

Abstract
Disorders of iron metabolism account for some of the most common human diseases. Cellular iron homeostasis is maintained by iron regulatory proteins (IRP)-1 and 2 through their binding to cis-regulatory iron-responsive elements (IREs) in target mRNAs. Mouse models with IRP deficiency have yielded valuable insights into iron biology, but the physiological consequences of gain of IRP function in mammalian organisms have remained unexplored. Here, we report the generation of a mouse line allowing conditional expression of a constitutively active IRP1 mutant (IRP1*) using Cre/Lox technology. Systemic activation of the IRP1* transgene from the Rosa26 locus yields viable animals with gain of IRE-binding activity in all the organs analyzed. IRP1* activation alters the expression of IRP target genes and is accompanied by iron loading in the same organs. Furthermore, mice display macrocytic erythropenia with decreased hematocrit and hemoglobin levels as well as impaired erythroid differentiation. Thus, inappropriately high IRP1 activity causes disturbed body iron distribution and erythropoiesis. This new mouse model further highlights the importance of appropriate IRP regulation in central organs of iron metabolism. Moreover, it opens novel avenues to study diseases associated with abnormally high IRP1 activity, such as Parkinson’s disease or Friedreich’s ataxia.
Electronic supplementary material
The online version of this article (doi:10.1007/s00109-013-1008-2) contains supplementary material, which is available to authorized users.
Keywords: Iron metabolism, IRP/IRE, Erythropoiesis, Conditional mouse mutant
Introduction
Dysregulation of iron metabolism is causative of some of the most common human diseases worldwide. While iron deficiency mostly engenders anemia, systemic iron excess, e.g., in hemochromatotic patients, can result in cirrhosis, cardiomyopathy, and diabetes. Localized iron accumulation has also been associated with and implicated in the pathogenesis of common neurodegenerative diseases, such as Alzheimer’s and Parkinson’s diseases [1].
To avoid the detrimental consequences of both iron deficiency and excess, homeostatic mechanisms have evolved at the systemic and cellular level. At the cellular level, iron homeostasis is secured mostly by the coordinate action of the RNA-binding proteins IRP-1 and 2 on mRNAs containing IREs within their 5′- or 3′ untranslated regions (UTRs) [2, 3]. Typical targets of IRP-mediated post-transcriptional regulation are proteins involved in iron uptake (e.g., TFRC or transferrin receptor 1 (TfR1)), utilization (e.g., erythroid 5′ aminolevulinic acid synthase 2 (ALAS2)), storage (e.g., FTL1 and FTH1 or ferritin light and heavy chains (FTL and FTH)), and export (SLC40A1 or ferroportin (FPN)). In turn, the cellular iron pool regulates IRE-binding of the two IRPs through distinct mechanisms: iron–sulfur (Fe-S) cluster assembly on IRP1, causing its conversion to a cytosolic aconitase, and iron-targeted degradation of IRP2 and apo-IRP1 [2].
In humans, deregulation of the IRP/IRE regulatory system has been shown to be associated with various pathological settings. Mutations in the IRE sequences of FTL have been detected in patients suffering from the autosomal dominant hyperferritinemia cataract syndrome [3]. In addition, a mutation of FTH-IRE has been implicated in autosomal dominant iron overload in four members of a Japanese family [3]. These observations indicate that altered IRP-IRE recognition could represent the underlying cause of those pathologies. In addition, polymorphisms in the IRP2 promoter and coding sequence have been associated with increased susceptibility to chronic obstructive pulmonary and Alzheimer’s disease, respectively [3]. Furthermore, abnormally high IRP1 activity has been found in patients affected by diseases such as sideroblastic anemia linked to Glrx5 deficiency [4], Friedreich’s ataxia [5], or Parkinson’s disease [6].
To explore the in vivo functions of the IRP/IRE system, mouse lines with constitutive and tissue-specific loss of IRP function have been analyzed during the last decade [7–10]. The early embryonic lethality of constitutive combined ablation of both IRPs has shown the fundamental importance of this cellular homeostatic machinery [11–13]. Restricted IRP deletions have also unraveled its crucial role for intestinal [12] and liver function [13], respectively.
Despite the link of abnormally high IRP1 activity to human diseases [4–6], the consequences of gain of IRP1 function per se in a mammalian organism have not yet been investigated due to the lack of a suitable model system. Here, we present the generation of a mouse model allowing conditional gain of IRP1 function using Cre/Lox technology. We targeted the Rosa26 locus [14] with a Cre-activatable transgene encoding a FLAG-tagged, stabilized IRP1 mutant (IRP1*) which escapes Fe-S cluster-mediated regulation, being constitutively active in its IRE-binding form. We show that IRP1* is expressed in different organs thereby generating increased IRE-binding activity. The resulting systemic gain of IRP1 function causes altered body iron distribution and altered expression of IRP target genes. Furthermore, we show that appropriate IRP1 expression is critical for normal erythropoiesis, as mice with gain of IRP1 function suffer from macrocytic anemia associated with impaired maturation of red blood cells.
Materials and methods
Gene targeting and mice
The plasmids pBigT and pROSA26PA were obtained from S. Srinivas [15]. The PGK-Neo cassette from pBigT was modified to an IRESbgeo [9]. The KpnI linearization site of pROSA26PA was replaced by a SfiI/FseI/PmeI multi-cloning site. Irp1 cDNA was cloned from mouse Sv129 ES cells. By step PCR, three cysteine residues required for Fe-S cluster assembly, C437, C503, C506, were mutated to serine and a fourth one, C118, to alanine, resulting in stabilization of the apoprotein in the presence of heme (Vasanthakumar and Eisenstein, unpublished findings). The resulting mutant Irp1 cDNA was fused to a DYKDDDDK (FLAG) tag (designated Irp1*) and inserted into pBigTIRESbgeo to produce the pBigTIRESbgeoIRP1* plasmid. PacI-AscI digested pBigTIRESIRP1* was inserted into the modified pROSA26PA plasmid.
E14 ES cells were electroporated with the XhoI-linearized targeting construct and cultivated in the presence of G418 (Invitrogen). Resistant ES cell clones were analyzed using RT-PCR and Southern blotting. Targeted ES cell clones were injected into C57BL6/J embryos to obtain chimeras that were backcrossed to C57BL6/J to check for germline transmission of the knock-in Rosa26 locus. Animals carrying the targeted allele have been crossed with a Hprt-Cre deletor strain [16]. Mice bearing the recombined Rosa26 locus were backcrossed to C57BL6/J for five generations and heterozygotes were then intercrossed to obtain wild type, heterozygous, and homozygous littermates. Animals were kept on constant light/dark cycle and food was supplied ad libitum. Mice were sacrificed at 8–10 weeks of age by CO2 inhalation. Heparinized blood was collected by cardiac puncture and pieces of tissues were flash-frozen. For molecular analyses of duodenal samples, mice were euthanized by cervical dislocation to avoid sample degradation. Animal handling was in accordance with European Molecular Biology Laboratory (EMBL) guidelines.
Southern blotting and PCR analyses
Proper targeting of the Rosa26 locus was verified by Southern blotting using 32P-labeled PCR probes obtained with primers PB1_fwd/PB1_rev and PB2_fwd/PB2_rev. Mice were routinely genotyped by duplex PCR using primers P1, P2, and P3 (Fig. 1a). The Cre transgene was detected using primers Cre_fwd and Cre_rev. Cre-mediated removal of the stop cassette was assessed by RT-PCR using primer pairs P4/P5 and P6/P7 (Fig. 1a). All primers are listed in Table S1.
Fig. 1.
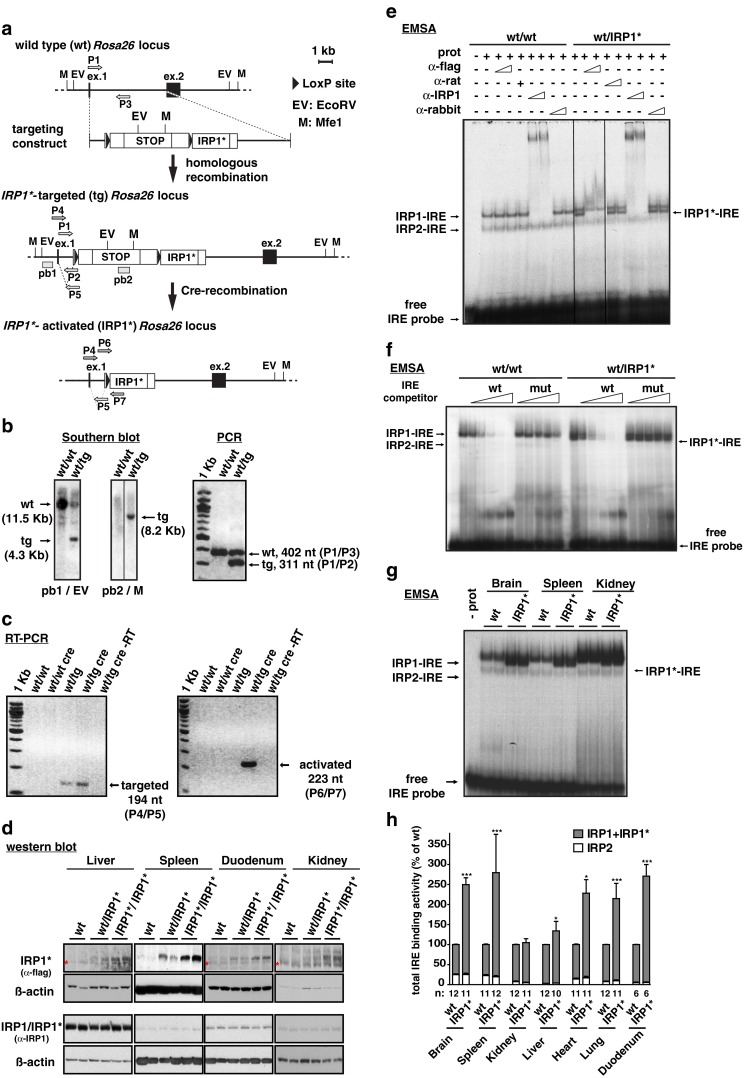
Targeting of the Rosa26 locus with a Cre/Lox inducible IRP1* expression construct and ensuing gain of IRP1 activity in mouse tissues. a Schematic representation of the wild type, IRP1*-targeted and IRP1*-activated Rosa26 locus and of the targeting construct. Restriction sites for EcoRV (EV) and Mfe1 (M) are indicated. The external probe, pb1 (recognizing the first exon of the Rosa26 locus) and the internal one, pb2 (recognizing the stop cassette) are shown. Primers used to assess Cre-mediated recombination of the targeted allele and for routine genotyping are indicated. b Southern blot to verify bona fide targeting of the Rosa26 locus (left panel). Restriction enzymes and probes used are specified below the panels; genotypes are indicated above each lane. Multiplex-PCR for routine detection of the targeted Rosa26 locus (right panel); primer pairs used are indicated on the right. c RT-PCR to confirm Cre-mediated recombination of the Rosa26 locus using cDNA from duodenum. Primer pair P4/P5 is specific for the targeted locus (left panel), but does not discriminate the recombination event. P6/P7 is specifically designed to detect the activated allele (right panel). d Representative western blot showing IRP1* or IRP1 + IRP1* (IRP1/IRP1*) expression in spleen, duodenum, kidney, and liver extracts. An anti-FLAG-tag antibody was used to detect IRP1*, one raised against IRP1 recognizes both IRP1 and IRP1*. In liver, duodenum, and kidney samples, a cross-reacting band is marked with an asterisk. β-actin was used as a standard. e EMSA analysis to assess the IRE-binding activity of IRP1*. Cytoplasmic extracts from ileum were used. An anti-FLAG antibody was used to identify the IRP1*-IRE complex by supershift. An antibody recognizing both IRP1 and IRP1* was used to supershift both IRP1- and IRP1*-IRE complexes. Normal rat and rabbit IgGs, respectively, were used as a negative control. f Competitive EMSA using cytoplasmic extracts from ileum. Molar excess of an unlabeled wild type or mutant FTH1 IRE competitor RNAs over the radiolabeled FTH1 IRE probe was used. g Representative EMSA showing the degree of gain of IRP1 activity in cytoplasmic extracts from the brain, spleen, and kidney. Wild type and IRP1* homozygous animals were analyzed; genotypes are indicated above each lane. h The histogram represents the relative quantification of a series of EMSAs performed on a larger number of animals using cytoplasmic extracts from the brain, spleen, kidney, liver, heart, lung, and duodenum. Sample size is indicated (n). *p<0.05; ***p<0.001
RNA analyses
Total RNA was extracted and reverse-transcribed using Superscript II (Invitrogen, Darmstadt, Germany).
For qPCR analysis, two independent reverse transcription reactions were performed on each sample, using 1 μg of RNA. The resulting cDNA was used for quantitative real-time PCR, using SYBR Green and an ABI 7500 sequence-detection system instrument and software (Applied Biosystems, Darmstadt, Germany). For each target gene, the level of expression was calculated as average of the duplicates and then calibrated to α-tubulin (TUBB5) mRNA levels. Similar results were obtained when using β-actin (ACTB) as reference gene (not shown).
Protein analyses
Total protein extracts were prepared and western blot analysis was performed using antibodies against IRP1, FPN, TfR1, FTL, and β-actin, as previously described [17]. A rat anti-FLAG-tag antibody (BioLegend, San Diego, USA) was used for specific detection of IRP1*. Relative protein expression was assessed by densitometric analysis of immunoblot staining intensity using the ImageJ software (National Institutes of Health, Bethesda, MD, US).
Electrophoretic mobility shift assays (EMSA) were performed as previously described [9] using 8 μg of cytoplasmic extracts and a 32P-labeled human FTH1 IRE probe. For supershift experiments, antibodies (0.5 and 1.5 μg) against IRP1, FLAG, and appropriate rabbit and rat IgG controls were added to the reaction. Competitive EMSA were performed using a molar excess of unlabeled wild type FTH1-IRE probe or mutant version bearing a C deletion in the IRE loop which impairs the IRP/IRE interaction [18]. The bands corresponding to IRP/IRE complexes were quantified using a FLA2000 phosphorimager (Fujifilm, Tokyo, Japan).
Hematology and blood chemistry
Hematological and plasma iron parameters were determined as previously described [13]. Serum ferritin was measured by the Claude Bernard Institute Chemistry Laboratory (Paris, France) using an Olympus 400 analyzer (Olympus, Tokyo, Japan).
Tissue iron content and distribution
Nonheme iron content was measured in whole tissue lysates using the bathophenanthroline chromogen method [17].
To determine body iron distribution, mice were administered intravenously with 59Fe in isotonic HEPES-buffered saline (Fe(NO3)3 complexed with nitrotriacetic acid (1:2); labeled with ∼2 μCi 59Fe/animal; 0.2 μmol Fe/kg body weight). 59Fe activity in tissues was measured 2 weeks after injection using a well-type γ-counter (1282 Compugamma CS, LKB, Wallac, Finland). Results were normalized by subtraction of the amount of 59Fe calculated to be in the residual blood of each organ [19].
Flow cytometry analysis of primary erythroblasts
Bone marrow cells were obtained from femurs dissociated into ice-cold phosphate-buffered saline (PBS). Cells (2 × 106) were co-stained with PeCy5-labeled Ter119, FITC-conjugated CD44, and PE-conjugated CD71 antibodies (eBiosciences, Frankfurt, Germany) in 200 μl of 50 % 2.4G2/50 % (PBS 2 % FBS) at 4 °C for 20 min. Flow cytometry was performed with a MoFlo high-speed cell sorter (Dako, Eching, Germany). FlowJo software (Tree Star, Ashland, OR) was used for analysis.
Statistical analysis
Data are shown as mean values ± SEM. Statistical analysis was performed using two-tailed Student’s t test. P values <0.05 were considered statistically significant.
Results
Targeted expression of a conditional gain of IRP1 function allele from the mouse Rosa26 locus
Normally, IRP1 predominates in its cytosolic aconitase form but can switch to IRE-binding upon Fe-S cluster disassembly [20]. To generate a gain of IRP1 function expression construct, we substituted three cysteine residues required for Fe-S cluster assembly (C437S, C503S, and C506S) [21]. In addition, we introduced a C118A substitution that stabilizes the apoprotein preferentially against heme-mediated degradation (Vasanthakumar and Eisenstein, unpublished findings). The resulting mutant IRP1, referred to as IRP1*, which was also C-terminally FLAG-tagged, is stabilized and escapes Fe-S cluster-mediated regulation, being constitutively active in its IRE-binding form.
Given the potential toxicity of high level IRP1 expression, we generated a conditional mutant using Cre/Lox technology. The IRP1* cDNA was inserted into the permissive and ubiquitously expressed Rosa26 locus together with an upstream floxed β-geo stop cassette. The latter prevents IRP1* transcription from the Rosa26 promoter; its excision upon Cre-mediated recombination enables expression of IRP1* in a conditional manner (Fig. 1a).
The correct insertion of the targeting cassette was confirmed by Southern blot (Fig. 1b, left panel); routine genotyping was performed by PCR analysis (Fig. 1b, right panel). To achieve systemic expression of IRP1*, we crossed mice bearing the Irp1*-targeted Rosa26 allele with a Cre deletor strain expressing the Cre transgene under the control of the murine Hprt promoter [16]. The latter allows removal of the stop cassette early at the zygote stage and consequent IRP1* expression in the whole body. Figure 1c depicts the RT-PCR analysis to confirm Cre-mediated excision of the stop cassette.
Mice homozygous for the systemically activated Irp1*-Rosa26 locus were obtained in Mendelian ratios; they are viable, fertile, and reach adulthood without overt abnormalities. Immunoblot with an anti-FLAG antibody shows IRP1* expression in different organs, including the spleen, duodenum, kidney, and liver. Consistently, IRP1* expression is higher in homozygotes compared to heterozygous mice (Fig. 1d). The use of an anti-IRP1 antibody, reactive with IRP1* as well as endogenous IRP1, indicates that IRP1* protein is not highly overexpressed compared to the endogenous protein.
Mice expressing IRP1* display increased IRE-binding activity
We first tested the functionality of IRP1* as a specific IRE-binding protein by EMSA using cytoplasmic protein extracts from selected tissues. Typically, samples from wild type animals yield two RNA–protein complexes, representing IRP1- and IRP2-IRE, respectively (Fig. 1e). When samples from IRP1* mice were analyzed, a third RNA–protein complex appeared with intermediate mobility. The intermediate band is specifically supershifted by an anti-FLAG antibody and also, together with IRP1-IRE, by an anti-IRP1 antibody recognizing both IRP1 and IRP1*. Addition of control immunoglobulins does not elicit similar effects. This experiment shows that IRP1* is able to bind IRE sequences. Competitive EMSA experiments also demonstrate the specificity of the IRP1*-IRE interaction (Fig. 1f). IRP1* binding to the radiolabeled IRE probe is progressively competed by increasing molar excess of unlabeled wild type IRE sequence but not by a mutant IRE.
To quantify the gain of IRP1 activity, we systematically analyzed extracts from the brain, spleen, kidney, liver, heart, lung, and duodenum, by EMSA. Figure 1g depicts a representative result, demonstrating the increase in IRE-binding activity in organs expressing IRP1*. Quantification of the assays performed on a larger number of animals (Fig. 1h) revealed that, while IRP2 binding activity remains largely unchanged, the sum of IRP1 and IRP1* activity is significantly higher in IRP1* samples compared to wild type. This increase in IRE-binding activity displays tissue-dependent differences, ranging from a mild gain (10–30 %) in the kidney and liver, to a two- to three-fold increase in the brain, spleen, heart, lung, and duodenum.
These data document that IRP1* exerts specific IRE-binding activity and that its presence results in a significant gain of total IRP1 function in all organs analyzed.
Increased IRE-binding activity alters the expression of IRP targets in vivo
Increased IRE-binding activity would be predicted to translationally repress 5′IRE targets, such as FPN and FTL, and stabilize mRNAs bearing an IRE in their 3′UTR, such as TfR1. To assess the in vivo effects of gain of IRP1 function on the post-transcriptional regulation of IRP target genes, we determined RNA and protein levels of FPN, TfR1, and FTL in the liver, spleen, and duodenum (Fig. 2).
Fig. 2.

Impact of gain of IRP1 function on the expression of IRP target genes. Representative western blots of ferroportin (FPN), transferrin receptor 1 (TfR1), and ferritin L (FTL) using protein extracts from the liver, spleen, and duodenum. β-actin was used as a standard. Homozygous males were analyzed; genotypes are indicated above each lane. The histograms represent relative quantification of protein and RNA levels of each IRP target in the three organs. Protein levels were normalized to β-actin, RNA levels to α-tubulin mRNA. Wild type and IRP1* homozygous animals were analyzed; sample size is indicated (n). **p<0.01; ***p<0.001
In the liver, FPN protein levels are mostly unchanged, while FPN mRNA levels are diminished. In splenic samples, we observe a tendency towards decreased FPN protein levels without corresponding changes in mRNA levels. In the duodenum, FPN mRNA also appears unchanged, while the corresponding protein expression is slightly increased. TfR1 protein and mRNA levels tend to be upregulated in the spleen. In the duodenum, TfR1 protein levels are also elevated, while its mRNA is mostly unchanged. In the liver, TfR1 mRNA and protein levels are largely normal. Surprisingly, the expected decrease of FTL expression could not be observed in any of the three tissues. In the spleen, both FTL protein and mRNA are unchanged. FTL protein appears upregulated in the liver, while its mRNA is decreased. In the duodenum, we detected a marked and statistically significant increase of FTL protein without change of the corresponding mRNA.
Despite the apparent tissue-specific and interindividual variability observed, the gain of IRP1 function has a detectable impact on the expression of IRP target genes. This mainly results in the expected tendency towards FPN repression and TfR1 upregulation, together with sustained FTL expression.
Gain of IRP1 function promotes iron loading in the liver, spleen, and duodenum
IRE-binding activity is normally increased in conditions of cellular iron depletion. An artificially generated state of IRP activation, such as IRP1* expression, would thus be expected to trigger an increase in cellular iron content. To assess whether gain of IRP1 function affects iron levels in mouse tissues, we performed spectrophotometric measurements of the total non-heme iron content of the liver, spleen, and duodenum. This analysis revealed increased iron levels in all three organs, between 1.7-fold in the liver, 1.5-fold in the duodenum, and 1.3-fold in the spleen, respectively (Fig. 3a).
Fig. 3.

Gain of IRP1 function causes iron loading in the liver, spleen, and duodenum. a Non-heme iron content in total extracts from the liver, spleen, and duodenum. b Body distribution of 59Fe, 2 weeks after intravenous administration. Homozygous males were analyzed; sample size is indicated (n). *p<0.05; ***p<0.001
To determine whether gain of IRP1 function more broadly affects body iron distribution, we injected 59Fe intravenously and monitored its deposition in several tissues after 2 weeks (Fig. 3b). In agreement with the above results, this experiment shows preferential accumulation of 59Fe in the liver and spleen of mice expressing IRP1*. There was no detectable accumulation of 59Fe in the duodenum. This observation could reflect that the increased nonheme iron content observed in this tissue may result from higher dietary iron intake rather than increased serum iron uptake and/or the consequences of sloughing of duodenal enterocytes within 2 weeks after 59Fe injection.
Overall, these data show that gain of IRP1 function alters body iron distribution with preferential iron loading in the liver, spleen, and duodenum.
Abnormally high IRP1 activity causes macrocytic anemia
Iron is mainly used for the hemoglobinization of red blood cells. To evaluate whether gain of IRP1 function affects hematological parameters, we analyzed blood cell profiles as well as plasma iron values. IRP1* mice are erythropenic and have a lower hemoglobin content, lower hematocrit, and increased mean cell volume (MCV) with some differences between genders, i.e., IRP1* females display a bigger increase in MCV and MCH compared to males with the same genotype (Table 1).
Table 1.
Hematology and blood chemistry in IRP1* mice
| Males | Females | |||
|---|---|---|---|---|
| wt | IRP1* | wt | IRP1* | |
| RBC (×106/μL) | 11.0 ± 0.2 | 9.7 ± 0.2*** | 11.2 ± 0.2 | 11.1 ± 0.1*** |
| Hemoglobin (g/dL) | 15.7 ± 1.0 | 14.1 ± 0.3*** | 16.4 ± 0.2 | 15.6 ± 0.4 |
| Hematocrit (%) | 58.9 ± 0.4 | 51.8 ± 1.1** | 60.3 ± 1.2 | 55.3 ± 0.8** |
| MCV (fL) | 53.2 ± 0.1 | 53.8 ± 0.3 | 52.9 ± 0.2 | 54.9 ± 0.1*** |
| MCH (pg) | 14.3 ± 0.2 | 14.6 ± 0.1 | 14.6 ± 0.1 | 15.1 ± 0.2* |
| Serum Fe (mg/dL) | 122.4 ± 9.6 | 104.3 ± 13.4 | 149.5 ± 10.1 | 146.7 ± 1.6 |
| TIBC (mg/dL) | 373.6 ± 35.7 | 500.5 ± 38.8 | 346.5 ± 29.1 | 358.3 ± 30.5 |
| Tf saturation (%) | 36.0 ± 4.4 | 20.6 ± 1.9** | 46.8 ± 6.3 | 44.0 ± 4.2 |
| Ferritin (ng/L) | 136.8 ± 11.9 | 142.0 ± 13.4 | 153.9 ± 12.4 | 139.1 ± 6.4 |
Results are shown as mean ± SEM. Ten homozygous animals per gender and per group were analyzed.
RBC red blood cell, MCV mean corpuscular volume, MCH mean corpuscular hemoglobin, TIBC (total iron-binding capacity) = serum Fe + UIBC (unbound iron-binding capacity), Tf saturation serum Fe/TIBC × 100.
*p<0.05; **p<0.01; ***p<0.001
Serum iron and ferritin levels are unchanged, although transferrin saturation is decreased in males as a result of increased total iron-binding capacity (Table 1). To assess the state of activation of the systemic regulator of iron homeostasis, we measured hepcidin mRNA levels in the liver and found it to be largely unchanged (Fig. S1).
Altogether, supraphysiological IRP1 activity causes a relatively mild macrocytic anemia, without altered systemic iron availability.
IRP1 hyperactivity impairs erythroid maturation
To assess whether the macrocytic anemia affecting IRP1* mice results from impaired erythropoiesis, we analyzed the maturation state of bone marrow-derived erythroid progenitors by flow cytometry. Co-staining for Ter119 (erythroid lineage marker) and CD44 (adhesion receptor showing a defined expression pattern during erythroblastic maturation) allows separation of erythroblasts into five different subpopulations according to FSC (forward scatter) and CD44 levels (Fig. 4a) [22]. We found a significant increase (1.4- to 1.6-fold) in the relative abundance of the three earliest precursors, i.e., proerythroblasts, basophilic and polychromatophilic erythroblasts, together with a significant decrease of mature erythrocytes (∼30 %) (Fig. 4b). The latter is consistent with the decrease in RBC observed in peripheral blood. Co-staining for CD71 (TfR1) also revealed increased levels of the iron importer in all five subpopulations of bone marrow-derived erythroid precursors (Fig. 4c). We found a significant increase of TfR1 intensity, corresponding to ∼1.7-fold in subpopulation I, ∼2-fold in II, ∼2.8-fold in III, ∼3.2-fold in IV, and ∼1.3-fold in V (the latter not being apparent in the graph due to TfR1 approaching background levels in mature erythrocytes from both wild type and IRP1* mice) (Fig. 4d). These data suggest that excessive iron import via TfR1 may interfere with normal erythropoiesis, and that IRP1 activity critically influences erythroid maturation.
Fig. 4.
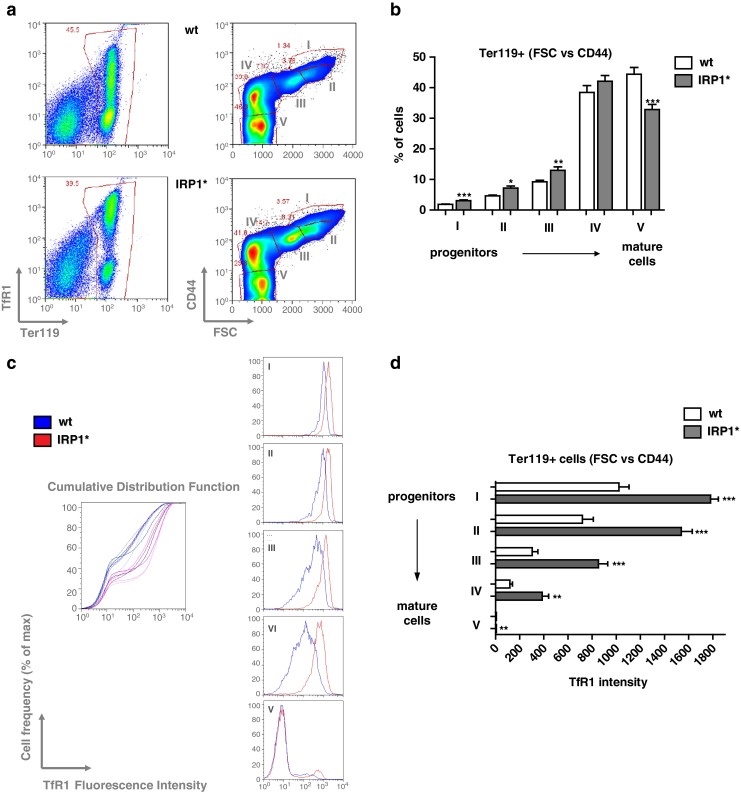
Gain of IRP1 function impairs normal erythropoiesis. a Representative FACS profiles of bone marrow-derived erythroid progenitor cells co-stained with Ter-119, CD71, and CD44. For each genotype, the panels on the left side show the classical Ter119/CD71 profiles allowing for separation of immature (Ter119+, CD71 high) and more mature (Ter119+, CD71 low) cells. The right panels represent CD44 vs. FSC (forward scatter) on the erythroid gate established in the Ter119/CD71 profile. Populations I, II, III, IV, and V represent distinct and progressively more mature erythroid populations [22]. Upper panels correspond to wild type sample, the lower ones to IRP1*. b Relative quantification of the frequencies of each erythroid population. c Histogram representing the cumulative cell frequency distribution as a function of TfR1 intensity of erythroblasts (left panel). Histograms showing the peaks of cell frequency distribution as a function of TfR1 intensity in the distinct five subpopulations of erythroid progenitors (right panels). d Relative quantification of TfR1 intensity in each erythroid subpopulation. Six homozygous females per each genotype were analyzed. *p<0.05; **p<0.01; ***p<0.001
Discussion
The IRP/IRE regulatory system has been extensively studied in cultured cells, forming the basis for the molecular understanding of its central importance in cellular iron homeostasis. The generation of murine in vivo models of loss of IRP function has unraveled its function for life [11–13]. To complement the study of IRP KO models and to be able to understand its involvement in human diseases associated with abnormally high IRP activity, we generated a mouse model of gain of IRP1 function. While this model should prove valuable for future studies, we here also report that appropriate IRP1 activity is important to maintain physiological body iron distribution and normal erythropoiesis.
IRP1 overexpression has previously been reported to be toxic in cells [23]. In flies, ubiquitous and muscle-specific overexpression of IRP1A, the only IRP homolog possessing IRE-binding activity of the two cytosolic aconitases, has been shown to cause pre-adult lethality [24]. To prevent the potential risk of in vivo toxicity/lethality of expressing a gain of IRP1 function mutant, we generated a conditional allele by Cre/Lox technology. For moderate rather than excessive levels of expression of IRP1*, we targeted the ubiquitously expressed Rosa26 locus [14] with a promoterless construct. This strategy proved to be successful in obtaining, for the first time, live animals expressing a gain of IRP1 function protein (IRP1*).
IRP1* protein is moderately expressed, but even the low expression of IRP1* significantly augments the total level of IRE-binding activity in mouse tissues. Increased IRP1 activity is apparent in all tissues; the kidney and, to a lower extent, the liver are among the organs where IRP1 is normally expressed at the highest levels [8]. This most probably explains why the contribution of IRP1* in these two organs is relatively smaller than the gain of IRP1 function in tissues with lower basal IRP1 activity, such as the brain, spleen, heart, lung, and duodenum.
IRP1* alters the expression of IRP target genes, although we noticed considerable interindividual and tissue-dependent variability. Tissue-specific responses to elevated IRP1 activity, but also the different degree of gain of IRP1 function in different organs, may account for the inter-tissue variability of the expression of IRP targets. Future work will have to dissect the molecular mechanisms underlying the unexpected responses, such as the increased expression of FTL in the liver and duodenum in response to IRP1*.
Expression of IRP1* causes iron accumulation in the liver, spleen, and duodenum. While the molecular mechanisms underlying iron deposition in IRP1* mice remain to be understood in detail, forced cellular iron accumulation entirely meets the predicted phenotype. Contrary to IRP2 KO mice where the spleen is iron-depleted, the splenic iron content of IRP1* mice is elevated, which correlates with a decreased expression of the iron exporter FPN. The latter is likely explained by stronger translational repression of the FPN mRNA, which is in agreement with high IRP activity and unchanged hepcidin expression. Similarly, iron accumulation in liver macrophages could account for the hepatic iron loading in IRP1* mice, although Perl staining of liver sections did not reveal preferential iron accumulation in these cells (not shown). When IRP2 is ablated, liver and duodenum are iron-loaded and this is associated with higher ferritin levels [12, 17]. In gain of IRP1 function, hepatic and duodenal iron accumulation is also associated with FTL upregulation, which is somewhat counterintuitive considering the IRP/IRE regulatory paradigm. Translational repression of 5′IRE targets is normally achieved by IRP-mediated inhibition of the cap structure-dependent recruitment of the small ribosomal subunit to the mRNA [25]. Interestingly, lack of FTL repression in a context of high IRP1 activity has been observed before [26–29] and could possibly be explained by cap-independent translation via the internal ribosomal entry site recently found in the 5′UTR of FTL mRNA [30].
Mice with gain of IRP1 function display macrocytic anemia associated with impaired erythroid maturation. Although increased cell death could contribute to the decrease in mature erythroid cells, the concurrent expansion of early erythroid progenitors rather suggests a maturation block. Thus, abnormally high IRP1 activity compromises normal erythropoiesis, which can in turn explain the macrocytosis. The detailed mechanism underlying this hematological phenotype remains to be elucidated, but its independence from changes in systemic iron availability suggests that it likely is a cell-autonomous defect. In IRP2 KO mice, the microcytic anemia is associated with lower TfR1 levels [17] and depletion of bone marrow iron stores [31]. Conversely, TfR1 expression on the cell membrane of erythroid progenitors from IRP1* mice is increased. This finding points toward the possibility that the defect could result from excessive iron uptake by erythroid precursors. This explanation could also account for the macrocytosis. While alternative or additional explanations are possible, iron toxicity may represent one of the causes of defective erythropoiesis in IRP1* mice. FPN has recently been hypothesized to export iron also from erythroid cells [32] and our data suggest that FPN tends to be diminished in IRP1* tissues. If reduced FPN expression also applies to erythroid precursors, diminished iron export could further contribute to intracellular iron loading and toxicity. However, erythroid expression of a FPN variant (FPN1b) lacking the 5′IRE [33] could possibly bypass the potential repression of FPN1A (the IRE-containing isoform) by IRP1*.
Erythroid 5-aminolevulinate synthase (eALAS), the enzyme that catalyzes the rate-limiting step in heme biosynthesis, could also be implicated in the anemia of IRP1* mice. eALAS mRNA is translationally regulated by the IRP/IRE system and it has been shown to be upregulated in erythroid precursors from IRP2 KO mice [31]. If eALAS was translationally repressed via its 5′IRE in erythroblasts from IRP1* mice, an ensuing block of heme synthesis could contribute to iron accumulation.
Repression of HIF2α (EPAS-1) via its 5′IRE is also conceivable in a setting of high IRP1 activity. This may in turn impair HIF2α-mediated induction of erythropoietin expression in the kidney with a consequent reduced stimulus for erythropoiesis. The 5′IRE target mitochondrial aconitase (mAco), an enzyme involved in the TCA cycle, could also be downregulated by a gain of IRP1 activity. Interestingly, when mAco is pharmacologically inhibited by fluoroacetate, mice develop a similar hematological phenotype as IRP1* mice [34]. Although feline leukemic virus receptor (FLCVR) is not a direct IRP target, the hematological phenotype of IRP1* mice is somewhat reminiscent of the FLCVR KO model. There, heme toxicity has been hypothesized to cause an early erythropoietic blockade [35]. In principle, all of these responses, involving different IRP targets, could act in isolation or in combination to cause the macrocytic anemia of IRP1* mice.
A secondary increase of IRP expression and activity has recently also been reported as a consequence of disruption of the main mechanism of iron-mediated degradation of IRP2. F box and leucine-rich repeat protein 5 (FBXL5) is a subunit of an E3 ubiquitin ligase complex regulating IRP2 stability [3]. Systemic or liver-specific ablation of FBXL5 causes fatal accumulation of ferrous iron [36]. Yet, this model does not allow discrimination between the direct consequences of FBXL5 ablation per se and those directly due to increased IRP expression.
Secondary gain of IRP1 function has been described, for instance, as a consequence of dysfunctions of Fe-S cluster biosynthesis. Due to the ensuing reduced availability of cytosolic Fe-S clusters, regulation of IRP1 is impaired, resulting in abnormally high levels of active IRP1. This has been shown in animal models with ablation of frataxin (Fxn), glutaredoxin 5 (Glrx5), and the mitochondrial ATP-binding cassette transporter (Abcb7) [37, 38]. Those mutations are also causative of human diseases, namely Friedreich’s ataxia, sideroblastic anemia with iron overload, and X-linked sideroblastic anemia with ataxia, respectively [37, 38]. With the final aim of dissecting the molecular mechanisms underlying the pathogenesis of those diseases, our model with inducible primary gain of IRP1 function could serve to pinpoint the abnormalities directly caused by elevated IRP1 activity.
The IRP1* mouse line embodies the first model of primary and inducible gain of IRP1 function in a mammalian organism. This mouse thus represents a useful model system to expand our understanding of IRP regulation in vivo. Moreover, it could prove to be of great value to better understand the molecular etiology of human diseases associated with abnormally high IRP1 activity, such as sideroblastic anemia linked to Glrx5 deficiency [4], Friedreich’s ataxia [5], or Parkinson’s disease [6].
Electronic supplementary material
Below is the link to the electronic supplementary material.
(DOC 165 kb)
Acknowledgments
We are grateful to Prof. H-J. Gröne for expert histological analyses, to Dunja Ferring-Appel for technical assistance, and to S. Srinivas (Columbia University, New York, USA) for the plasmids pBigT and pROSA26PA. We also thank the EMBL transgenic mouse service, the staff of the EMBL laboratory animal resources, and the EMBL Flow Cytometry Core Facility for their contributions to this work. This work was supported by the Hildegard-Grunow-Foundation Munich to K.S. and by an EEC FP6 grant (LSHM-CT-2006-037296 Euroiron1) and a Virtual Liver Network grant (funding number 315740) to M.W.H..
Conflict of interest
The authors declare that they have no competing financial interests.
References
- 1.Fleming RE, Ponka P. Iron overload in human disease. N Engl J Med. 2012;366:348–359. doi: 10.1056/NEJMra1004967. [DOI] [PubMed] [Google Scholar]
- 2.Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to tango: regulation of mammalian iron metabolism. Cell. 2010;142:24–38. doi: 10.1016/j.cell.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 3.Anderson CP, Shen M, Eisenstein RS, Leibold EA. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim Biophys Acta. 2012;1823:1468–1483. doi: 10.1016/j.bbamcr.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Camaschella C, Campanella A, De Falco L, Boschetto L, Merlini R, Silvestri L, Levi S, Iolascon A. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood. 2007;110:1353–1358. doi: 10.1182/blood-2007-02-072520. [DOI] [PubMed] [Google Scholar]
- 5.Lobmayr L, Brooks DG, Wilson RB. Increased IRP1 activity in Friedreich ataxia. Gene. 2005;354:157–161. doi: 10.1016/j.gene.2005.04.040. [DOI] [PubMed] [Google Scholar]
- 6.Faucheux BA, Martin M-E, Beaumont C, Hunot S, Hauw J-J, Agid Y, Hirsch EC. Lack of up-regulation of ferritin is associated with sustained iron regulatory protein-1 binding activity in the substantia nigra of patients with Parkinson’s disease. J Neurochem. 2002;83:320–330. doi: 10.1046/j.1471-4159.2002.01118.x. [DOI] [PubMed] [Google Scholar]
- 7.LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, et al. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet. 2001;27:209–214. doi: 10.1038/84859. [DOI] [PubMed] [Google Scholar]
- 8.Meyron-Holtz EG, Ghosh MC, Iwai K, LaVaute T, Brazzolotto X, Berger UV, Land W, Ollivierre-Wilson H, Grinberg A, Love P, et al. Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J. 2004;23:386–395. doi: 10.1038/sj.emboj.7600041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galy B, Ferring D, Benesova M, Benes V, Hentze MW. Targeted mutagenesis of the murine IRP1 and IRP2 genes reveals context-dependent RNA processing differences in vivo. RNA. 2004;10:1019–1025. doi: 10.1261/rna.7220704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galy B, Ferring D, Hentze MW. Generation of conditional alleles of the murine iron regulatory protein (IRP)-1 and -2 genes. Genesis. 2005;43:181–188. doi: 10.1002/gene.20169. [DOI] [PubMed] [Google Scholar]
- 11.Smith SR, Ghosh MC, Ollivierre-Wilson H, Hang Tong W, Rouault TA. Complete loss of iron regulatory proteins 1 and 2 prevents viability of murine zygotes beyond the blastocyst stage of embryonic development. Blood Cells Mol Dis. 2006;36:283–287. doi: 10.1016/j.bcmd.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 12.Galy B, Ferring-Appel D, Kaden S, Gröne H-J, Hentze MW. Iron regulatory proteins are essential for intestinal function and control key iron absorption molecules in the duodenum. Cell Metab. 2008;7:79–85. doi: 10.1016/j.cmet.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Galy B, Ferring-Appel D, Sauer SW, Kaden S, Lyoumi S, Puy H, Kölker S, Gröne H-J, Hentze MW. Iron regulatory proteins secure mitochondrial iron sufficiency and function. Cell Metab. 2010;12:194–201. doi: 10.1016/j.cmet.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 14.Zambrowicz BP, Imamoto A, Fiering S, Herzenberg LA, Kerr WG, Soriano P. Disruption of overlapping transcripts in the ROSA βgeo 26 gene trap strain leads to widespread expression of β-galactosidase in mouse embryos and hematopoietic cells. Proc Natl Acad Sci U S A. 1997;94:3789–3794. doi: 10.1073/pnas.94.8.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srinivas S, Watanabe T, Lin C-S, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang S-HE, Silva FJ, Tsark WMK, Mann JR. A cre/loxP-deleter transgenic line in mouse strain 129S1/SvImJ. Genesis. 2002;32:199–202. doi: 10.1002/gene.10030. [DOI] [PubMed] [Google Scholar]
- 17.Galy B, Ferring D, Minana B, Bell O, Janser HG, Muckenthaler M, Schümann K, Hentze MW. Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2) Blood. 2005;106:2580–2589. doi: 10.1182/blood-2005-04-1365. [DOI] [PubMed] [Google Scholar]
- 18.Gray NK, Pantopoulos K, Dandekar T, Ackrell BA, Hentze MW. Translational regulation of mammalian and Drosophila citric acid cycle enzymes via iron-responsive elements. Proc Natl Acad Sci U S A. 1996;93:4925–4930. doi: 10.1073/pnas.93.10.4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schümann K, Szegner B, Kohler B, Pfaffl MW, Ettle T. A method to assess 59Fe in residual tissue blood content in mice and its use to correct 59Fe-distribution kinetics accordingly. Toxicology. 2007;241:19–32. doi: 10.1016/j.tox.2007.08.082. [DOI] [PubMed] [Google Scholar]
- 20.Rouault TA. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol. 2006;2:406–414. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 21.Clarke SL, Vasanthakumar A, Anderson SA, Pondarré C, Koh CM, Deck KM, Pitula JS, Epstein CJ, Fleming MD, Eisenstein RS. Iron-responsive degradation of iron-regulatory protein 1 does not require the Fe-S cluster. EMBO J. 2006;25:544–553. doi: 10.1038/sj.emboj.7600954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen K, Liu J, Heck S, Chasis JA, An X, Mohandas N. Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis. Proc Natl Acad Sci U S A. 2009;106:17413–17418. doi: 10.1073/pnas.0909296106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeRusso PA, Philpott CC, Iwai K, Mostowski HS, Klausner RD, Rouault TA. Expression of a constitutive mutant of iron regulatory protein 1 abolishes iron homeostasis in mammalian cells. J Biol Chem. 1995;270:15451–15454. doi: 10.1074/jbc.270.26.15451. [DOI] [PubMed] [Google Scholar]
- 24.Lind MI, Missirlis F, Melefors Ö, Uhrigshardt H, Kirby K, Phillips JP, Söderhäll K, Rouault TA. Of two cytosolic aconitases expressed in Drosophila, only one functions as an iron-regulatory protein. J Biol Chem. 2006;281:18707–18714. doi: 10.1074/jbc.M603354200. [DOI] [PubMed] [Google Scholar]
- 25.Muckenthaler M, Gray NK, Hentze MW. IRP-1 binding to ferritin mRNA prevents the recruitment of the small ribosomal subunit by the cap-binding complex eIF4F. Mol Cell. 1998;2:383–388. doi: 10.1016/S1097-2765(00)80282-8. [DOI] [PubMed] [Google Scholar]
- 26.Cairo G, Recalcati S, Montosi G, Castrusini E, Conte D, Pietrangelo A. Inappropriately high iron regulatory protein activity in monocytes of patients with genetic hemochromatosis. Blood. 1997;89:2546–2553. [PubMed] [Google Scholar]
- 27.Cairo G, Tacchini L, Pietrangelo A. Lack of coordinate control of ferritin and transferrin receptor expression during rat liver regeneration. Hepatol (Baltimore, MD) 1998;28:173–178. doi: 10.1002/hep.510280123. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Pantopoulos K. Conditional derepression of ferritin synthesis in cells expressing a constitutive IRP1 mutant. Mol Cell Biol. 2002;22:4638–4651. doi: 10.1128/MCB.22.13.4638-4651.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen G, Fillebeen C, Wang J, Pantopoulos K. Overexpression of iron regulatory protein 1 suppresses growth of tumor xenografts. Carcinogenesis. 2007;28:785–791. doi: 10.1093/carcin/bgl210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daba A, Koromilas AE, Pantopoulos K. Alternative ferritin mRNA translation via internal initiation. RNA. 2012;18:547–556. doi: 10.1261/rna.029322.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cooperman SS, Meyron-Holtz EG, Olivierre-Wilson H, Ghosh MC, McConnell JP, Rouault TA. Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2. Blood. 2005;106:1084–1091. doi: 10.1182/blood-2004-12-4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang D-L, Senecal T, Ghosh MC, Ollivierre-Wilson H, Tu T, Rouault TA. Hepcidin regulates ferroportin expression and intracellular iron homeostasis of erythroblasts. Blood. 2011;118:2868–2877. doi: 10.1182/blood-2011-01-330241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang D-L, Hughes RM, Ollivierre-Wilson H, Ghosh MC, Rouault TA. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metabol. 2009;9:461–473. doi: 10.1016/j.cmet.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Talbot AL, Bullock GC, Delehanty LL, Sattler M, Zhao ZJ, Goldfarb AN. Aconitase regulation of erythropoiesis correlates with a novel licensing function in erythropoietin-induced ERK signaling. PLoS One. 2011;6:e23850. doi: 10.1371/journal.pone.0023850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keel SB, Doty RT, Yang Z, Quigley JG, Chen J, Knoblaugh S, Kingsley PD, De Domenico I, Vaughn MB, Kaplan J, et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science. 2008;319:825–828. doi: 10.1126/science.1151133. [DOI] [PubMed] [Google Scholar]
- 36.Moroishi T, Nishiyama M, Takeda Y, Iwai K, Nakayama Keiichi I. The FBXL5-IRP2 axis is integral to control of iron metabolism in vivo. Cell Metabol. 2011;14:339–351. doi: 10.1016/j.cmet.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 37.Sheftel A, Stehling O, Lill R. Iron–sulfur proteins in health and disease. Trends Endocrin Metab. 2010;21:302–314. doi: 10.1016/j.tem.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 38.Ye H, Rouault TA. Human iron–sulfur cluster assembly, cellular iron homeostasis, and disease. Biochemistry. 2010;49:4945–4956. doi: 10.1021/bi1004798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 165 kb)