

Abstract
G-protein β subunits perform essential neuronal functions as part of G-protein βγ and Gβ5-regulators of G-protein signaling (RGS) complexes. Both Gβγ and Gβ5-RGS are obligate dimers that are thought to require the assistance of the cytosolic chaperonin CCT and a cochaperone, phosducin-like protein 1 (PhLP1) for dimer formation. To test this hypothesis in vivo, we deleted the Phlp1 gene in mouse (Mus musculus) retinal rod photoreceptor cells and measured the effects on G-protein biogenesis and visual signal transduction. In the PhLP1-depleted rods, Gβγ dimer formation was decreased 50-fold, resulting in a >10-fold decrease in light sensitivity. Moreover, a 20-fold reduction in Gβ5 and RGS9–1 expression was also observed, causing a 15-fold delay in the shutoff of light responses. These findings conclusively demonstrate in vivo that PhLP1 is required for the folding and assembly of both Gβγ and Gβ5-RGS9.
Introduction
Seven-transmembrane G-protein-coupled receptors (GPCRs) and their associated heterotrimeric G-proteins constitute a major means by which eukaryotic cells respond to external signals. As a result, G-protein signaling regulates numerous aspects of cellular physiology (Wettschureck and Offermanns, 2005). This is especially true in neurons, where G-proteins modulate synaptic transmission (Betke et al., 2012), synaptic plasticity (Vellano et al., 2011), and sensory signals (Wettschureck and Offermanns, 2005; Han and Simon, 2011). The process begins when a signaling molecule binds the GPCR, inducing a conformational change that activates the heterotrimeric G-protein by driving exchange of GTP for GDP on the α subunit (Gα). GTP binding causes dissociation of the G-protein complex into Gα-GTP and the G-protein βγ dimer (Gβγ), each of which interacts with effector enzymes and ion channels to mediate the cellular response to the signal. The lifetime of Gα-GTP is determined by regulators of G-protein signaling (RGS) proteins that accelerate the hydrolysis of GTP by Gα, allowing reassociation of Gα with Gβγ in the inactive Gαβγ heterotrimer (Kimple et al., 2011). In this manner, the G-protein cycles between the active and inactive state, continually reassessing the activation state of the receptor.
To perform its signaling functions, the individual subunits of the G-protein must be synthesized and assembled into functional heterotrimers. A key event in this process of G-protein biogenesis is the folding of the Gβ subunit and its dimerization with Gγ. Gβγ is an obligate dimer in which neither subunit is stable in the absence of the other (Higgins and Casey, 1994), which raises the question of how the dimer could form when the individual subunits are unable to achieve a stable conformation on their own. Investigations into the mechanism of Gβγ assembly have pointed to important roles for the molecular chaperone cytosolic chaperonin-containing T-complex polypeptide 1 (CCT; also called TRiC) and a cochaperone phosducin-like protein 1 (PhLP1) in the assembly process (McLaughlin et al., 2002; Humrich et al., 2005; Knol et al., 2005; Lukov et al., 2005, 2006; Wells et al., 2006). Subsequent studies of the neuronal RGS complex, another obligate dimer of the Gβ5 isoform and RGS proteins of the R7 family (Martemyanov and Arshavsky, 2009), indicated that CCT was required for dimer formation while PhLP1 appeared to play a lesser role, suggesting that the mechanism of Gβ5-RGS assembly might be different than that of Gβγ assembly (Howlett et al., 2009).
To test the proposed role of CCT and PhLP1 in Gβγ and Gβ5-RGS assembly in vivo, we created a conditional PhLP1 knock-out in retinal rod photoreceptors and measured the effects on G-protein-mediated phototransduction in these cells. We found that the loss of PhLP1 has profound effects on transducin (Gt) heterotrimer formation in rods, resulting in equally striking effects on visual function. Unexpectedly, Gβ5-RGS9 assembly was just as dependent on PhLP1 as was Gt. These findings conclusively demonstrate an essential role for PhLP1 in both Gβγ and Gβ5-RGS9 assembly in vivo.
Materials and Methods
Generation of rod-specific PhLP1 knock-out mice.
All experiments with mice were performed in accordance with National Institutes of Health policy on animal use and were approved by the Brigham Young University and Washington University Institutional Animal Care and Use Committees. The construction of the targeting vector for the Phlp1 gene was based on an established protocol (Wu et al., 2008). In brief, recombineering and traditional cloning techniques were used to capture the genomic fragment of the Phlp1 gene from a BAC plasmid and assemble the targeting vector. The first loxP site was introduced in intron 1 of the Phlp1 gene upstream of the translation initiation site in exon 2. A Frt-Neo-Frt cassette and the second LoxP site were inserted in intron 3. Two thymidine kinase genes were added flanking the final targeting vector using Gateway recombination (Fig. 1A). Approximately 30 μg of the linearized targeting vector were electroporated into G4-derived ES cells (a gift from A. Nagy, Samuel Lunenfeld Research Institute, Toronto, Canada) and the cells were subjected to positive/negative selection using G418 and FIAU selection media. Positive clones were confirmed by Southern blotting. These cells were karyotyped and microinjected into mouse embryos. The embryos were implanted into surrogate mothers, and the chimeric offspring were bred with ROSA-FLP transgenic C57BL/6 mice to determine germ-line transmission while at the same time removing the neo gene that was used for positive selection. The mice containing the fully processed Phlp1-loxP gene were then bred with rhodopsin-Cre (iCre75) mice and further inbred to produce homozygous PhLP1-loxP+/+ iCre75+/− mice, designated as PhLP1F/FCre+ mice. Germline transmission in F1 and in subsequent generations was confirmed by PCR of ear clips using primers P3 and P4 for the PhLP1-LoxP allele and the wild-type allele. The forward P3 primer (5′ GATCACTTTGACTGGGGAATGATTTTAGGT) was just upstream of the targeting Neo site inside intron 3. The reverse P4 primer (5′ GAGGTGGTAAGCAGGTGTACTGGCTGGTTT) was downstream of the Neo targeting site and inside intron 3 (Fig. 1A). Genotyping for determination of the presence of Flp and Cre genes in these mice were performed according to the Jackson Laboratory Web protocol [http://jaxmice.jax.org; strain 129S4/SvJaeSor-Gt(ROSA) 26Sortm1(FLP1)Dym/J] and Li et al. (2005), respectively. Mice were kept under the standard 12 h light/dark cycle. Experiments were performed on age-matched 4- to 5-week-old animals of either sex unless indicated otherwise.
Figure 1.

Generation and characterization of the rod photoreceptor-specific PhLP1 knock-out mouse. A, The scheme for constructing the floxed Phlp1 gene is shown. Sketch 1 shows the targeting vector with the short and long homology arm regions, the neomycin selection cassette flanked by frt recombination sites (green triangles) placed in intron 3 and the loxP recombination sites in introns 1 and 3 (red triangles). Crossover lines indicate the regions of homologous recombination. Sketch 2 shows the structure of the wild-type Phlp1 locus. Sketch 3 shows the targeted Phlp1 locus after recombination. Sketch 4 shows the floxed Phlp1 gene after removal of the neomycin cassette by FLP recombination. Sketch 5 shows the disrupted gene after Cre recombination. P3 and P4 represent the forward and reverse primer sites used for genotyping the flox-Phlp1 mice. B, PCR genotyping results using the P3 and P4 primers. The flox-Phlp1 gene produced a 704 bp product, while the wild-type phlp1 gene produced a 600 bp product. C, Immunoblot detection of PhLP1 in rod outer segment preparations from 1-month-old PhLP1+/+Cre+, PhLP1F/+Cre+, and PhLP1F/FCre+ mice. D, Immunolocalization of PhLP1 in retinal cross-sections from 1-month-old PhLP1+/+Cre+, PhLP1F/+Cre+, and PhLP1F/FCre+ mice. E, Time course of Cre-mediated loss of PhLP1 expression. Retinal extracts from PhLP1F/FCre+ and PhLP1F/FCre− mice were immunoblotted for PhLP1 at the indicated postnatal days. The PhLP1 bands were quantified and normalized relative to the PhLP1F/FCre− control. Symbols represent the average ± SEM from three mice of each genotype. Representative immunoblots are shown below the graph.
Antibodies.
Rabbit antibodies sc-389-Gαt1, sc-381-Gβ3, sc-382-Gβ4, sc-373-Gγ1, sc-15382-rhodopsin, and sc-47778 β-actin were from Santa Cruz Biotechnology. Rabbit antibodies PAB-06800-G against PDEαβγ2 were from CytoSignal Research Products. Rabbit antibodies AB5405-Cone-opsin and AB5585-recoverin were from Millipore. A rabbit antibody ab6046-β-tubulin was from Abcam. A rabbit antibody PA1–731-visual arrestin was from Affinity Bioreagents. A rat antibody MCA2178-CCTε was from Serotec. Rabbit antibodies against GC1, GC2, GCAP1, and GCAP2 were from the Baehr laboratory. Rabbit antibodies against rhodopsin kinase and RGS9 were from the Chen laboratory. A rabbit antibody against Gβ5 was a gift from W. Simonds (National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD), and rabbit antibodies against R9AP were from K. Martemyanov (Scripps Research Institute, Jupiter, FL) and V. Arshavsky (Duke University, Durham, NC). Rabbit antibodies against Gβ1, PhLP1, PhLP2A, PhLP3, and phosducin were from the Willardson laboratory (Thulin et al., 1999; Lee et al., 2004).
Retinal morphology and immunolocalization.
The superior hemisphere of eyes from PhLP1F/FCre+ and control mice was cautery marked for orientation. These eyes were enucleated under ambient illumination without dark adaptation and were immersion fixed for 2 h using freshly prepared 4% paraformaldehyde in phosphate buffer (0.1 m phosphate, pH 7.4) and cryoprotected overnight in 30% sucrose in phosphate buffer. The cornea and lens were then removed, and the eyecups were embedded in optimal cutting temperature (OCT) compound. Cryosections of 12 μm were cut through the optic nerve head along the vertical meridian and were placed on Superfrost microscope slides. Cryosections with intact morphology were used to characterize photoreceptor degeneration. These sections were washed with phosphate buffer to remove excess OCT and labeled with 100 ng/ml of 4′, 6-diamidino-2-phenylindole dihydrochloride (DAPI) nuclear stain for 30 min in the dark. Sections were then mounted and coverslipped. Digital images were acquired and were used to measure the outer nuclear layer thickness in the superior central retina in proximity to the edge of the optic nerve head. The number of nuclei in the nuclear stacks was counted in 12 different places and the average was calculated and reported.
For immunohistochemistry, cryosections were washed in phosphate buffer to remove excess OCT and blocked for 30 min using blocking buffer (10% normal donkey serum, 1.0% bovine serum albumin, 0.1% Triton X-100, and 0.03% NaN3 in phosphate buffer, pH 7.4). Primary antibodies were applied to sections in a humidified chamber overnight at 4°C. After washing in phosphate buffer, fluorescein-conjugated secondary antibodies were applied for 30 min at room temperature. The sections were viewed using an Olympus Fluoview FV300 confocal laser-scanning microscope.
For retinal histology determinations, mouse eyecups were fixed overnight in 2% glutaraldehyde-1% paraformaldehyde in 0.1 m cacodylate buffer (pH 7.4), postfixed 1 h in 1% osmium tetroxide in 0.1 m cacodylate buffer, dehydrated by passing through an ascending series of ethanols, and embedded in Spurr's epoxy resin (Ted Pella). Sections 1 μm thick were cut and contrasted using Richardson's stain (methylene blue-azure II).
Immunoblotting.
Whole retinal extracts were prepared from eyes of PhLP1F/FCre+ mice and controls under ambient illumination. The retinas were harvested and placed in ice-cold extraction buffer (11.9 mm phosphate, 137 mm NaCl, 2.7 mm KCl, 2% NP-40, and 6 μg/ml Sigma Protease inhibitor cocktail). The retinas were disrupted by passing through an 18 gauge needle, and the extracts were centrifuged to remove cellular debris. The isolation of rod outer segments was performed as described previously (Chen et al., 2012) with minor modifications. Briefly, retinas were removed from dark-adapted mice under dim red light. Sucrose (47%) in PBS was added, and the suspension was vortexed for 45–60 s. The disrupted retinas were centrifuged and the supernatants were decanted and diluted one-to-one with PBS. The rod outer segments were collected by centrifugation, and the supernatant was discarded. The final outer segment pellet was resuspended in extraction buffer. Protein concentrations of whole retinal extracts and outer segment preparations were determined, and equal amounts of protein were immunoblotted for the indicated proteins. Immunoblots were quantified using a LICOR Odyssey near-infrared imaging scanner.
Gβγ dimer determination.
Approximately 5 nmol of purified His6-tagged rat phosducin (Pdc), 30 μl of cobalt slurry, and 400 μl of equilibration buffer (20 mm HEPES, 500 mm NaCl, 10 mm imidazole, pH 8.0) were added to retina extracts containing 150 μg of protein. Samples were incubated on ice for 45 min. The cobalt beads were washed three times with the equilibration buffer, and Pdc-containing complexes were released with elution buffer (20 mm HEPES, 500 mm NaCl, 500 mm imidazole, pH 7.0). The coprecipitates were immunoblotted for Gβ1 and Pdc, and the band intensities were quantified on the LICOR scanner.
Association of Gβ1 with CCT was measured by coimmunoprecipitation. Retinal extracts containing 800 μg of protein were immunoprecipitated using 0.5 μg of a CCTε antibody. The immunoprecipitates were immunoblotted for Gβ1, Gβ5, and CCTε, and the band intensities were quantified using a LICOR scanner. The Gβ1/CCTε and Gβ5/CCTε band intensity ratio was calculated and normalized to that of the wild type.
Quantitative RT-PCR.
Total RNA from whole retinas was isolated using RNA-STAT60 according to the manufacturer's protocol (TelTest). Approximately 2 μg of the isolated RNA was then treated with DNase (Promega) and reverse transcribed using dT oligo primers (Invitrogen) and SuperScript III reverse transcriptase (Invitrogen) according to the manufacturer's instructions to synthesize cDNA. The RT-PCR was performed using the TaqMan gene expression assay primers and probe mix (Applied Biosystems). Mouse GAPDH (Mm99999915_g1), PDCL (Mm01327170_m1), GNAT1 (Mm01229120_m1), GNB1 (Mm00515003_m1), GNG1 (Mm00802676_g1), GNB5 (Mm00515871_m1), and RGS9 (Mm01250426_m1) mRNAs were measured on an Applied Biosystems 7500 Real-Time PCR System. The PCR was performed under the following conditions: one cycle at 50°C for 2 min, one cycle at 95°C for 10 min, and 40 cycles at 95°C for 15 s and at 60°C for 1 min. The data were normalized to the GAPDH reference. The RT-PCR results were analyzed using the ΔΔCt method to compare the mRNA levels of PhLP1F/FCre+ mice to controls. At least three animals of each genotype were used, and the RT-PCR analysis was repeated three times for each animal.
Optomotor responses.
Visual acuity and contrast sensitivity of PhLP1F/FCre+ and control mice were measured using a two-alternative forced-choice protocol (Umino et al., 2008). The Optomotry system (Cerebral Mechanics) consisted of a square array of four computer monitors with a pedestal in the center where the mouse was placed. An infrared-sensitive television camera and a round array of six infrared LEDs mounted above the animal were used to observe the mouse but not the monitors. Using a staircase paradigm, rotating stimuli (sine-wave vertical gratings) were applied on the monitors where they formed a virtual cylinder around the mouse (Prusky et al., 2004). The mouse responded to the stimuli by reflexively rotating its head in the corresponding direction. Optomotor responses were measured under two background illumination conditions: scotopic (−4.45 log cd m−2) or photopic (1.85 log cd m−2). For scotopic conditions, background monitor luminance was controlled by neutral density film filters.
Visual acuity was defined as the threshold for spatial frequency (Fs) of gratings with 100% contrast and measured at the speed (Sp) of 6.3°/s for both illumination conditions. In this mode, Fs was gradually increased by the computer protocol until its threshold was determined. Temporal frequency (Ft) was automatically adjusted by the computer program, based on the following equation: Ft = Sp · Fs (Umino et al., 2008). Contrast sensitivity was defined as the inverse of contrast threshold for optomotor responses. In this mode, contrast of the stimuli was gradually decreased by the computer protocol until its threshold was determined. Fs was fixed at 0.128 cycles/degree, Ft was set to 0.8 Hz, and Sp was set to 6.3°/s for both illumination conditions. Data were analyzed using independent two-tailed Student's t test, with an accepted significance level of p < 0.05.
Electroretinogram responses.
Mice were dark adapted overnight and anesthetized using isofluorane. Pupils were dilated by adding a drop of 1% tropicamide for 15 min to the eyes. A recording electrode was placed on the cornea with a reference electrode inserted subdermally in the cheek and a ground electrode subdermally at the base of the tail. Electroretinogram (ERG) responses were measured using an Ocuscience HMsERG system. Full-field scotopic ERG recordings of both PhLP1F/FCre+ and controls were performed with flashes of increasing white light intensities from 4 × 10−4 to 102 cd·s m−2. The amplitudes of the a-wave and b-wave at different light intensities were then compared between the PhLP1F/FCre+ and control mice. The intensity–response data were fitted to the Naka–Rushton hyperbolic function:
 |
where R is the response amplitude, Rmax is the maximal response amplitude, I is the flash intensity, n is the Hill coefficient (exponent), and I1/2 is the half-saturating light intensity. Statistical significance was determined using an independent two-tailed Student's t test comparing PhLP1F/FCre+ and PhLP1+/+Cre+ mice with an accepted significance level of p < 0.05.
Single-cell suction recordings.
PhLP1F/FCre+ and control mice were dark adapted overnight and killed by CO2 asphyxiation. Retinas were removed under infrared illumination, chopped into small pieces, and transferred to a perfusion chamber located on the stage of an inverted microscope. A single rod outer segment was drawn into a glass microelectrode filled with a solution containing the following (in mm): 140 NaCl, 3.6 KCl, 2.4 MgCl2, 1.2 CaCl2, 3 HEPES, pH 7.4, 0.02 EDTA, and 10 glucose. The perfusion Locke's solution containing (in mm) 112.5 NaCl, 3.6 KCl, 2.4 MgCl2, 1.2 CaCl2, 10 HEPES, pH 7.4, 20 NaHCO3, 3 Na succinate, 0.5 Na glutamate, 0.02 EDTA, and 10 glucose was bubbled with a 95% O2/5% CO2 mixture and heated to 37–38°C.
Light stimuli were applied as 20 ms test flashes of calibrated 500 nm light. Photoresponses were amplified, low-pass filtered (30 Hz, eight-pole Bessel), and digitized (1 kHz). Integration time (Tintegr) was calculated as the integral of the dim flash response with the transient peak amplitude normalized to unity. Normalized dim flash fractional sensitivity (Sf(n)) was calculated from the linear region of the intensity–response curve as the ratio of the response amplitude to flash strength and then normalized by the amplitude of the saturating response. Half-saturating light intensity (I1/2) was calculated from the intensity–response relationship as the test flash intensity required to produce a response of half maximal amplitude. The intensity–response relationships were fitted to the Naka–Rushton hyperbolic function as described above. The time constant for the dim flash response recovery (τrec) was derived from the best single-exponential fit to the late declining phase of the response. The dominant recovery time constant (τD) was determined from supersaturating flashes (Pepperberg et al., 1992) by using a 10% criterion for photocurrent recovery from saturation. The amplification of the rod phototransduction cascade was evaluated from test flash intensities that produced identical rising phases of dim flash responses. Data were analyzed with Clampfit 10.2 and Origin 8.5 software. Statistical significance was determined using an independent two-tailed Student's t test comparing PhLP1F/FCre+ and PhLP1+/+Cre+ mice with an accepted significance level of p < 0.05.
Results
Progressive retinal degeneration in PhLP1-deficient mice
To ablate PhLP1 activity in rod photoreceptor cells, we generated a conditional allele of the Phlp1 gene with loxP sites flanking exons 2 and 3 (Fig. 1A, sketch 4). This floxed-Phlp1 allele (PhLP1F) was detected by PCR, with a 704 base pair PCR product for the Phlp1F allele and a 600 bp product for the wild-type allele (Fig. 1B). The Phlp1F gene was specifically disrupted in rod photoreceptor cells by generating mice that were homozygous for the Phlp1F allele and heterozygous for the rod-specific Cre driver, iCre75 (Li et al., 2005). After Cre-mediated recombination in rods, the disrupted gene lacks exons 2 and 3 (Fig. 1A, sketch 5) and is thus missing the translation initiation site in exon 2. PhLP1 was nearly undetectable in immunoblots of purified rod outer segment preparations from homozygous mutant PhLP1-Cre mice (PhLP1F/FCre+), while it was clearly present in outer segment preparations from heterozygous mutant (PhLP1F/+Cre+) or wild-type (PhLP1+/+Cre+) mice (Fig. 1C). The loss of PhLP1 expression in rods was confirmed by immunolocalization of PhLP1 in retinal cryosections. Wild-type and PhLP1F/+Cre+ retinas showed PhLP1 expression in the inner segment and nuclear region of the photoreceptors as well as in the cells of the inner retina, whereas PhLP1 expression was largely lost in the photoreceptor layer but not in the inner retina of the homozygous knock-out mice (Fig. 1D). Weak residual PhLP1 staining could be observed in the photoreceptor layer and likely stems from PhLP1 expression in cones. Together, these data demonstrate that PhLP1 was specifically deleted from rod photoreceptors.
Cre recombination in iCre75 mice starts at approximately postnatal day 7 and is complete by postnatal day 18 (Li et al., 2005). Accordingly, loss of PhLP1 began over this same time period and reached an 80% reduction in whole retinal extracts by 35 d (Fig. 1E). At this point, the 20% residual PhLP1 in the retina extracts must come from the cells of the inner retina because it was no longer found in the rods (Fig. 1C). The loss of PhLP1 in rods resulted in progressive photoreceptor degeneration, as measured by the decrease in photoreceptor nuclei with age (Fig. 2A). There was no loss of photoreceptor nuclei at 1 month of age, but loss became apparent at 2 months and beyond, with half of the nuclei gone by 3–4 months. Nearly all the rod photoreceptors were lost by 9 months with just one layer of presumably cone nuclei left. Importantly, Cre expression alone did not cause photoreceptor degeneration over this time period, as shown by the PhLP1+/+Cre+ control mice.
Figure 2.

Progressive photoreceptor degeneration in PhLP1-deficient rods. A, Age-dependent loss of photoreceptors in PhLP1F/FCre+ mice. The number of photoreceptors was measured by counting rows of DAPI-stained nuclei in the outer nuclear layer from mice at increasing ages. Data points at 1 month represent the average ± SEM from three mice of each genotype. (Error bars are smaller than the symbols.) Other data points are from single mice. B, Retinal morphology of PhLP1F/FCre+ compared to wild-type mice at 5 weeks and 6 months. Retinal sections were contrasted using Richardson's stain (methylene blue-azure II). OS, Outer segment; IS, inner segment; ONL, outer nuclear layer; INL, inner nuclear layer.
Closer examination of the retinal morphology at 35 d showed relatively normal rod structures in the PhLP1F/FCre+ mice with an ∼25% shortening of the rod outer segment compared to the wild type (Fig. 2B), whereas at 6 months most of the photoreceptors were gone and the remaining rods were malformed with almost no outer segments (Fig. 2B). These morphological changes were similar over all regions of the retina. Thus, once Cre-induced disruption of the Phlp1 gene was complete, the rods began to degenerate. Consequently, experiments to assess the effects of PhLP1 deletion on G-protein expression and phototransduction were performed on 4- to 5-week-old mice after the loss of PhLP1, but before significant photoreceptor degeneration had occurred.
Deletion of PhLP1 dramatically reduces expression of Gt and Gβ5-RGS9 in rods
We next measured the effect of the loss of PhLP1 in rods on the expression of G-protein subunits and RGS9 in the retina by immunohistochemistry (Fig. 3). There was a significant reduction of Gαt1, Gβ1, Gγ1, Gβ4, Gβ5, and RGS9 in the photoreceptor layer of PhLP1F/FCre+ mice. In contrast, there was no loss of Gβ4 and Gβ5 in the inner nuclear layer of these animals, where PhLP1 expression was normal (Fig. 1D). In addition, rhodopsin expression in rods was unchanged, suggesting the effects of PhLP1 deletion were specific for G-protein and Gβ5-RGS9 complexes and not a result of photoreceptor degeneration. These results show that deletion of PhLP1 in rods severely reduces the expression of G-protein subunits and that the effect is confined to rods as expected.
Figure 3.
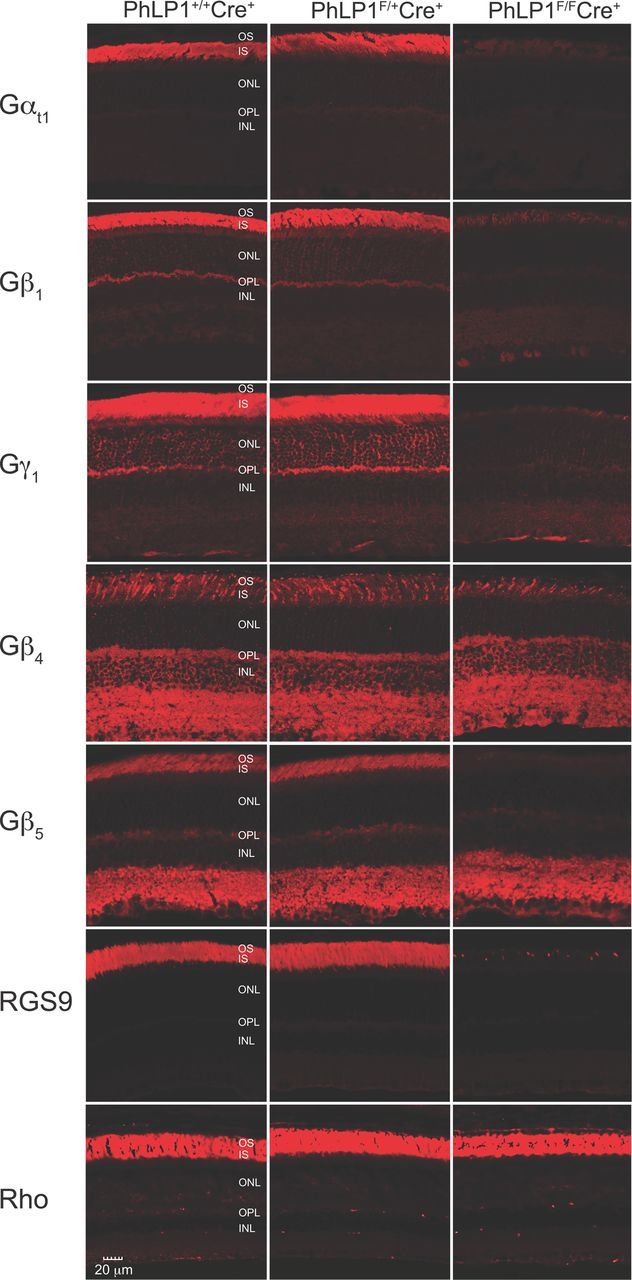
Immunolocalization of G-protein subunits in PhLP1-deficient rods. Retinal cross-sections from 1-month-old PhLP1+/+Cre+, PhLP1F/+Cre+, and PhLP1F/FCre+ mice were probed with primary antibodies to Gαt1, Gβ1, Gγ1, Gβ4, Gβ5, RGS9, and rhodopsin and detected with fluorescein-conjugated secondary antibodies.
To further assess the effects of PhLP1 deletion on protein expression, whole retinal extracts and isolated rod outer segment preparations were immunoblotted for G-protein subunits, RGS9, and other photoreceptor proteins (Fig. 4). PhLP1 deletion in rods caused an 80% decrease in the total amount of PhLP1 in the retina, with the residual levels reflecting the fraction of PhLP1 expressed in other retinal cell types (Fig. 4A,B). This decrease caused similar reductions in Gαt, Gβ1, Gβ4, Gγ1, Gβ5 long isoform (Gβ5-L), and RGS9. In contrast, levels of two G-protein β subunits that are not detectable in rods, Gβ3 and the Gβ5 short isoform (Gβ5-S), were not affected by the loss of PhLP1, confirming that this was a rod-specific effect. The largest changes were observed with Gβ5-L and RGS9, which both were decreased by 90%. The smallest change was a 68% decrease in Gβ1, which was unexpected given the proposed role of PhLP1 in Gβγ assembly (Lukov et al., 2005). These decreases stemmed from posttranslational events because quantitative RT-PCR analysis showed no changes in Gαt, Gβ1, Gγ1, Gβ5-L, or RGS9 mRNA levels upon PhLP1 deletion in rods (Fig. 4C).
Figure 4.

Protein expression in PhLP1-deficient rods. A, Immunoblots of whole retinal extracts for PhLP1, various G-protein subunits, RGS9, and R9AP are shown for 1-month-old PhLP1+/+Cre+, PhLP1F/+Cre+, and PhLP1F/FCre+ mice. B, Quantification of the immunoblot bands in A relative to the wild type. C, Levels of the indicated mRNAs in whole retinal extracts were determined by quantitative RT-PCR. The primer for PhLP1 detection was in exon 2 so no RT-PCR product would be expected from the Cre-deleted gene. D, Immunoblots of other rod and cone proteins are shown for 1-month-old PhLP1+/+Cre+, PhLP1F/+Cre+, and PhLP1F/FCre+ mice. E, Quantification of the immunoblot bands in D relative to the wild type. F, Immunoblots of rod outer segment preparations from 1-month-old PhLP1+/+Cre+, PhLP1F/+Cre+, and PhLP1F/FCre+ mice are shown for the indicated proteins. G, Quantification of the immunoblot bands in F relative to the wild type. In all panels, bars represent the average ± SEM from three to five different preparations. *p < 0.05; **p < 0.01; ***p < 0.001.
Expression levels of a number of other rod and cone proteins were also tested and none of them showed changes in the PhLP1F/FCre+ retinas except for Pdc, which was decreased by 70% (Fig. 4D,E). The lack of change in these proteins upon PhLP1 deletion shows that the effects were specific to the Gt and Gβ5-RGS9 complexes, supporting the notion from the morphometric analysis that rod degeneration was not yet apparent in 1-month-old mutant animals (Fig. 2). The decrease in Pdc expression was not unexpected as a similar decrease was observed in two different Gγ1 knock-out mouse lines where Gβ1γ1 expression was ablated (Lobanova et al., 2008; Kolesnikov et al., 2011). Moreover, Gβ1 and Gγ1 expression was decreased similarly in a Pdc knock-out mouse (Krispel et al., 2007). Thus, Pdc and Gβ1γ1 appear to stabilize each other in rods, and the loss of Gβ1γ1 upon PhLP1 deletion would be expected to result in a secondary loss of Pdc. Expression of other members of the Pdc family PhLP2A and PhLP3 was also tested. There was no detectable expression of PhLP2A in the retina (data not shown). In contrast, PhLP3 was found in the retina, but there was no change in its expression upon PhLP1 deletion (Fig. 4D,E). Similarly, expression of the CCTε subunit in the retina was unchanged with loss of PhLP1 as was expression of two important CCT substrates, β-actin and β-tubulin.
To assess more closely protein expression in the rods themselves, isolated rod outer segment preparations were also analyzed by immunoblotting (Fig. 4F,G). PhLP1 was reduced in PhLP1F/+Cre+ mice and was completely lost in PhLP1F/FCre+ mice. As in whole retinas, isolated outer segments showed significant reductions in Gαt1, Gβ1, Gβ4, Gγ1, Gβ5-L, RGS9, and Pdc. The magnitudes of these changes were similar to those observed in whole retinas except that Gβ5-L and RGS9 were almost undetectable in homozygous rods. By comparison, expression of rhodopsin, PDEγ, CCTε, and the CCT substrate β-actin was unchanged in the PhLP1 knock-out rods, again indicating that the effects of PhLP1 deletion were specific to G-proteins. A second CCT substrate, β-tubulin, showed a small but statistically significant decrease (23%), suggesting that CCT function was diminishing at 5 weeks of age in the absence of PhLP1. There was no detectible PhLP2A or PhLP3 in these preparations for any of the PhLP1 genotypes (data not shown), ruling out the possibility of compensation by other PhLP isoforms. Interestingly, R9AP expression was increased by nearly twofold in the mutant rods (Fig. 4F,G), perhaps as a compensatory response to preserve as much of the Gβ5-RGS9 complex as possible, given that R9AP is known to stabilize Gβ5-RGS9 (Keresztes et al., 2004).
Gβ1γ1 assembly is severely compromised in PhLP1-deficient rods
Of all the subunits of the Gt and Gβ5-RGS9 complexes, the Gβ1 level was the least affected (Fig. 4B,G). This result was surprising considering the strong dependence of Gβ1γx assembly on PhLP1 observed in cultured cells (Lukov et al., 2005; Howlett et al., 2009), and it raised the possibility that the remaining Gβ1 in rods might not be in a Gβγ complex. To address this issue, we determined the relative amount of Gβ1γx dimers in whole retina and in rod outer segment preparations by measuring Gβ1γx binding to Pdc, taking advantage of the fact that Pdc only binds Gβ in Gβγ complexes, while PhLP1 binds both Gβ-CCT and Gβγ complexes (McLaughlin et al., 2002; Lukov et al., 2005, 2006). Using this method, we found an 85% reduction in Pdc-associated Gβ1 in whole retinas and a 98% loss of Pdc-associated Gβ1 in rod outer segment preparations from PhLP1F/FCre+ mice (Fig. 5A,B), indicating that very little Gβγ dimer formation was occurring in PhLP1-depleted rods. The residual amount of Gβ1 associated with Pdc in whole retinas in this assay reflects the Gβ1 found in the inner retina (Lobanova et al., 2008), whose Gβγ assembly is not affected by PhLP1 deletion in the rods. The nearly complete loss of Gβ1γx dimers in the rod outer segment preparations indicates that the remaining 32% of Gβ1 observed in the rods of PhLP1F/FCre+ mice (Fig. 4G) was not assembled into Gβγ dimers. Previous in vitro work has shown that Gβ1 does not release from CCT in the absence of PhLP1 (Lukov et al., 2006), suggesting that the remaining Gβ1 was associated with CCT and unable to assemble with Gγ in the absence of PhLP1. To test this idea, the amount of Gβ1 bound to CCT in whole retina extracts was measured by coimmunoprecipitation (Fig. 5C). As predicted, there was a significant increase (fivefold) in Gβ1 bound to CCT in the PhLP1F/FCre+ retinas compared to the wild type (Fig. 5D). In addition, there was a twofold increase in Gβ1 bound to CCT in the heterozygous PhLP1F/+Cre+ retinas, suggesting that even a partial loss of PhLP1 (Fig. 4G) resulted in some accumulation of Gβ1 on CCT. Together, these data show that in the absence of PhLP1, Gβ1 accumulates on CCT and is unable to release and form Gβγ dimers.
Figure 5.

Gβγ dimer determination in PhLP1-deficient rods. The amount of Gβγ dimers in whole retina or rod outer segment preparations from 1-month-old PhLP1+/+Cre+, PhLP1F/+Cre+, and PhLP1F/FCre+ mice was determined in a Pdc-binding assay. A, The amount of Gβγ in Pdc precipitates was measured by blotting for Gβ1. B, Quantification of the Gβ1 bands in A relative to the wild type. C, The amount of Gβ1 and Gβ5 associated with CCT in whole retinas was determined by coimmunoprecipitation. CCT complexes were immunoprecipitated with an antibody to CCTε, and Gβ1 and Gβ5 in the immunoprecipitate were determined by immunoblotting. D, Quantification of the Gβ1/CCTε and Gβ5/CCTε band ratios relative to the wild type. In all panels, bars represent the average ± SEM from three to five different preparations. *p < 0.05; ***p < 0.001.
In the case of Gβ5, PhLP1 has been reported to have the opposite effect. Instead of mediating release from CCT, PhLP1 was found to increase Gβ5 binding to CCT (Howlett et al., 2009). To test this possibility, the coimmunoprecipitation of Gβ5-L with CCT was also examined. Indeed, there was a 50% decrease in Gβ5 binding to CCT in retinas from PhLP1F/FCre+ mice compared to the wild type (Fig. 5D). This result emphasizes the differences in the role of PhLP1 in Gβγ and Gβ5-RGS9 assembly in vivo (see Fig. 8).
Figure 8.

Effects of PhLP1 deletion on Gβγ and Gβ5-RGS9 assembly. A, B, The scheme summarizes the proposed mechanisms for G-protein heterotrimer (A) and Gβ5-RGS9-R9AP assembly (B) and the effects of PhLP1 deletion on the assembly process (shown in red).
Scotopic vision is largely deteriorated in the absence of rod PhLP1
The substantial loss of Gt and Gβ5-RGS9 expression in rods upon PhLP1 deletion would be expected to have a profound effect on visual performance. To test this possibility, we compared the visual acuity and contrast sensitivity of PhLP1F/FCre+ and wild-type mice by measuring optomotor head-turning responses to rotating grating stimuli (Prusky et al., 2004; Umino et al., 2008). We found that PhLP1F/FCre+ mice had a threefold lower visual acuity than wild-type animals under dim light (scotopic) conditions in which vision is dominated by rods (Fig. 6A). Contrast sensitivity of the PhLP1F/FCre+ mice was even more impaired (by10-fold) under scotopic conditions (Fig. 6B). However, under bright light (photopic) conditions in which vision is dominated by cones, the PhLP1F/FCre+ mice performed just as well as wild-type controls (Fig. 6C,D). These results demonstrate that rod vision is severely impaired in the rod-specific PhLP1 knock-out mice as would be expected from the observed reductions in Gt and Gβ5-RGS9 expression. The fact that photopic vision was normal also indicates that the loss of rod function had not affected cone function in the predegenerate retina.
Figure 6.
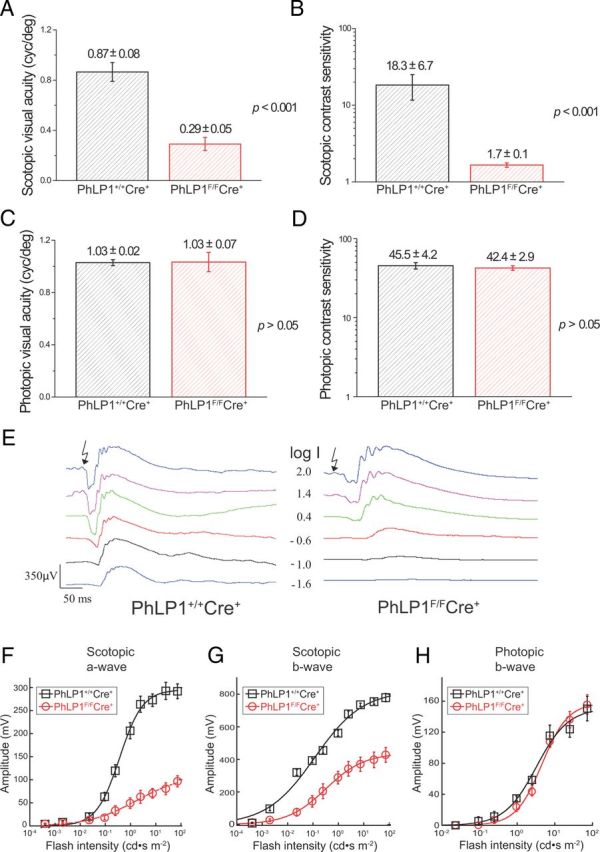
Impairment of scotopic vision in PhLP1-deficient mice. Data were derived from mouse optomotor responses to rotating gratings under both scotopic (−4.45 log cd m−2) and photopic (1.85 log cd m−2) background illumination conditions. In contrast sensitivity measurements, Ft was fixed at its optimal value of 0.8 Hz for both conditions. A, Scotopic visual acuity. B, Scotopic contrast sensitivity. C, Photopic visual acuity. D, Photopic contrast sensitivity. All data are means ± SEM. E, Families of ERG responses from 1-month-old PhLP1+/+Cre+ and PhLP1F/FCre+ mice are shown. Light intensity values (I) are in log candela seconds per square meter. F, Intensity–response relationships for scotopic a-waves from PhLP1+/+Cre+ (n = 7) and PhLP1F/FCre+ (n = 12) mice. Data were fit to a hyperbolic Naka–Ruston function that yielded fit parameters shown in Table 1. G, Intensity–response relationships for scotopic b-waves. Data were fit as in F, and fit parameters are shown in Table 1. H, Intensity–response relationships for photopic b-waves from five mice are shown. Data were fit as in F.
To further assess visual function in the rod PhLP1 knock-out mice, a full-field ERG analysis was performed. Scotopic ERG responses to a series of light flashes of increasing intensity showed that deletion of PhLP1 in rods caused a sizeable reduction in light sensitivity and response amplitude (Fig. 6E). An intensity–response relationship for the scotopic a-wave showed a 17-fold desensitization and a nearly twofold decrease in maximal response amplitude in PhLP1F/FCre+ mice (Fig. 6F; Table 1). The changes were similar for the scotopic b-wave, which is dominated by rod ON bipolar cells (Fig. 6G; Table 1). In contrast, the intensity–response relationship for cones, as measured by the photopic ERG b-wave, was unaffected by the loss of PhLP1 (Fig. 6H). These ERG results are consistent with the optometry data, confirming that the loss of Gt upon PhLP1 deletion in rods severely impairs rod but not cone photoresponses.
Table 1.
ERG intensity–response parameters
| Scotopic a-wave |
Scotopic b-wave |
|||||
|---|---|---|---|---|---|---|
| I1/2 (cd · s m−2) | Rmax (μV) | n (I1/2) | I1/2 (cd · s m−2) | Rmax (μV) | n (I1/2) | |
| PhLP1+/+Cre+ (n = 7) | 0.60 ± 0.11 | 337 ± 26 | 0.87 ± 0.10 | 0.14 ± 0.04 | 806 ± 48 | 0.48 ± 0.03 |
| PhLP1F/FCre+ (n = 12) | 10.7 ± 3.1** | 178 ± 23*** | 0.59 ± 0.05* | 0.86 ± 0.19** | 494 ± 41*** | 0.90 ± 0.10** |
Half-saturating light intensity (I1/2), maximal response amplitudes (Rmax), and Hill coefficients (n) from fits of the data in Figure 6, F and G, are given for the scotopic a-wave and b-wave for PhLP1+/+Cre+ and PhLP1F/FCre+ mice. Values are means ± SEM.
*p < 0.05;
**p < 0.01;
***p < 0.001 all compared to wild type values.
Phototransduction is compromised in PhLP1-deficient rods
To examine more closely the effect of PhLP1 deletion on the phototransduction cascade, we measured photoresponses of individual dark-adapted mouse rods using suction electrode recordings. Responses to a series of light flashes of increasing intensity were measured (Fig. 7A,B). Several conspicuous features of the responses are noteworthy. First, sensitivity was markedly reduced in the PhLP1-deficient rods. This difference can be seen by comparing the responses to 49.5 photons μm−2 (marked in red) from the wild-type and PhLP1F/FCre+ rods. Stimulus–response curves further illustrate this decrease in sensitivity (Fig. 7C), exhibiting a 12-fold increase in the intensity required to produce a half-saturating response (I1/2; Table 2). This reduction in sensitivity is similar to what was seen in the ERG responses and can be attributed to the loss of Gt expression in the PhLP1-deficient rods. Second, there was an ∼25% decrease in the average response amplitude from 14.4 to 10.6 pA in PhLP1-depleted rods (Idark; Table 2), which most likely stems from the shortening of the rod outer segments to a similar extent in the PhLP1F/FCre+ mice (Fig. 2B).
Figure 7.
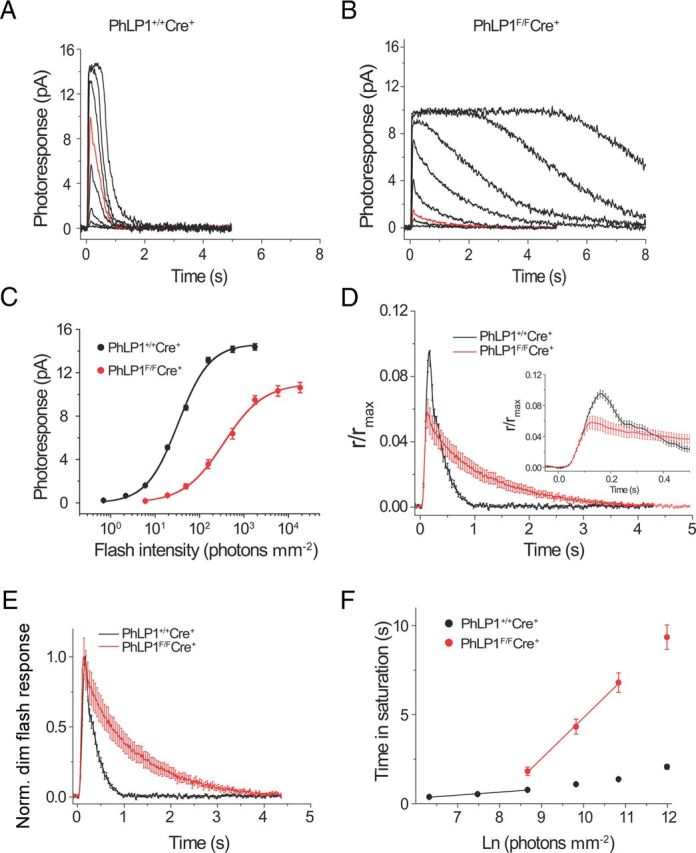
Single-cell photoresponses of PhLP1-deficient rods. A, B, Representative families of flash responses from 1-month-old PhLP1+/+Cre+ (A) and PhLP1F/FCre+ (B) mice are shown. Test flashes of 500 nm light with intensities of 0.7, 2.2, 6.0, 19.0, 49.5, 157, 557, and 1764 photons μm−2 (for control rods) or 6.0, 19.0, 49.5, 157, 557, 1764, 5811, and 18415 photons μm−2 (for PhLP1F/FCre+ rods) were delivered at time 0. The red traces show responses to an identical light intensity (49.5 photons μm−2). C, Intensity–response functions for rods from PhLP1+/+Cre+ (n = 22) and PhLP1F/FCre+ (n = 23) mice. Symbols represent the average ± SEM. Data were fit with a hyperbolic Naka–Rushton function and fit parameters are shown in Table 2. D, Amplification of the phototransduction cascade in mouse rods. Population-averaged dim flash responses to light intensities of 49.5 photons μm−2 for PhLP1F/FCre+ rods and 6.0 photons μm−2 for wild-type rods were normalized to their corresponding maximum dark currents, rmax. Then the PhLP1F/FCre+ fractional response was scaled down by the factor of 2.2 to make its initial rising phase coincide with that of wild-type response. The light intensity corresponding to this scaled PhLP1F/FCre+ response was thus determined as 49.5/2.2 = 22.5 photons μm−2. The inset shows the rising phase of the responses on an extended time scale. E, Kinetics of the dim flash responses. Normalized population-averaged dim flash responses to light intensities of 6.0 photons μm−2 for PhLP1+/+Cre+ (n = 22) and 49.5 photons μm−2 for PhLP1F/FCre+ (n = 23), demonstrating the decelerated photoresponse inactivation in PhLP1-deficient rods. F, Determination of the dominant time constant of recovery (τD) from a series of supersaturating flashes. Linear fits throughout the data yielded τD values indicated in Table 2. Values are means ± SEM.
Table 2.
Rod single-cell recording parameters
| Idark (pA) | Tpeak (ms) | Tintegr (ms) | Sf(n) (μm2/ph) | I1/2 (ph/μm2) | n (I1/2) | τrec (ms) | τD (ms) | |
|---|---|---|---|---|---|---|---|---|
| PhLP1+/+Cre+ (n = 22) | 14.4 ± 0.3 | 161.0 ± 2.7 | 268.7 ± 12.3 | 1.6 × 10−2 ± 7.7 × 10−4 | 32.2 ± 1.1 | 1.31 ± 0.02 | 220.7 ± 17.0 | 169.1 ± 6.7 |
| PhLP1F/FCre+ (n = 23) | 10.6 ± 0.5*** | 125.0 ± 4.5*** | 915.5 ± 63.7*** | 2.9 × 10−3 ± 4.4 × 10−4*** | 378.7 ± 59.5*** | 1.24 ± 0.05 NS | 1107.9 ± 95.7*** | 2354.2 ± 168.8*** |
The following parameters are from the data in Figure 7. Idark, Dark current measured from saturated responses. Time-to-peak (Tpeak) and integration time (Tintegr) refer to responses whose amplitudes were <0.2 Idark and fell within the linear range. Sf(n), normalized dim flash fractional sensitivity (amplitude of dim flash response divided by flash strength and then normalized for the amplitude of saturating response); I1/2, half-saturating light intensity; n (I1/2), Hill coefficient in the Naka–Rushton equation; τrec, time constant of single-exponential decay of dim flash response recovery phase; τD, dominant time constant of recovery after supersaturating flashes determined from the linear fit to time in saturation versus intensity semilog plot (the “Pepperberg constant”). Values are means ± SEM.
NS, p > 0.05 (not significant);
***p < 0.001 (all compared to wild-type values).
Examining the flash response kinetics revealed two additional major effects of the loss of PhLP1 on rod phototransduction. First, the amplification rate of phototransduction was evaluated by directly comparing the light intensities required to produce identical response activation phases. We compared population-averaged fractional responses in the linear range that corresponded to 49.5 photons μm−2 for PhLP1F/FCre+ rods and 6.0 photons μm−2 for PhLP1+/+Cre+ rods. To obtain the best match of the rising phases, the fractional dim flash PhLP1F/FCre+ response required additional scaling down by a factor of 2.2 (Fig. 7D). The ratio of the two light intensities corrected for the scaling factor (49.5 photons μm−2/2.2 scaling factor/6.0 photons μm−2) yielded a 3.8-fold reduction in the signal amplification rate in the PhLP1-deficient rods. This reduction corresponded closely to the decrease in Gαt levels observed in the mutant rods (Fig. 4G).
The second and the most striking change in the response kinetics in PhLP1-deficient rods was their substantially decelerated falling phase. The dim flash recovery time constant τrec was increased by fivefold, on average, from 0.22 to 1.1 s (Fig. 7E; Table 2), and the dominant time constant of recovery from saturating flashes (τD), determined from a Pepperberg plot, was increased by 14-fold, on average, from 0.17 to 2.4 s (Fig. 7F; Table 2). This conspicuously slower recovery rate can be readily explained by the 20-fold decrease in Gβ5-RGS9 expression in the PhLP1-deficient rods (Fig. 4G) and is qualitatively consistent with the slow recovery rates observed in Gβ5 and RGS9 knock-out mice (Chen et al., 2000; Krispel et al., 2003).
Discussion
PhLP1 and Gt expression
The specific deletion of PhLP1 from mouse rod photoreceptor cells demonstrates an essential role for PhLP1 in maintaining normal photoreceptor physiology. The absence of PhLP1 caused a dramatic decrease in the expression of all three Gt subunits and the Gβ5-RGS9 complex, resulting in a peculiar perturbation of the phototransduction cascade coupled with progressive photoreceptor degeneration. The loss of Gt subunits can be directly attributed to an inability of Gβ and Gγ to form heterodimers because almost no Gβγ was found in PhLP1-depleted rods (Fig. 5A,B). In addition to the loss of Gβ1 and Gγ1, there was also a fourfold decrease in Gαt expression. This decrease in Gαt is not surprising since it was also observed in two different Gγ1 knock-out mouse lines that were also deficient in Gβ1γ1 (Lobanova et al., 2008; Kolesnikov et al., 2011). It is interesting to note that Gβ1 expression was decreased by only 68% in PhLP1-deficient rods (Fig. 4G), yet almost no Gβ1γx dimers were detectible (Fig. 5A,B). It appears that the residual Gβ1 accumulates on CCT (Fig. 5C,D). Based on these results, a scheme depicting the effects of the loss of PhLP1 on Gβγ assembly is shown in Figure 8A. In the absence of PhLP1, Gβ1 cannot release from CCT to assemble with Gγ1. The resulting uncomplexed Gαt and Gγ1 subunits either remain bound to chaperones or free in the cytosol where they are targeted for degradation by the proteasome or other cellular proteases.
PhLP1 and retinal degeneration
The accumulation of Gβ on CCT is the probable cause for the progressive retinal degeneration observed in the PhLP1F/FCre+ mice. There are several reasons to suspect that this is the case. First, like the PhLP1F/FCre+ mice, both of the previously characterized Gγ1 knock-out mouse strains exhibited photoreceptor degeneration and showed residual Gβ1 in rods before the onset of degeneration (Lobanova et al., 2008; Kolesnikov et al., 2011). Second, transgenic overexpression in rods of an N-terminally truncated PhLP1 isoform, which acts as a dominant-negative inhibitor of Gβγ assembly and causes considerable accumulation of Gβ1 on CCT (Lukov et al., 2006), resulted in very rapid photoreceptor death (Posokhova et al., 2011). Third, the accumulation of Gβ1 on CCT would be expected to trigger degeneration by compromising the ability of CCT to fold other protein substrates that are essential for maintaining photoreceptor structure and function. The small decrease in β-tubulin expression (Fig. 4G) and the corresponding shortening of the rod outer segment (Fig. 2B) in the knock-out mice suggest that CCT function is beginning to deteriorate at 1 month of age. There are mechanisms for the clearance of CCT substrates that cannot be folded by shuttling them to the proteasome for degradation (McClellan et al., 2005). Despite these protective mechanisms, the inability of Gβ1 to release from CCT in the absence of PhLP1 appears to eventually overwhelm the chaperonin system and compromise CCT function to the point that photoreceptor degeneration occurs.
PhLP1 and phototransduction
The visual behavior tests and electrophysiological measurements further support the idea that Gt function is severely impaired in the absence of PhLP1. The fourfold decrease in the amplification rate in mutant rods (Fig. 7D) closely corresponds to their reduced Gαt levels (Fig. 4G). This reduction is, however, far less than the ∼50-fold decrease in Gβ1γ1 levels in rods (Fig. 5). The preservation of this higher-than-expected phototransduction gain could likely be attributed to catalytic action of the residual Gβ1γ1 in which each Gβ1γ1 dimer participates in the activation of multiple Gαt subunits by photo-activated rhodopsin. This observation was first described in vitro (Fung, 1983), but evidence for a catalytic function of Gβγ in vivo has been lacking. A catalytic effect of the residual Gβγ could explain why the reduction in signal amplification in PhLP1-depleted rods was less than the 33- to 40-fold drop observed in the two Gγ1 knock-out mouse lines, which had similar amounts of Gαt as our PhLP1F/FCre+ mouse but no Gβ1γ1 (Lobanova et al., 2008; Kolesnikov et al., 2011). In those studies, the weak residual signal was attributed to inefficient Gαt coupling to light-activated rhodopsin in the absence of Gβ1γ1. In comparison, the ∼10-fold higher signal amplification in PhLP1-depleted rods must arise from more than just Gβγ-independent Gαt activation. It seems unlikely that compensation by Gβγ dimers other than Gβ1γ1 is responsible for the remaining signaling because all Gβγ combinations depend on PhLP1 for assembly (Howlett et al., 2009), an observation that is consistent with the reduced expression of Gβ4 in addition to Gβ1 in the absence of PhLP1 (Fig. 4). Whatever the source of this signaling, it is clear that the loss of PhLP1 in rods has a profound effect on rod phototransduction through the impairment of Gβγ assembly.
PhLP1 and Gβ5-RGS9 expression
The most surprising result of PhLP1 deletion in rods is the 95% loss of Gβ5-L and RGS9 proteins (Fig. 4F,G) and the corresponding slow rate of photoresponse recovery following both dim and saturating flashes (Fig. 7E,F). This loss of expression must be associated with an inability to form Gβ5-RGS9 complexes because genetic deletion of either subunit results in complete loss of the other in rods (Chen et al., 2000, 2003). Thus, if the Gβ5-RGS9 dimer cannot form, the subunits are degraded. A previous study investigating the role of PhLP1 in the assembly of Gβ5 and RGS7, another member of the R7 family of RGS proteins, suggested that the formation of the Gβ5-RGS7 complex was only partially dependent on PhLP1 (Howlett et al., 2009). In that study, an 80% siRNA-mediated PhLP1 knockdown resulted in only a twofold reduction in assembly rate of overexpressed Gβ5 and RGS7. In contrast, the 95% loss of Gβ5-L and RGS9 in PhLP1-deficient rods indicates that PhLP1 is essential for Gβ5-RGS9 assembly in vivo. Thus, genetic deletion of PhLP1 in an otherwise normal physiological environment in rods was needed to demonstrate the very strong dependence of Gβ5-RGS9 dimer formation on PhLP1.
A scheme showing the effects of PhLP1 deletion on Gβ5-RGS9-R9AP complex formation is shown in Figure 8B. The scheme shows that in the absence of PhLP1, nascent Gβ5 cannot form a productive folding complex with CCT. This idea stems from the fact that PhLP1 increased the binding of Gβ5 to CCT by tenfold in overexpression experiments (Howlett et al., 2009). This stabilization of the Gβ5-CCT interaction by PhLP1 is in stark contrast to the effects of PhLP1 on the Gβ1-CCT interaction in which PhLP1 is required for the release of folded Gβ1 from CCT (Fig. 8A). The inability to fold Gβ5 in the absence of PhLP1 leaves RGS9 without its obligate binding partner, resulting in its rapid degradation.
The loss of Gβ5-RGS9 caused a dramatic decrease in the photoresponse recovery rate as reflected in the τrec and τD values of 1.1 and 2.4 s, respectively. Interestingly, these recovery rates are not as slow as in the RGS9 knock-out mouse, with τrec and τD values of 2.6 and 9.0 s, respectively (Chen et al., 2000). These differences suggest that there are residual Gβ5-RGS9 complexes in the PhLP1-deficient rods acting on the remaining Gαt to set the recovery rate. The residual Gβ5-RGS9 may arise from minimal Gβ5-RGS9 assembly in the absence of PhLP1.
In summary, the deletion of PhLP1 results in an inability of G-protein subunits to form Gβγ and Gβ5-RGS9 dimers, demonstrating conclusively that PhLP1 plays an essential role in the assembly of these complexes in vivo. This inability to form dimers causes a marked reduction of Gt and Gβ5-RGS9 complexes in PhLP1-deficient rods, which results in severely compromised scotopic vision from substantially reduced rod light sensitivity and greatly retarded photoresponse recovery. The presence of unassembled G-protein subunits causes stress to the CCT chaperonin and possibly other chaperone systems in rods, eventually resulting in photoreceptor death. These findings emphasize the importance of molecular chaperones in the assembly of obligate dimers such as Gβγ and Gβ5-RGS9 and have important implications in all neurons and other cell types where G-protein signaling plays a vital role in cell physiology.
Footnotes
This work was supported by National Institutes of Health Grants EY012287 (B.M.W.); EY08123, EY019298, and EY014800-039003 (W.B.); EY019312 and EY021126 (V.J.K.); and EY013811 (C.-K.C.). Further funding came from a Foundation Fighting Blindness grant to W.B. We recognize Susan Tamowski and the University of Utah Core Transgenic/Gene-targeting Mouse Facility for excellent technical assistance in generating Phlp1F germ line chimeras and for providing the ROSA26-Flp mice. We thank Sen Wu and Houbin Zhang of the University of Utah for assistance with the construction of the targeting vector for the PhLP1 gene and immunohistochemistry, respectively.
The authors declare no competing financial interests.
References
- Betke KM, Wells CA, Hamm HE. GPCR mediated regulation of synaptic transmission. Prog Neurobiol. 2012;96:304–321. doi: 10.1016/j.pneurobio.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CK, Burns ME, He W, Wensel TG, Baylor DA, Simon MI. Slowed recovery of rod photoresponse in mice lacking the GTPase accelerating protein RGS9-1. Nature. 2000;403:557–560. doi: 10.1038/35000601. [DOI] [PubMed] [Google Scholar]
- Chen CK, Eversole-Cire P, Zhang H, Mancino V, Chen YJ, He W, Wensel TG, Simon MI. Instability of GGL domain-containing RGS proteins in mice lacking the G protein beta-subunit Gbeta5. Proc Natl Acad Sci U S A. 2003;100:6604–6609. doi: 10.1073/pnas.0631825100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CK, Woodruff ML, Chen FS, Chen Y, Cilluffo MC, Tranchina D, Fain GL. Modulation of mouse rod response decay by rhodopsin kinase and recoverin. J Neurosci. 2012;32:15998–16006. doi: 10.1523/JNEUROSCI.1639-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung BK. Characterization of transducin from bovine retinal rod outer segments. J Biol Chem. 1983;258:10495–10502. [PubMed] [Google Scholar]
- Han SK, Simon MI. Intracellular signaling and the origins of the sensations of itch and pain. Sci Signal. 2011;4:pe38. doi: 10.1126/scisignal.2002353. [DOI] [PubMed] [Google Scholar]
- Higgins JB, Casey PJ. In vitro processing of recombinant G protein gamma subunits. Requirements for assembly of an active beta gamma complex. J Biol Chem. 1994;269:9067–9073. [PubMed] [Google Scholar]
- Howlett AC, Gray AJ, Hunter JM, Willardson BM. Role of molecular chaperones in G protein β5/regulator of G protein signaling dimer assembly and G protein βγ dimer specificity. J Biol Chem. 2009;284:16386–16399. doi: 10.1074/jbc.M900800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humrich J, Bermel C, Bünemann M, Härmark L, Frost R, Quitterer U, Lohse MJ. Phosducin-like protein regulates G-protein βγ folding by interaction with TCP-1α. J Biol Chem. 2005;280:20042–20050. doi: 10.1074/jbc.M409233200. [DOI] [PubMed] [Google Scholar]
- Keresztes G, Martemyanov KA, Krispel CM, Mutai H, Yoo PJ, Maison SF, Burns ME, Arshavsky VY, Heller S. Absence of the RGS9.Gbeta5 GTPase-activating complex in photoreceptors of the R9AP knockout mouse. J Biol Chem. 2004;279:1581–1584. doi: 10.1074/jbc.C300456200. [DOI] [PubMed] [Google Scholar]
- Kimple AJ, Bosch DE, Giguère PM, Siderovski DP. Regulators of G-protein signaling and their Galpha substrates: promises and challenges in their use as drug discovery targets. Pharmacol Rev. 2011;63:728–749. doi: 10.1124/pr.110.003038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knol JC, Engel R, Blaauw M, Visser AJ, van Haastert PJ. The phosducin-like protein PhLP1 is essential for Gβγ dimer formation in dictyostelium discoideum. Mol Cell Biol. 2005;25:8393–8400. doi: 10.1128/MCB.25.18.8393-8400.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesnikov AV, Rikimaru L, Hennig AK, Lukasiewicz PD, Fliesler SJ, Govardovskii VI, Kefalov VJ, Kisselev OG. G-protein betagamma-complex is crucial for efficient signal amplification in vision. J Neurosci. 2011;31:8067–8077. doi: 10.1523/JNEUROSCI.0174-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krispel CM, Chen CK, Simon MI, Burns ME. Prolonged photoresponses and defective adaptation in rods of Gbeta5−/− mice. J Neurosci. 2003;23:6965–6971. doi: 10.1523/JNEUROSCI.23-18-06965.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krispel CM, Sokolov M, Chen YM, Song H, Herrmann R, Arshavsky VY, Burns ME. Phosducin regulates the expression of transducin βγ subunits in rod photoreceptors and does not contribute to phototransduction adaptation. J Gen Physiol. 2007;130:303–312. doi: 10.1085/jgp.200709812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BY, Thulin CD, Willardson BM. Site-specific phosphorylation of phosducin in intact retina—dynamics of phosphorylation and effects on G protein βγ dimer binding. J Biol Chem. 2004;279:54008–54017. doi: 10.1074/jbc.M405669200. [DOI] [PubMed] [Google Scholar]
- Li S, Chen D, Sauvé Y, McCandless J, Chen YJ, Chen CK. Rhodopsin-iCre transgenic mouse line for Cre-mediated rod-specific gene targeting. Genesis. 2005;41:73–80. doi: 10.1002/gene.20097. [DOI] [PubMed] [Google Scholar]
- Lobanova ES, Finkelstein S, Herrmann R, Chen YM, Kessler C, Michaud NA, Trieu LH, Strissel KJ, Burns ME, Arshavsky VY. Transducin γ-subunit sets expression levels of α- and β-subunits and is crucial for rod viability. J Neurosci. 2008;28:3510–3520. doi: 10.1523/JNEUROSCI.0338-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukov GL, Hu T, McLaughlin JN, Hamm HE, Willardson BM. Phosducin-like protein acts as a molecular chaperone for G protein βγ dimer assembly. EMBO J. 2005;24:1965–1975. doi: 10.1038/sj.emboj.7600673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukov GL, Baker CM, Ludtke PJ, Hu T, Carter MD, Hackett RA, Thulin CD, Willardson BM. Mechanism of assembly of G protein βγ subunits by protein kinase CK2-phosphorylated phosducin-like protein and the cytosolic chaperonin complex. J Biol Chem. 2006;281:22261–22274. doi: 10.1074/jbc.M601590200. [DOI] [PubMed] [Google Scholar]
- Martemyanov KA, Arshavsky VY. Biology and functions of the RGS9 isoforms. Progress in molecular biology and translational science. 2009;86:205–227. doi: 10.1016/S1877-1173(09)86007-9. [DOI] [PubMed] [Google Scholar]
- McClellan AJ, Scott MD, Frydman J. Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell. 2005;121:739–748. doi: 10.1016/j.cell.2005.03.024. [DOI] [PubMed] [Google Scholar]
- McLaughlin JN, Thulin CD, Hart SJ, Resing KA, Ahn NG, Willardson BM. Regulatory interaction of phosducin-like protein with the cytosolic chaperonin complex. Proc Natl Acad Sci U S A. 2002;99:7962–7967. doi: 10.1073/pnas.112075699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepperberg DR, Cornwall MC, Kahlert M, Hofmann KP, Jin J, Jones GJ, Ripps H. Light-dependent delay in the falling phase of the retinal rod photoresponse. Vis Neurosci. 1992;8:9–18. doi: 10.1017/S0952523800006441. [DOI] [PubMed] [Google Scholar]
- Posokhova E, Song H, Belcastro M, Higgins L, Bigley LR, Michaud NA, Martemyanov KA, Sokolov M. Disruption of the chaperonin containing TCP-1 function affects protein networks essential for rod outer segment morphogenesis and survival. Mol Cell Proteomics. 2011;10:M110.000570. doi: 10.1074/mcp.M110.000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusky GT, Alam NM, Beekman S, Douglas RM. Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Invest Ophthalmol Vis Sci. 2004;45:4611–4616. doi: 10.1167/iovs.04-0541. [DOI] [PubMed] [Google Scholar]
- Thulin CD, Howes K, Driscoll CD, Savage JR, Rand TA, Baehr W, Willardson BM. The immunolocalization and divergent roles of phosducin and phosducin-like protein in the retina. Mol Vis. 1999;5:40. [PubMed] [Google Scholar]
- Umino Y, Solessio E, Barlow RB. Speed, spatial, and temporal tuning of rod and cone vision in mouse. J Neurosci. 2008;28:189–198. doi: 10.1523/JNEUROSCI.3551-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellano CP, Lee SE, Dudek SM, Hepler JR. RGS14 at the interface of hippocampal signaling and synaptic plasticity. Trends Pharmacol Sci. 2011;32:666–674. doi: 10.1016/j.tips.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells CA, Dingus J, Hildebrandt JD. Role of the chaperonin CCT/TRiC complex in G protein betagamma-dimer assembly. J Biol Chem. 2006;281:20221–20232. doi: 10.1074/jbc.M602409200. [DOI] [PubMed] [Google Scholar]
- Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol Rev. 2005;85:1159–1204. doi: 10.1152/physrev.00003.2005. [DOI] [PubMed] [Google Scholar]
- Wu S, Ying G, Wu Q, Capecchi MR. A protocol for constructing gene targeting vectors: generating knockout mice for the cadherin family and beyond. Nature protocols. 2008;3:1056–1076. doi: 10.1038/nprot.2008.70. [DOI] [PubMed] [Google Scholar]