, San Jose, CA). For analysis of stress fiber formation, F-actin was visualized by phalloidin-TRITC staining (Sigma-Aldrich, St. Luis, MO), and samples mounted in DAPI I (Abbott, Abbott Park, IL) were imaged using an AxioImager Z1 microscope (Carl Zeiss, Göttingen, Germany).
Cell invasion
A549 cells transfected with miR-296 mimics were seeded (5×104 cells) after 48 h in serum-free medium in Matrigel-coated chambers (8.0 µm pores, BD Biosciences). When siRNA or gene overexpression was performed, HBEC3 or the indicated lung cancer cell type were seeded in Matrigel-coated inserts after 24 h. FBS-containing medium was used as chemoattractant in the bottom chamber, and cells were allowed to migrate for 24 h, as described (17, 22). Cells that had invaded the lower surface of the membrane were fixed with methanol, stained with DAPI, and quantified by fluorescence microscopy (Nikon E600, Nikon Instruments Inc., Melville, NY).
In vivo liver metastasis model
All experiments involving animals were approved by an Institutional Animal Care and Use Committee. Female SCID/beige mice (6–8 weeks of age) were anesthetized with ketamine hydrochloride, the abdominal cavity was exposed by laparotomy, and animals were injected in the spleen with 4×106 H460 or A549 cells previously transfected with control non-targeting or Numbl-directed siRNA. To avoid potential confounding effects due to variable growth of a primary tumor, the spleen was removed 24 h after injection of the tumor cells, as previously described (23). On d. 11, all mice in the two groups were sacrificed. Liver samples were formalin-fixed, paraffin embedded, and analyzed histologically by hematoxylin-eosin staining.
Patient material
A series of 209 consecutive patients surgically treated for non-small cell lung cancer (NSCLC) at Fondazione IRCCS Ca’ Granda Hospital (Milan, Italy) between 2000 and 2004 was available for this study. This patient series included 149 cases of adenocarcinoma (AdCa) and 60 cases of squamous cell carcinoma (SCC) of the lung. Clinical outcome data were available for 172 patients (82%). NSCLC cases were staged according to the current TNM classification of malignant tumors (International Union Against Cancer, UICC, 7th edition, 2009). The clinical characteristics of the patient series analyzed in this study are summarized in Supplementary Table 1. An informed consent was obtained from all patients enrolled, and the study was approved by an Institutional Review Board (IRB) of the Fondazione IRCCS Ca’ Granda, Milan, Italy. The follow-up period ranged from 0 to 132 months (average 55.2 months). At the last follow-up (January 2011), 100 patients were deceased for progression of NSCLC, whereas 72 patients were alive. For four patients, matched distant metastatic lesions were available as well. Frozen tissues were available from an independent series of NSCLC patients previously described (24).
Statistical analysis and clinical validation
Differences among sample groups were analyzed using one-sided Student’s t-test, or Wilcoxon signed rank test. For survival analysis, the Kaplan–Meier method was used. To examine a potential association of Numbl staining and overall survival, NSCLC patients were assigned to two groups according to target expression. Cases whose immunoreactivity was scored below Numbl median value of immunoreactivity fell in the “Numbllow” group, whereas cases whose immunoreactivity levels were above the median value fell in the “Numblhigh” group. The two-sided log-rank test was used to compare survival curves. Statistical analyses were performed using GraphPad Prism version 4 for Windows or Ministat 2.1 software. A p<0.05 was considered as statistically significant.
RESULTS
miR-296-mediated tumor suppression
We began this study by looking at additional targets of miR-296 potentially implicated in tumor suppression (17). In addition to Scribble (17), transfection of model A549 non-small cell lung cancer (NSCLC) cells with miR-296 inhibited the mRNA (Fig. 1A) and protein (Fig. 1B) expression of Numb-like (Numbl), a polarity protein originally described in neuronal progenitors (25, 26). In contrast, transfection of tumor cells with anti-miR-296 had no effect on Scribble or Numbl levels, compared to control cultures (Fig. 1A, B). Two putative miR-296-responsive sites were predicted in the Numbl 3’UTR (Fig. 1C), and their combined mutagenesis reversed miR-296 repression of Numbl regulatory sequences (Fig. 1D), indicating that Numbl is a direct gene target of miR-296. Functionally, expression of miR-296 inhibited lung cancer cell migration in a wound closure assay (Fig 1E), blocked tumor cell invasion across Matrigel-coated inserts (Fig. 1F), and suppressed colony formation in soft agar (Fig. 1G). In reciprocal experiments, transfection of anti-miR-296 increased tumor cell migration, invasion, and colony formation (Fig. 1E–G). Consistent with a tumor-suppressive function, miR-296 levels were downregulated in lung cancer cells, compared to normal human bronchial epithelial HBEC3 cells (Fig. 1H), and were progressively lost during stepwise tumorigenesis in transgenic mice that express the mutant k-Ras-G12D oncogene in the lung (27) (Fig. 1I). In addition, miR-296 expression was significantly reduced in NSCLC patients, compared to normal lung (Fig. 1J).
Figure 1.

miR-296 regulation of Numbl. A, B, A549 cells transfected with miR-296 antagonist (anti-miR-296) or precursor (pre-miR-296) were analyzed by quantitative PCR (A, RQ) or Western blotting (B). C, Sequence of Numbl 3’UTR, and positions of two putative miR-296 sites that were subject to mutagenesis (Mut-1 and Mut-2). D, HEK293 cells expressing wild-type (WT), single (Mut-1) or double (Mut-2) Numbl 3’UTR mutant reporter construct were transfected with miR-296 precursor and analyzed for luciferase activity. Mean±SEM (n=3). * p<0.05. E, A549 cells were transfected with control or miR-296 mimics, and analyzed in a wound healing assay after 24 h. Bar graph, quantification of cell migration. Mean±SEM (n=3). ***, p<0.001. U, arbitrary units. F, G. A549 cells were transfected as in (A) and analyzed for cell invasion across Matrigel-coated inserts after 24 h (F), or colony formation in soft agar (G). Mean±SEM (n=3). ** p<0.01; *** p<0.001. H, Quantification of miR-296 expression in normal human bronchial epithelial cells (HBEC3), or lung cancer cell lines (A549, H460), by quantitative PCR. I, Expression of miR-296 in lung tissues from k-Ras-G12D transgenic mice at the indicated disease stages, by quantitative PCR. **, p<0.001; *, p<0.01. J, Comparative expression of miR-296 in a series of 67 patients with diagnosis of non-small cell lung cancer (NSCLC) and 64 cases of non-neoplastic lung parenchyma (Lung), by quantitative PCR. *, p<0.05.
Numbl regulation of tumor cell motility
To determine whether Numbl functioned in the miR-296 tumor suppressor network, we next silenced its expression by small-interfering RNA (siRNA). Transfection of lung cancer cells with Numbl-directed siRNA suppressed Numbl mRNA (Fig. 2A) and protein (Fig. 2B) levels, compared to non-targeting siRNA. Numbl knockdown reproduced the effect of miR-296 expression, and suppressed the migration (Fig. 2C), and invasion (Fig. 2D) of lung cancer cells (Supplementary Fig. S1A), compared to control transfectants. Similarly, siRNA knockdown of Numbl in breast adenocarcinoma MDA-MB231 cells (Supplementary Fig. S1B) inhibited tumor cell migration (Fig. 2E) and invasion (Supplementary Fig. S1C). In contrast, silencing of Numbl in prostate adenocarcinoma PC3 cells had no effect (Supplementary Fig. S1D), suggesting a tumor-specific response. Consistent with a potential loss of polarity function, silencing of Numbl in lung cancer cells resulted in extensive cytoskeletal changes, with reduced content of oriented actin fibers, compared to control transfectants (Fig. 2F). In addition, loss of Numbl impaired directional cell migration, characterized by defective Golgi orientation relative to nuclei and the migration front of tumor cells (Fig. 2G).
Figure 2.
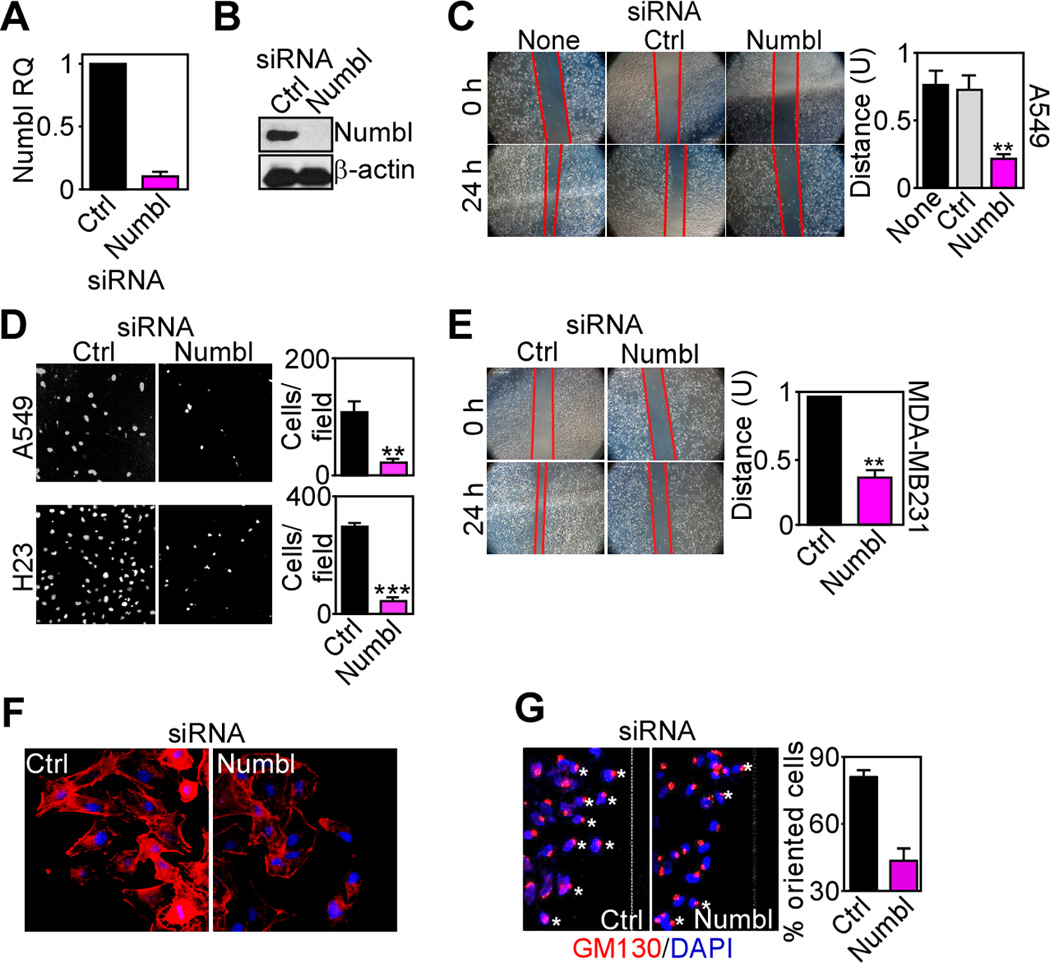
Control of tumor cell motility by Numbl. A, B. A549 cells were transfected with control non-targeting (Ctrl) or Numbl-directed siRNA and analyzed by quantitative PCR (A) or Western blotting (B). C, Monolayers of A549 cells transfected with control (Ctrl) or Numbl siRNA were analyzed for wound closure after 24 h. ** p<0.01. Bar graph, quantification of migration distance. U, units. None, non-transfected cells. D, The indicated siRNA-transfected lung cancer cell types were analyzed for invasion across Matrigel inserts after 24 h. Images correspond to DAPI-stained nuclei of invaded cells. Bar graph, quantification of cell invasion. E, Breast adenocarcinoma MDA-MB231 cells were transfected with control non-targeting (Ctrl) or Numbl-directed siRNA and analyzed for cell migration in a wound closure assay at the indicated time intervals. Bar graph, quantification of migration distance. U, units. F, siRNA-transfected A549 cells were stained for rhodamine-phalloidin (F-actin) and analyzed by fluorescence microscopy. Nuclei were stained with DAPI. G, siRNA transfected A549 cells were labeled for the Golgi marker GM130, and analyzed by fluorescence microscopy. DNA was stained with DAPI. Dotted line, migration edge (wound). Bar graph, quantification of cells with maloriented Golgi.
The biochemical requirements of Numbl regulation of tumor cell motility were next investigated. First, Numbl silencing in lung cancer cell types was associated with decreased expression of fibronectin, β-catenin and vimentin (Fig. 3A) thus differently from conventional hallmarks of epithelial-mesenchymal transition (10). Conversely, loss of Numbl induced dephosphorylation, i.e. inactivation, of focal adhesion kinase (FAK) on Tyr397 (Fig. 3B), and of Src on Tyr416 (Fig. 3C), two pivotal cell motility effectors.
Figure 3.
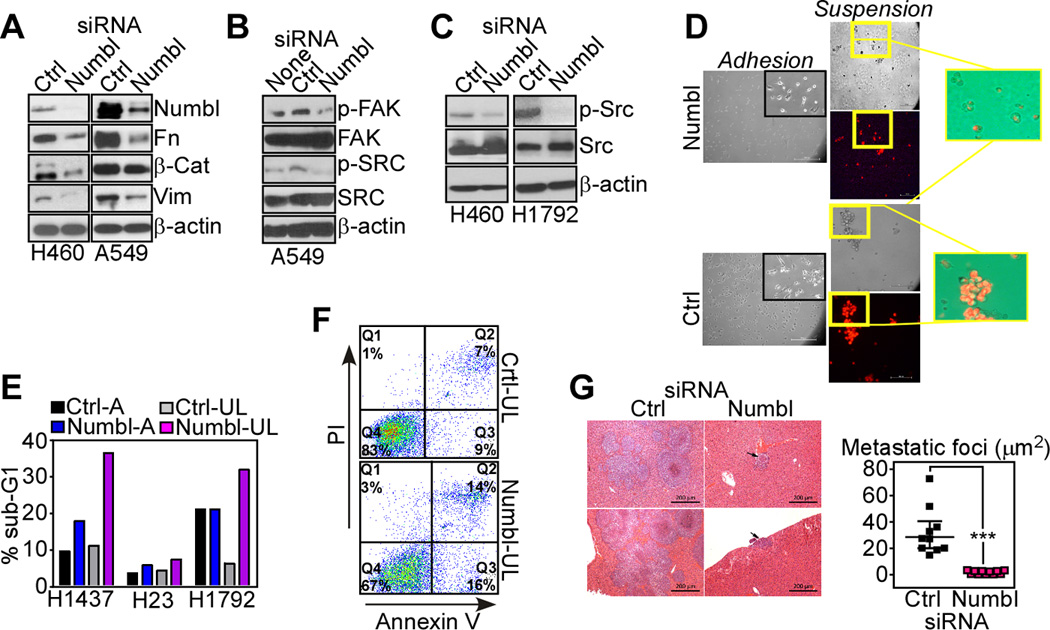
Numbl regulation of tumor cell invasion and metastasis. A–C. The indicated lung cancer cell lines were transfected with control non-targeting siRNA (Ctrl) or Numbl-directed siRNA and analyzed by Western blotting. p-, phosphorylated. None, untransfected cells. D, PKH26-labeled A549 cells (red), were transfected with the indicated siRNA and analyzed after 24 h for anoikis using ultra-low attachment conditions (suspension) Insets, image merge of bright field and fluorescence photomicrographs. E, The indicated siRNA-transfected lung cancer cell types were maintained attached (A) or plated under ultra-low attachment conditions (UL) and analyzed for DNA content and flow cytometry. The percentage of cells with hypodiploid, sub-G1 DNA content is indicated. Representative experiment out of two independent determinations. F, siRNA transfected A549 cells maintained in ultra-low (UL) attachment conditions were stained with fluorescein-Annexin V plus propidium iodide (PI), and analyzed by multiparametric flow cytometry. The percentage of cells in each quadrant is indicated. G, Representative images of hematoxylin-eosin-stained liver sections from animals injected intrasplenically with H460 cells transfected with control non-targeting or Numbl-directed siRNA. Scale bar, 200 µm. The surface area of metastatic foci was quantified. *** p<0.001. Mean±SEM. Each symbol corresponds to an individual animal.
Role of Numbl in lung cancer metastasis
Next, we asked whether Numbl-directed random cell motility influenced the metastatic propensity of tumor cells, and we first looked at potential changes in anoikis. This is a form of cell death caused by detachment of epithelial cells from the extracellular matrix, and considered a key determinant of metastasis (28). Numbl knockdown in lung cancer cells maintained in suspension was associated with nearly completely loss of cell viability, whereas adherent cells were not significantly affected, by PKH26 vital staining and fluorescence microscopy (Fig. 3D). Consistent with these data, siRNA silencing of Numbl in attached lung cancer cell types had negligible effects on cell viability, by flow cytometry quantification of hypodiploid, i.e. sub-G1 DNA content (Fig. 3E). In contrast, knockdown of Numbl under conditions of ultra-low cell attachment resulted in considerably increased cell death, compared to control transfectants (Fig. 3E). These cells exhibited increased reactivity for Annexin V, consistent with apoptosis, by multiparametric flow cytometry (Fig. 3F). To test whether this pathway was important, in vivo, we next injected lung cancer cells transfected with control or Numbl-directed siRNA in the spleen of immunocompromised mice and looked at their ability to form liver metastasis within an 11-d interval. In these experiments, control transfectants formed large metastatic foci in the liver of all reconstituted animals (Fig. 3G). In contrast, siRNA silencing of Numbl nearly completely abolished the ability of lung cancer cells to form liver metastasis in this model (Fig. 3G).
Numbl modulation of Klf4-dependent transcription
To elucidate the mechanism(s) of Numbl regulation of tumor cell motility and invasion, we next used an inducible luciferase reporter array of 45 transcription factors (Fig. 4A). siRNA silencing of Numbl under these conditions resulted in increased activity of seven transcription factors (Fig 4B) implicated in pluripotency and differentiation (Klf4, VDR or c-Myc) (29), or stress-sensing (Nrf1-2, AP-1, ATF6 or YY1) responses (30). In particular, de-repression of Kruppel-like factor-4 (Klf4)-dependent transcription was the most significant change observed in response to Numbl knockdown (Fig. 4B). To validate these changes, we next looked at potential modulation of Klf4 target gene expression in response to Numbl targeting. In these experiments, transfection of lung cancer cells with Numbl-directed siRNA, but not control non-targeting siRNA, increased the expression of p21Cip1/WAF1 mRNA (Fig. 4C) and protein (Fig. 4D), and downregulated Sparc levels (31) (Fig. 4C), two known Klf4 target genes. Overall Klf4 protein levels did not change and a control siRNA had no effect (Fig. 4C, D). To further test the specificity of Numbl regulation of Klf4-dependent transcription, we next looked at the levels of several other “stemness” molecules under conditions of Numbl-Klf4 targeting. In these experiments, siRNA silencing of Klf4, Numbl or the potentially related molecule, Numb (see below), did not significantly affect the mRNA levels of receptor-ligand pairs in the Notch (Notch-Jagged 1) or Sonic Hedgehog (SHH-Gli1) pathway (Fig. 4E). Overall levels of c-Myc were also not significantly different between the variously silenced cells and control cultures (Fig. 4E). We next studied a potential reciprocal relationship between Numbl and Klf4 in normal and lung cancer cells. Endogenous Numbl was present more abundantly in nuclei of tumor cells, with a smaller pool of the molecule expressed in their cytosol (Fig. 4F). By contrast, endogenous Klf4 was almost undetectable in lung cancer cells, and became expressed solely in nuclei, but not cytosol, upon plasmid transfection (Fig. 4F). In gene silencing studies, knockdown of Numbl or Klf4 did not affect the expression or subcellular localization of the other molecule (Fig. 4G). In contrast, expression of Klf4 in normal or tumor cell types was associated with Numbl co-localization in nuclei of transfected cells, by confocal microscopy (Fig. 4H).
Figure 4.

Klf4-Numbl regulation in lung cancer. A, Heat map representing changes in activity of 45 transcription factors in A549 cells transfected with control (Ctrl) or Numbl-directed siRNA. B, Quantification of transcription factor (TF) activity expressed as ratio between Numbl-silenced A549 cells and control transfectants. ** p<0.01; * p<0.05. C, Transfected A549 cells were analyzed by quantitative PCR. D, The indicated transfected lung cancer cell types were analyzed by Western blotting. E, A549 cells were transfected with control non-targeting siRNA (Ctrl) or siRNA directed to Numbl, Numb or Klf4 and analyzed for changes in expression of the indicated gene products by quantitative PCR. Mean±SEM (n=3). F, G. Nuclear (Nuclei) or cytosolic (Cyto) fractions isolated from H460 (F) or A549 (G) cells transfected with Klf4 (HA-Klf4), or siRNA (si) to Klf4 or Numbl were analyzed by Western blotting. Laminin A (Lam A) and β-tubulin were used as markers for nuclear or cytosolic fractions, respectively. Ctrl, non-targeting siRNA. H, A549 (top) or HBEC3 (bottom) cells were transfected with Klf4 cDNA (HA-Klf4) and probed with an antibody to HA or Numbl. Cells were imaged using differential interference contrast microscopy (DIC) before merging DIC with fluorescent images. Nuclei were stained with DAPI. Scale bar, 15 µm.
Independent regulation and distinct functions of Numb and Numbl in lung cancer
Numbl has been proposed as a protein related to Numb, a regulator of cell fate specification and Notch1 signaling (32), and this potential relationship in lung cancer was next investigated. siRNA silencing of Numbl did not affect the expression of Numb in lung cancer cells (Fig. 5A). At variance with the results obtained with Numbl, silencing of Numb mRNA (Fig. 5B) or protein expression (Fig. 5C) increased lung cancer cell invasion across Matrigel-coated inserts (Fig. 5D). The response to anoikis was also different between the two molecules, as siRNA knockdown of Numb did not affect the viability of lung cancer cells under attachment conditions or forced to remain in suspension, by DNA content analysis and flow cytometry (Fig. 5E). With respect to potential modulation of Klf4 target genes, knockdown of Numb did not affect p21Cip1/WAF1 levels, whereas Sparc expression was actually increased (Fig. 5F).
Figure 5.

Functional characterization of Numb. A, The indicated lung cancer cells were transfected with control (Ctrl) or Numbl-directed siRNA and analyzed by Western blotting. B, C. The indicated lung cell types were transfected with control or Numb-directed siRNA and analyzed by quantitative PCR (B), or Western blotting (C). D, Transfected lung cell types as in (B and C) were analyzed for invasion across Matrigel-coated inserts after 24 h. Mean±SEM (n=3). **, p<0.001. E, A549 cells transfected with control or Numb-directed siRNA were grown in adhesion (A) or suspension (ultra-low, UL) conditions and analyzed after 24 h for DNA content by propidium iodide staining and flow cytometry. The percentage of cells with sub-G1 DNA content is shown. Bar graph, quantification of cells with sub-G1 DNA content. FC, fold change. F, A549 cells were transfected with control (Ctrl) or Numb-directed siRNA and analyzed by quantitative PCR.
Numbl-Klf4 signaling controls the pool of lung cancer-initiating, progenitor cells
Klf4 orchestrates a pivotal “stemness” transcriptional program (33), which may participate in context-specific tumor suppression (31), including in lung cancer (34). Consistent with a potential tumor suppressive function, Klf4 was nearly undetectable in lung cancer cells, compared to normal HBEC3 (Fig. 6A), and its re-introduction in tumor cells by plasmid transfection (Supplementary Fig. S2A) was sufficient to inhibit cell invasion across Matrigel inserts (Fig. 6B). siRNA silencing of Klf4 (Supplementary Fig. S2B, C) had no effect in tumor cells (Fig. 6B). In contrast, Klf4 knockdown enhanced the invasive potential of normal HBEC3 cells (Supplementary Fig. S2C and Fig. 6C). In parallel experiments, transfection of Numbl (Supplementary Fig. S2A, C) had no effect on tumor cell motility (Fig. 6B), and modestly increased HBEC3 invasion (Fig. 6C).
Figure 6.

Numbl-Klf4 modulation of a lung cancer-initiating progenitor phenotype. A, The indicated normal (HBEC3) or lung cancer cell lines were examined by Western blotting. B, C. A549 (B) or HBEC3 (C) cells were transfected with the indicated siRNA or plasmid and analyzed in a Matrigel invasion assay after 24 h. Mean±SEM (n=3). *** p<0.001. D, A549 cells transfected with control (Ctrl) or Klf4-directed siRNA, or Klf4 or Numbl cDNA were stained with Hoechst 33342 without (None, top) or with (bottom) 50 µM reserpine, and analyzed by multiparametric flow cytometry. The side population (SP) compartment was identified as the cell fraction abolished by reserpine. Numbers quantify the SP as percentage of the viable population. Bar graph, quantification of SP. * p<0.05. E, F. SP and non-SP fractions sorted from A549 cells were analyzed by Western blotting (E) or quantitative PCR (F). Left, Numbl; Right, miR-296. G, Aliquots of sorted side population (SP) or non-side population (non-SP) A549 cells were analyzed for expression of the indicated gene products by quantitative PCR. Mean±SEM (n=3). H, siRNA transfected A549 cells were analyzed for SP as in (D). Bar graph, quantification of SP. ** p<0.01. I, The sorted SP from A549 transfectants was analyzed in a Matrigel invasion assay after 24 h. Images correspond to DAPI-stained nuclei of invaded cells. Bar graphs, quantification of cell invasion. Mean±SEM (n=3). **, p<0.001.
To determine whether Numbl-Klf4 signaling affects a tumor-initiating, progenitor phenotype, we next examined the “side population” of lung cancer types, which is enriched in stem-like cells (20). Transfection of Klf4 significantly reduced the side population of A549 cells (Fig. 6D). In contrast, transfection of these cells with a Numbl cDNA had the opposite effect, and expanded the side population compartment of A549 cells by approximately two-fold (Fig. 6D). Consistent with the results above, siRNA silencing of Klf4 had no effect on the side population of A549 cells, compared to control transfectants (Fig. 2H). Next, we looked at the individual cell populations isolated by fluorescence sorting. Numbl was preferentially expressed in the side population of A549 cells, by Western blotting (Fig. 6E) and quantitative RT-PCR (Fig. 6F). Mirroring these results, its upstream repressor, miR-296, was nearly exclusively present in the non-side population fraction of A549 cells (Fig. 6F). With respect to other developmentally regulated “stemness” markers, only c-Myc was preferentially enriched in the side population of A549 cells (Fig. 6G). In contrast, expression of Notch, SHH or Klf4 was not statistically different in side population or non-side population fractions of A549 cells (Fig. 6G). Functionally, siRNA knockdown of Numbl depleted the side population of A549 cells by up to 87%, compared to control transfectants (Fig. 6H). This was associated with nearly complete inhibition of tumor cell invasion across Matrigel inserts (Fig. 6I). Consistent with the data reported above, silencing of Numb did not modulate the side population of A549 cells (Supplementary Fig. S3).
Role of Numbl-Klf4 signaling in lung cancer progression
Based on the data above, we next looked at the potential impact of a Numbl-Klf4 signaling axis in human tumors. Consistent with its oncogenic properties, in vitro, the expression of Numbl was significantly increased in many human tumors, by immunohistochemistry (Fig. 7A, B, and Supplementary Fig. S4), and analysis of public databases (Supplementary Fig. S5). When compared to other “stemness” markers, Nanog expression was also found broadly upregulated in various patient-derived human tumors (Supplementary Fig. S6A), whereas the levels of Oct4 were more restricted to testicular and breast cancer (Supplementary Fig. S6B).
Figure 7.
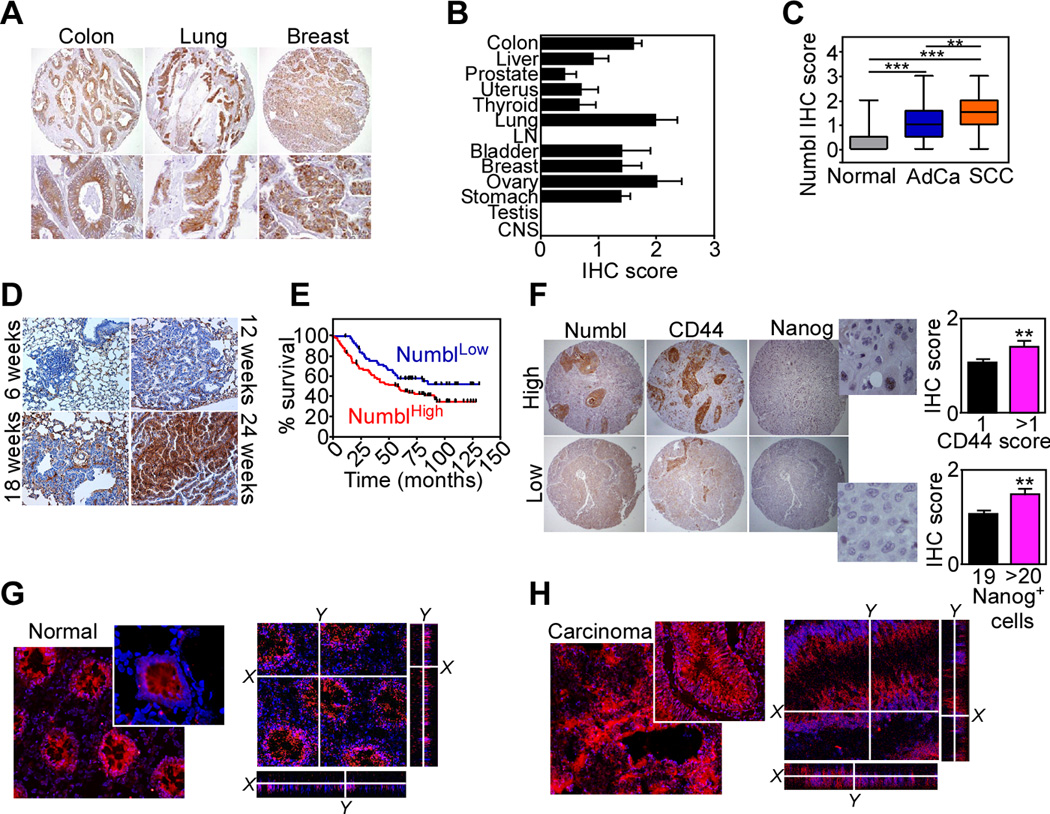
Numbl-Klf4 signaling in lung cancer progression. A, Representative samples of colon, lung and breast human cancers were stained with an antibody to Numbl by immunohistochemistry. B, Summary of Numbl expression in 13 human cancers by immunohistochemistry (IHC). C, Numbl expression in NSCLC (AdCa, n=149; SCC, n=60), or non-neoplastic lung (n=27), by IHC. ** p<0.01; *** p<0.001. D, Tissue sections of normal (6 weeks), hyperplastic (12 weeks), adenomatous (18 weeks) or adenocarcinoma (24 weeks) lung lesions from k-Ras-G12D transgenic mice were analyzed for Numbl expression by IHC. E, Kaplan-Meyer curves of overall survival of NSCLC patients according to Numbl IHC score. p=0.03 by Log-Rank test. F, NSCLC samples were analyzed for co-expression of Numbl and CD44 or Nanog-positive cells. Bar graphs, quantification of IHC for CD44 (top) or Nanog expression (bottom). Magnification ×50 (cores), ×600 (enlargements). G, H. Tissue samples from normal colonic mucosa (G) or colorectal AdCa (H) from the same patient were stained with an antibody to Numbl and analyzed by confocal microscopy. Nuclei were stained with DAPI. Magnification ×400, ×600 (insets).
With respect to NSCLC, the tumor cell population stained intensely positive for Numbl, compared to the adjacent non-neoplastic parenchyma (Supplementary Fig. S7A), with higher levels in squamous cell carcinoma (SCC), than adenocarcinoma (AdCa, Fig. 7C). In both human samples (Supplementary Fig. S7B), and mouse k-Ras mutant lung tumors (27) (Fig. 7D), Numbl levels increased with disease progression, from localized lesions to metastatic foci. In terms of disease outcome, Numbl was associated with shorter overall survival in NSCLC patients (n=209; HR= 1.7, 95% CI= 1.037–2.439, p=0.03; Fig. 7E), and its expression in these cases correlated with the presence of other stem cell markers, including Nanog and CD44 (35) (Fig. 7F). In addition to deregulated expression, Numbl was subcellularly mislocalized in human tumors, from a membranous staining in the normal colonic mucosa (Fig. 7G) to a diffuse cytoplasmic and perinuclear distribution in colorectal adenocarcinoma (Fig. 7H), and matched liver metastasis (Supplementary Fig. S7C).
DISCUSSION
In this study, we have shown that a developmentally-regulated polarity protein originally described in neuronal progenitors, Numbl (25, 26), becomes subcellularly deregulated and over-expressed in various human cancers. This results from loss of a tumor suppressive miR, miR-296 (17), which actively represses Numbl expression. In turn, aberrantly increased levels of Numbl support multiple metastatic traits in lung cancer, including enhanced cell invasion, resistance to anoikis, maintenance of cancer-initiating, progenitor-like cells, and metastatic competency, in vivo. Mechanistically, this pathway involves Numbl inhibition of the Klf4 “stemness” transcriptional program (33, 36), resulting in shortened overall survival in lung cancer patients.
Polarity proteins are known for maintaining the integrity of epithelia and directional cell migration (18). Perturbation of their function in cancer, especially with respect to gap junction assembly, cell orientation, and cell invasion has also been postulated (37), suggesting that these molecules may participate in an evolutionary conserved pathway of tumor suppression (38). However, recent data point to a more complex scenario, as polarity proteins become broadly over-expressed and subcellularly mislocalized in various malignancies (39), and exploited for random, as opposed to directional cell migration, tumor cell invasion and disease maintenance (17). The results presented here further support the model of a general exploitation of polarity proteins for tumor progression, and identify Numbl as a novel effector of aberrant lung cancer cell motility, invasion and metastasis, in vivo. Although Numb (32) and Numbl have been proposed as related gene products with potentially overlapping functions in cell fate specifications (25), our results show that these molecules have instead entirely distinct functions in cancer, with no role for Numb in tumor cell motility.
One of the emerging roles of polarity proteins is in the regulation of stem cell-related functions (40), including cancer stem cells (41). The role of these rare, potentially cancer-initiating cells in disease maintenance is far from elucidated (42), but there is evidence from clinical correlates (9), and genetic disease models in mice (43), that they may play a role in the pathogenesis and progression of lung cancer. Here, Numbl expression emerged as a novel requirement to maintain, and potentially expand the side population of lung cancer cell types, a subset enriched in the stem cell phenotype (20) that carries elevated metastatic potential (44). A key mechanistic requirement of this pathway was the ability of Numbl to antagonize Klf4-dependent transcription, a pivotal “stemness” program (29, 33), implicated in “context-dependent” tumor suppression (31), including in lung cancer (34).
Although the molecular requirements of how Numbl inhibits Klf4-dependent gene expression need to be further elucidated, the two molecules were shown to co-localize in nuclei of tumor cells, and expression of Klf4, alone, was sufficient to shut off tumor cell motility and eliminate the progenitor-like population of lung cancer cells. Altogether, these data suggest a dual role of deregulated Numbl in lung cancer, promoting aberrant cell migration and invasion when localized in the cytosol, and antagonizing Klf4-dependent suppression of the cancer stem cell phenotype in the nucleus. Consistent with this model, Klf4 was virtually undetectable in lung cancer cell types, whereas it potently antagonized migration of normal bronchial epithelial cells. The ability of Numbl to prevent anoikis (28), and maintain phosphorylated levels of known survival kinases, including FAK and Src, may also elevate the anti-apoptotic threshold in the cancer-initiating stem-like compartment, thus further enhancing their metastatic propensity. Irrespectively, Numbl-directed tumorigenesis emerged here as a major determinant of unfavorable outcome in lung cancer patients, correlating with expression of other stem cell markers, Nanog and CD44 (8), but not Oct4, and overall supporting an important pathogenetic role of the stem/progenitor cell compartment in lung cancer progression (9).
In sum, this study identified novel molecular determinants of lung cancer metastasis, uncovering a broad oncogenic role for the Numbl polarity protein in tumor cell invasion coupled to the maintenance of progenitor-like, cancer-initiating cells (41). Despite gaps in our understanding of cancer stem cells (42), and their interplay with pathways of cell motility and drug resistance (10), targeting the Numbl pathway described here may open new therapeutic prospects to limit the metastatic potential of advanced lung cancer.
Supplementary Material
ACKNOWLEDGMENTS
We thank Frederick Keeney (Wistar) for help with imaging studies and Jeffrey Faust (Wistar) for help with flow cytometry-sorting analyses.
GRANT SUPPORT
This work was supported by NIH grants CA140043, HL54131, CA78810 and CA118005 (DCA), and by grants from Fondazione Cariplo (2010-0846), Fondazione Berlucchi and Ministero della Salute “5×1000” (SB). AF was supported by a fellowship of the Doctorate School of Molecular Medicine at Università degli Studi di Milano. Support for Core Facilities utilized in this study was provided by Cancer Center Support Grant (CCSG) CA010815 to The Wistar Institute.
Footnotes
The authors declare that no conflict of interest exists.
AUTHORS’ CONTRIBUTIONS
Conception and design: V. Vaira, S. Bosari. D. C. Altieri
Development of methodology: V. Vaira, N. M. Martin, A. Faversani.
Acquisition of data (provided animals, acquired and managed patients, provided facilities): D.S. Garlick, S. Ferrero, M. Nosotti, J.L. Kissil
Analysis and interpretation of data: V. Vaira, S. Bosari, D. C. Altieri
Writing, review and/or revision of manuscript: V. Vaira, S. Bosari, D. C. Altieri
Administrative, technical and material support: V. Vaira, S. Bosari, D. C. Altieri
Study supervision: S. Bosari, D. C. Altieri
Supplementary data for this article are available at Cancer Research Online.
REFERENCES
- 1.Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–292. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kasinski AL, Slack FJ. Epigenetics and genetics. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat Rev Cancer. 2011;11:849–864. doi: 10.1038/nrc3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kong YW, Ferland-McCollough D, Jackson TJ, Bushell M. microRNAs in cancer management. Lancet Oncol. 2012;13:e249–e258. doi: 10.1016/S1470-2045(12)70073-6. [DOI] [PubMed] [Google Scholar]
- 5.Wang L, Wang J. MicroRNA-mediated breast cancer metastasis: from primary site to distant organs. Oncogene. 2012;31:2499–2511. doi: 10.1038/onc.2011.444. [DOI] [PubMed] [Google Scholar]
- 6.Liu C, Tang DG. MicroRNA regulation of cancer stem cells. Cancer Res. 2011;71:5950–5954. doi: 10.1158/0008-5472.CAN-11-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saini S, Majid S, Shahryari V, Arora S, Yamamura S, Chang I, et al. miRNA-708 control of CD44(+) prostate cancer-initiating cells. Cancer Res. 2012;72:3618–3630. doi: 10.1158/0008-5472.CAN-12-0540. [DOI] [PubMed] [Google Scholar]
- 8.Zoller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011;11:254–267. doi: 10.1038/nrc3023. [DOI] [PubMed] [Google Scholar]
- 9.Peacock CD, Watkins DN. Cancer Stem Cells and the Ontogeny of Lung Cancer. J Clin Oncol. 2008;26:2883–2889. doi: 10.1200/JCO.2007.15.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corbetta S, Vaira V, Guarnieri V, Scillitani A, Eller-Vainicher C, Ferrero S, et al. Differential expression of microRNAs in human parathyroid carcinomas compared with normal parathyroid tissue. Endocrine-related cancer. 2010;17:135–146. doi: 10.1677/ERC-09-0134. [DOI] [PubMed] [Google Scholar]
- 13.Hong L, Han Y, Zhang H, Li M, Gong T, Sun L, et al. The prognostic and chemotherapeutic value of miR-296 in esophageal squamous cell carcinoma. Annals of surgery. 2010;251:1056–1063. doi: 10.1097/SLA.0b013e3181dd4ea9. [DOI] [PubMed] [Google Scholar]
- 14.Yu J, Li A, Hong SM, Hruban RH, Goggins M. MicroRNA alterations of pancreatic intraepithelial neoplasias. Clin Cancer Res. 2012;18:981–992. doi: 10.1158/1078-0432.CCR-11-2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wurdinger T, Tannous BA, Saydam O, Skog J, Grau S, Soutschek J, et al. miR-296 regulates growth factor receptor overexpression in angiogenic endothelial cells. Cancer Cell. 2008;14:382–393. doi: 10.1016/j.ccr.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei JJ, Wu X, Peng Y, Shi G, Basturk O, Yang X, et al. Regulation of HMGA1 expression by microRNA-296 affects prostate cancer growth and invasion. Clin Cancer Res. 2011;17:1297–1305. doi: 10.1158/1078-0432.CCR-10-0993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaira V, Faversani A, Dohi T, Montorsi M, Augello C, Gatti S, et al. miR-296 regulation of a cell polarity-cell plasticity module controls tumor progression. Oncogene. 2012;31:27–38. doi: 10.1038/onc.2011.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Etienne-Manneville S. Polarity proteins in migration and invasion. Oncogene. 2008;27:6970–6980. doi: 10.1038/onc.2008.347. [DOI] [PubMed] [Google Scholar]
- 19.Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. 1996;183:1797–1806. doi: 10.1084/jem.183.4.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho MM, Ng AV, Lam S, Hung JY. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007;67:4827–4833. doi: 10.1158/0008-5472.CAN-06-3557. [DOI] [PubMed] [Google Scholar]
- 21.Osmani N, Vitale N, Borg JP, Etienne-Manneville S. Scrib controls Cdc42 localization and activity to promote cell polarization during astrocyte migration. Curr Biol. 2006;16:2395–2405. doi: 10.1016/j.cub.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 22.Vaira V, Faversani A, Dohi T, Maggioni M, Nosotti M, Tosi D, et al. Aberrant overexpression of the cell polarity module scribble in human cancer. Am J Pathol. 2011;178:2478–2483. doi: 10.1016/j.ajpath.2011.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mehrotra S, Languino LR, Raskett CM, Mercurio AM, Dohi T, Altieri DC. IAP regulation of metastasis. Cancer Cell. 2010;17:53–64. doi: 10.1016/j.ccr.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nosotti M, Falleni M, Palleschi A, Pellegrini C, Alessi F, Bosari S, et al. Quantitative real-time polymerase chain reaction detection of lymph node lung cancer micrometastasis using carcinoembryonic antigen marker. Chest. 2005;128:1539–1544. doi: 10.1378/chest.128.3.1539. [DOI] [PubMed] [Google Scholar]
- 25.Rasin MR, Gazula VR, Breunig JJ, Kwan KY, Johnson MB, Liu-Chen S, et al. Numb and Numbl are required for maintenance of cadherin-based adhesion and polarity of neural progenitors. Nat Neurosci. 2007;10:819–827. doi: 10.1038/nn1924. [DOI] [PubMed] [Google Scholar]
- 26.Petersen PH, Zou K, Hwang JK, Jan YN, Zhong W. Progenitor cell maintenance requires numb and numblike during mouse neurogenesis. Nature. 2002;419:929–934. doi: 10.1038/nature01124. [DOI] [PubMed] [Google Scholar]
- 27.Kissil JL, Walmsley MJ, Hanlon L, Haigis KM, Bender Kim CF, Sweet-Cordero A, et al. Requirement for Rac1 in a K-ras induced lung cancer in the mouse. Cancer Res. 2007;67:8089–8094. doi: 10.1158/0008-5472.CAN-07-2300. [DOI] [PubMed] [Google Scholar]
- 28.Kenific CM, Thorburn A, Debnath J. Autophagy and metastasis: another double-edged sword. Curr Opin Cell Biol. 2010;22:241–245. doi: 10.1016/j.ceb.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 30.Li M, Baumeister P, Roy B, Phan T, Foti D, Luo S, et al. ATF6 as a transcription activator of the endoplasmic reticulum stress element: thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol Cell Biol. 2000;20:5096–5106. doi: 10.1128/mcb.20.14.5096-5106.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rowland BD, Peeper DS. KLF4, p21 and context-dependent opposing forces in cancer. Nat Rev Cancer. 2006;6:11–23. doi: 10.1038/nrc1780. [DOI] [PubMed] [Google Scholar]
- 32.Verdi JM, Schmandt R, Bashirullah A, Jacob S, Salvino R, Craig CG, et al. Mammalian NUMB is an evolutionarily conserved signaling adapter protein that specifies cell fate. Curr Biol. 1996;6:1134–1145. doi: 10.1016/s0960-9822(02)70680-5. [DOI] [PubMed] [Google Scholar]
- 33.Kim JB, Zaehres H, Wu G, Gentile L, Ko K, Sebastiano V, et al. Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature. 2008;454:646–650. doi: 10.1038/nature07061. [DOI] [PubMed] [Google Scholar]
- 34.Hu W, Hofstetter WL, Li H, Zhou Y, He Y, Pataer A, et al. Putative tumor-suppressive function of Kruppel-like factor 4 in primary lung carcinoma. Clin Cancer Res. 2009;15:5688–5695. doi: 10.1158/1078-0432.CCR-09-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leung EL, Fiscus RR, Tung JW, Tin VP, Cheng LC, Sihoe AD, et al. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS One. 2010;5:e14062. doi: 10.1371/journal.pone.0014062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghaleb AM, McConnell BB, Kaestner KH, Yang VW. Altered intestinal epithelial homeostasis in mice with intestine-specific deletion of the Kruppel-like factor 4 gene. Dev Biol. 2011;349:310–320. doi: 10.1016/j.ydbio.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Humbert PO, Grzeschik NA, Brumby AM, Galea R, Elsum I, Richardson HE. Control of tumourigenesis by the Scribble/Dlg/Lgl polarity module. Oncogene. 2008;27:6888–6907. doi: 10.1038/onc.2008.341. [DOI] [PubMed] [Google Scholar]
- 38.Wu M, Pastor-Pareja JC, Xu T. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature. 2010;463:545–548. doi: 10.1038/nature08702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ellenbroek SI, Iden S, Collard JG. Cell polarity proteins and cancer. Seminars in cancer biology. 2012;22:208–215. doi: 10.1016/j.semcancer.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 40.Martin-Belmonte F, Perez-Moreno M. Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer. 2012;12:23–38. doi: 10.1038/nrc3169. [DOI] [PubMed] [Google Scholar]
- 41.Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. 2011;147:759–772. doi: 10.1016/j.cell.2011.09.048. [DOI] [PubMed] [Google Scholar]
- 42.Vermeulen L, de Sousa e Melo F, Richel DJ, Medema JP. The developing cancer stem-cell model: clinical challenges and opportunities. Lancet Oncol. 2012;13:e83–e89. doi: 10.1016/S1470-2045(11)70257-1. [DOI] [PubMed] [Google Scholar]
- 43.Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 44.Akunuru S, Palumbo J, Zhai QJ, Zheng Y. Rac1 targeting suppresses human non-small cell lung adenocarcinoma cancer stem cell activity. PLoS One. 2011;6:e16951. doi: 10.1371/journal.pone.0016951. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.