

Abstract
This chapter provides a method for reprogramming human dermal fibroblasts into induced pluripotent stem cells (iPSCs) using three lentiviruses containing cDNAs for OCT4 and SOX2, KLF4 and C-MYC, and NANOG and LIN28, respectively. Lentiviral vectors are based on the human immunodeficiency virus (HIV) and provide an effective means for the delivery, integration, and expression of exogenous genes in mammalian cells. Lentiviruses are attractive gene delivery vehicles as they are able to infect both proliferating and nonproliferating cells. Lentiviruses stably integrate into the genome without incurring cellular toxicity and can maintain sustained transgene expression during prolonged host cell proliferation and differentiation. In this protocol, we describe how to prepare lentiviruses, stably transduce human fibroblasts, and identify bona fide iPSC colonies based on morphological similarity to human embryonic stem cell (ESC) colonies and live-cell immunological staining using cell-surface markers of human PSCs such as Tra-1-60 and Tra-1-81.
Keywords: induced pluripotent stem cells, lentivirus, live-cell imaging
1. Introduction
It is now possible to induce pluripotency in human somatic cells through the ectopic expression of a small number of transcription factors. The initial breakthrough was reported by the Yamanaka group at Kyoto University in 2006 when they demonstrated that induced expression of only four transcription factors, Oct3/4, Sox2, c-Myc, and Klf4, in mouse fibroblast cells resulted in the formation of embryonic stem cell (ESC)-like cells, termed induced pluripotent stem cells (iPSCs) (1). Subsequently, two independent groups demonstrated that human somatic cells can also be reprogrammed into iPSCs. Yamanaka’s group successfully applied the same technique they used in mice. They used human dermal fibroblasts (HDFs) and two other human fibroblast populations from different human donors and transduced them with retroviral vectors carrying human cDNAs for OCT4, SOX2, C-MYC, and KLF4 (2). The Thomson group at the University of Wisconsin discovered a new combination of factors sufficient for the generation of human iPSCs. They showed that, out of 14 genes, OCT4, SOX2, NANOG, and LIN28 represented a core set of genes able to reprogram human somatic cells with a mesenchymal phenotype to iPSCs following lentiviral (LV) transduction (3). These iPSCs exhibited the essential characteristics of human ESCs including their ability to differentiate into lineages of all three germ layers.
It is now evident that the combination of a small number of specific transcription factors can reprogram fully differentiated human somatic cells (4). The enforced expression of specific combinations of transcription factors can override and modulate existing gene networks and epigenetic marks. Indeed, an ability to induce pluripotency in somatic cells was previously demonstrated in elegant studies of the transfer of somatic cell nuclei into enucleated oocytes and the fusion of pluripotent stem cells with differentiated cells (5, 6).
Successful reprogramming requires expertise in a number of techniques from microbiology, molecular biology, and virology, to stem cell biology. Here, we present a protocol for (1) the production of rhabdoviral vesicular stomatitis virus G (VSV-G) envelope protein pseudotyped lentiviruses (LVs), (2) the transduction of HDFs, (3) a method for isolating iPSCs using live-cell immunocytochemistry, and (4) the culture and propagation of iPSC lines. This protocol emphasizes controlled steps for streamlining the iPSC derivation process.
The LV strategy for reprogramming detailed here provides a method to titer virus prior to transduction and to screen for the presence of human immunodeficiency virus (HIV) and replication-competent retroviruses (RCRs), both before and after transduction, using a single ELISA procedure.
Please note that Institutional Biosafety Committee approval is required before starting LV production and use.
2. Materials
2.1. Bacterial Cell Culture
-
Bacterial stabs:
pSIN4-EF2-O2S (Addgene, #21162). OCT4 and SOX2 genes.
pSIN4-EF2-N2L (Addgene, #21163). NANOG and LIN28 genes.
pSIN4-CMV-K2M (Addgene, #21164). KLF4 and C-MYC genes.
pMD2.G (Addgene, #12259). Contains gene for VSV-G envelope protein.
psPAX2 (Addgene, #12260). Lentiviral backbone, gag-pol genes.
1,000× Ampicillin: 100 mg/mL Ampicillin in sterile water. Filter sterilize 1,000× Ampicillin solution through a 0.22 μm filter. Store at −20°C.
Luria Broth (LB) Medium: 2% (w:v) LB (Sigma, L3022) in distilled water. Adjust pH of LB Medium to 7.4 with 5M NaOH solution and/or 1N HCl solution. Autoclave for 30 min at 121°C. Store at room temperature.
LB Agar + Ampicillin plates: 3.5% (w:v) LB Agar (Sigma-Aldrich, L2897) in distilled water. Autoclave for 30 min at 121°C. Cool to ~55°C. Using a clean air bench or, alternatively, a laboratory bench top with an open flame, add Ampicillin to a final concentration of 1×. Transfer 20 mL of LB Agar + Ampicillin solution into sterile, untreated 100-mm Petri dishes. Allow the LB Agar + Ampicillin solution to form a gel at room temperature. Store the plates at 4°C, out of the light, and as dry as possible.
50% Glycerol Solution: 1:1 (v:v) solution of 100% glycerol: distilled water. Autoclave for 30 min at 121°C. Store at room temperature.
Cryopreservation vials (Nalgene, 5000-1020).
Inoculating loop.
37°C shaking incubator.
Dry-air incubator.
2.2. Isolation of Purified Plasmids
Endofree Plasmid Mega-Kit (Qiagen, 12381).
100% Ethanol (Sigma, E7023).
45 mm-Neck glass bottles (Pyrex, 1395-1L or equivalent).
Tris-EDTA (TE) buffer (included in Endofree Plasmid Mega-Kit).
UV spectrophotometer.
2.3. Lentivirus Production
293FT cells (Invitrogen, R700-07).
293FT medium: Dulbecco’s modified essential medium (DMEM, Invitrogen, 12430), 10% (v:v) defined fetal bovine serum (FBS, Hyclone SH30070.03), 4 mM L-glutamine, 1× minimum essential medium nonessential amino acids (NEAA, Hyclone, SH30238.01), 1 mM sodium pyruvate (GibcoBRL 11360-070), 50 μg/mL geneticin.
100-mm TPP Tissue culture dishes.
15-mL Polypropylene tubes.
Opti-Mem I reduced-serum medium (Invitrogen, 31985-070).
Transfection medium: Opti-Mem I reduced-serum medium (Invitrogen, 31985-070), 4% (v:v) defined FBS (Hyclone SH30070.03), 4 mM L-glutamine, 1× MEM non-essential amino acids (NEAA).
Lipofectamine 2000 (Invitrogen, 11668).
Purified pMD2.G.
Purified psPAX2.
Purified pSIN4-EF2-O2S.
Purified pSIN4-EF2-N2L.
Purified pSIN4-CMV-K2M.
0.45-μm PVDF bottle filters (Millipore, SCHVU01RE).
Cryopreservation vials (Nalgene, 5000-1020).
Optiseal polyallomer ultracentrifuge tubes (Beckman Coulter, 361625).
XL-80 Ultracentrifuge.
Type 70 Ti Ultracentrifuge rotor.
2.4. Titering by ELISA
HIV-1 p24 Antigen Capture Assay (Advanced BioScience Laboratories, 5421).
MicroELISA plate reader, capable of absorbance readings at 450 nm.
2.5. Human Dermal Fibroblast Culture
Human dermal fibroblasts (HDFs).
HDF medium: Dulbecco’s modified Eagle medium (DMEM, Invitrogen #12430054), 10% (v:v) defined FBS (Hyclone SH30070.03), 4 mM L-glutamine, 1× NEAA, 1 mM sodium pyruvate.
Recombinant human basic fibroblast growth factor (bFGF, Millipore, GF003).
Dulbecco’s phosphate buffered saline without Ca2+ or Mg2+ (DPBS).
0.05% Trypsin/EDTA solution.
Trypsin neutralizer solution (Cascade Biologicals, R-002-100).
0.1% (w:v) Gelatin (Millipore, ES-006-B).
T-75 Tissue culture flasks.
6-Well dishes.
Bright-Line Hemacytometer.
2.6. LV Transduction of HDFs
Lentiviral stocks (concentrated and titered).
Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, 12430).
Dulbecco’s phosphate buffered saline with Ca2+ and Mg2+ (DPBS +/+) (Hyclone, SH30264.01).
0.1% (w:v) Gelatin (Millipore, ES-006-B).
6-Well dishes (BD, 353046).
Bright-Line Hemacytometer.
Polybrene (Sigma, H9268).
2.7. Pluripotent Stem Cell Culture
PSC medium: DMEM/F12 (Invitrogen, 10565-018), 20% knockout serum replacement (KSR, Invitrogen, 10828-028), 2 mM GlutaMax-I (Invitrogen, 35050), 100 μM beta- mercaptoethanol (Invitrogen, 21985-023), 1× minimum essential medium NEAA (Hyclone, SH30238.01), 20 ng/mL bFGF (Millipore, GF003).
Mitotically inactive CF-1 mouse embryonic fibroblasts (MEFs) (see Chapter 8 for details).
MEF medium: Dulbecco’s modified Eagle medium (DMEM, Invitrogen, 12430), 10% (v:v) defined FBS (Hyclone SH30070.03), 4 mM L-glutamine, 1× minimum essential medium NEAA (Hyclone, SH30238.01), 1 mM sodium pyruvate.
0.1% (w:v) Gelatin (Millipore, ES-006-B).
6-Well dishes (BD, 353046).
Y-27632 (ROCK inhibitor, Stemgent, 04-0012).
Optional: Microscope object marker (such as Nikon, MBW10020).
2.8. Live-Cell Imaging of iPSC Colonies
Dulbecco’s phosphate buffered saline with Ca2+ and Mg2+ (DPBS +/+) (Hyclone, SH30264.01).
PSC medium.
PSC medium without phenol red: Use DMEM/F12 without phenol red (Invitrogen, 11039).
Tra-1-60-Alexa Fluor 647 antibody (BD Pharmingen, 560122).
Tra-1-81-Alexa Fluor 488 antibody (BD Pharmingen, 560174).
Hoechst 33342 (Invitrogen, H3570), see Note 1.
Fluorescence microscope in BSL2+ facility.
Optional: Microscope object marker (such as Nikon, MBW10020).
3. Methods
3.1. Lentiviral Vector Plasmids
The LV vectors to be used for the production of the iPSCs are self-inactivating (SIN), HIV-1-based vectors. These LV systems offer increased biosafety to users. The pSin4-promoter-gene vectors contain a 400-bp deletion in the U3 region (ΔU3) of the 3′ LTR, which results in the transcriptional inactivation of the 5′ LTR following reverse transcription and chromosomal integration. The inability to efficiently transcribe full-length vector RNA by these SIN-HIV-1 vectors in transduced target cells minimizes the possibility of the formation of RCRs. In addition, these vectors contain a central polypurine tract within the pol gene (cPPT) which facilitates nuclear translocation of the HIV preintegration complex and improves lentiviral transduction efficiency in several types of cells. Expression of the Oct-4 with Sox-2 and Nanog with Lin28 transgenes are driven by the human elongation factor 2 (EF2) promoters in plasmids pSin4-EF2-O2S and pSin4-EF2-N2L, respectively. Expression of KLF-4 with c-Myc is driven by the CMV promoter in pSIN4-CMV-K2M vector. Each of these three vectors will be individually packaged in 293FT cells by co-transfection with the pMD2.G-envelope plasmid encoding the VSV-G envelope glycoprotein and the psPAX2 2nd generation LV backbone packaging vector that is optimal for virus production.
3.2. Bacterial Cell Culture
Addgene ships plasmids as individual Escherichia coli (E. coli) bacterial stab cultures. A stab culture is made by inoculating (or transferring) bacteria into a vial containing LB agar with the appropriate antibiotic. Store bacterial stabs at 4°C for no longer than 2 weeks. The subsequent protocol describes how to streak bacterial stabs on LB agar plates, isolate a single colony from your stab culture, and create bacterial glycerol stocks for long-term storage. Please note that the bacteria containing psPAX2 and pMD2.G are “high-copy,” while the bacteria containing pSIN4-EF2-O2S, pSIN4-EF2-N2L, and pSIN4-CMV-K2M are “low-copy” (see Note 2).
Over an open flame, sterilize an inoculating loop by holding its tip in the flame until it turns red. Wait for the loop to cool to prevent killing the bacterial sample.
For each bacterial stab obtained from Addgene, touch the end of the cooled inoculating loop to the bacterial sample you wish to transfer. Transfer bacterial cells to the LB Agar + Ampicillin plate by gently brushing the loop back-and-forth over the surface of the plate in a zig-zag fashion. Do not cross over previous streaks.
In separate dishes, repeat steps 1–2 for all five bacterial stabs obtained.
Cover the LB Agar + Ampicillin Petri plate, invert it, and incubate at 37°C in a dry-air incubator overnight.
The next morning, the plates should appear to have solid streaks of cells as well as isolated colonies. At this point, bacterial plates can be stored for several weeks at 4°C, if plates are inverted and sealed with parafilm wrap.
To make a LB liquid starter bacterial culture, aseptically transfer 6 mL of LB medium to a sterile 15-mL tube and add 6 μL of 1,000× Ampicillin stock. For multiple start-up cultures, the LB broth and antibiotic can be scaled up and aliquoted appropriately in 15-mL tubes.
Using a sterile inoculating loop, pick and transfer a single, well-isolated colony from a streaked bacterial plate to the tube containing LB medium + Ampicillin.
Close the top of the 15-mL tube and flame sterilize the inoculating loop before repeating the inoculations of the remaining four bacterial culture plates. These are your starter cultures.
Incubate at 37°C in a shaking incubator at 300 rpm for 12–16 h. These starter cultures will be used to inoculate larger volumes of LB for plasmid isolation and purification, and to create long-term glycerol stocks of E. coli harboring the respective plasmids.
For each high-copy bacterial culture, prepare 1 L of autoclaved LB Broth. For low-copy bacterial cultures, prepare 2.5 L of autoclaved LB Broth (see Note 2).
Just before inoculating LB with the respective E. Coli cultures, add fresh 1,000× Ampicillin solution to the LB to a final concentration of 1×. To ensure good aeration of the growing cultures, split the culture to a maximum volume of 500 mL contained within individual 2 L or larger clearly labeled Pyrex Flasks.
Inoculate each prelabeled LB + Ampicillin flask with 1 mL of its respective starter culture from step 9. Incubate at 37°C in a shaking incubator at ~300 rpm for 12–16 h.
With the remaining 5 mL of starter culture, make bacterial glycerol stocks. Under open-flame/bench-top sterile conditions, mix 500 μL of the overnight starter culture with 500 μL of 50% glycerol solution in a cryopreservation vial. Freeze and store the glycerol stock vial at −80°C. Glycerol stocks may be stored indefinitely. New LB Agar + Ampicillin plates may be streaked again from these glycerol stocks when needed.
Continuing from step 12, harvest the bacterial cell culture using centrifugation at 6,000 × g for 15 min at 4°C. Decant the supernatant and dispose of it in accordance with institutional guidelines. Bacterial pellets can be stored at this point, at −20°C.
To recover the plasmids, we recommend using the Qiagen EndoFree Plasmid Mega-Kit (Cat. no. 12381). This kit produces high-quality purified endotoxin-free plasmids, which are necessary to ensure high transfection efficiencies. Qiagen makes continuous improvements to this kit, so please refer to their most recent EndoFree Plasmid Purification Handbook for instructions.
Re-dissolve the air-dried purified plasmid DNA pellets in approximately 500–600 μL of endotoxin-free TE buffer (included in the kit). Calculate the concentration of the isolated plasmids using UV spectrophotometry at 260 nm. For reliable spectrophotometric DNA quantitation, A260nm readings should lie between 0.1 and 1.0. At a minimum, the recovered plasmid concentration should be 1 μg/mL. Concentrated preparations of plasmid aid in achieving high transduction efficiencies. If plasmid recovery is low, repeat the protocol and resuspend the DNA pellet in a smaller volume.
3.3. Lentivirus Production and Concentration
The following protocol is adapted from Invitrogen’s ViraPowerTM Lentiviral Expression System. Each LV is produced separately in 100-mm tissue culture dishes.
Day 1: Plate four 100-mm dishes with 5 × 106 293FT cells/ dish in 10 mL 293FT medium. 293FT cells should be 90–95% confluent at the time of transfection. Record the time of plating. As antibiotics can negatively affect transfection efficiency, do not include any antibiotics in this medium.
Day 2: Approximately 24 h after plating the 293FT cells, aspirate the medium and replace with 5 mL transfection medium. As above, do not include any antibiotics in the medium.
-
For each 100-mm dish, prepare DNA–Lipofectamine 2000 complexes as follows:
Tube A (sterile): Combine the following: 1.5 mL of Opti-MEM I medium without serum, 7.5 μg psPAX2 plasmid, 2.5 μg pMD2.G, and 6 μg of one of the target clone vectors (pSIN4-EF2-O2S, pSIN4-EF2-N2L, or pSIN4-CMV-K2M). Mix together gently. Do not vortex.
In Tube B (sterile): Combine the following: 1.5 mL of Opti-MEM I medium without serum and 36 μL of Lipofectamine 2000. Gently mix the Lipofectamine 2000.
Incubate Tube B at RT for 5 min before proceeding.
After the 5-min incubation, combine the contents of Tube A with Tube B. Mix gently.
Incubate for 20 min at RT to allow the DNA–Lipofectamine 2000 complexes to form. The solution may appear cloudy, but this will not impede the transfection.
-
Drop-wise, add the DNA–Lipofectamine 2000 complexes to each plate of 293FT cells. Mix by gently rocking the plate back-and-forth. Incubate the cells overnight at 37°C in a humidified 5% CO2 incubator. Record the time of transfection.
Caution: Remember that you are working with potentially infectious virus following this stage. Therefore, all manipulations of virus and virus-transduced cells must be carried out with appropriate PPE under BSL-2+ conditions using a certified, Class II, biological safety cabinet (BSC) and all virus-contaminated media, serological pipettes, barrier pipette tips, and tissue culture-ware must be deactivated in 10% (v:v) fresh sodium hypochlo-rite (household bleach) solution for at least 15 min prior to disposal. Follow the recommended NIH guidelines that are contained in the Biosafety in Microbiological and Biomedical Laboratories (BMBL) Manual and the NIH Guidelines for Research involving Recombinant DNA Molecules (http://www.cdc.gov/od/ohs/biosfty/bmbl4/bmbl4toc.htm, http://www.oba.od.nih.gov/oba/rac/guidelines_02/NIH_Guidelines_Apr_02.htm). In addition, consult your institution’s policies and procedures and receive approval from your Institutional Biosafety Committee prior to initiating LV experiments.
Note: Expression of the VSV-G glycoprotein causes 293FT cells to fuse, resulting in the appearance of large, multinucleated cells known as syncytia. This morphological change is normal and does not affect LV production.
Day 3: Remove the medium containing the DNA–Lipofectamine complexes and replace with 10 mL 293FT medium. Do not include any antibiotics in the medium.
Day 4/5: Harvest LV-containing supernatants 48–72 h post-transfection. Differences in titer are minimal when LV is harvested at 48 or 72 h.
Pass the collected LV-containing supernatant through a 0.45-μm PVDF bottle filter (Millipore, SCHVU01RE) to clear cellular debris.
Transfer the LV-containing supernatant into a sterile Optiseal Beckman Coulter ultracentrifuge tube. Seal the ultracentrifuge tube in strict accordance with the manufacturer’s instructions. In the BSC, load the ultracentrifuge rotor with the samples and seal the lid securely before removing from the BSC to the ultracentrifuge. Centrifuge at 70,000 × g for 90 min at 4°C.
-
Caution: Be sure the ultracentrifuge tube is completely filled with medium and make sure the cap and tube balancer is secured tightly.
Note: If you do not have access to an ultracentrifuge or you do not wish to concentrate your viral stock, pipette 1 mL aliquots of LV supernatants into screw-capped cryovials, place in a secondary container, and store viral stocks at −80°C.
Remove supernatant by aspirating or decanting the supernatant. Aspirate away from the bottom of the tube so as not to aspirate the pellet. You may see a pellet, but LV pellets are often clear. For each 100 mm dish, add 200 μL of sterile DMEM to the pellet. Scale accordingly. Do not triturate the pellet. Allow the pellet to slowly dissolve into the medium overnight at 4°C.
At this point, you may titer or freeze the virus, or transduce your HDFs directly. Viruses in general do not tolerate freeze–thaw well. We recommend freezing the prepared LV stocks in 20-μL aliquots in cryopreservation vials. In addition, freeze 1 vial with 10 μL of lentivirus. You will use this 10 μL vial for titering the virus by ELISA. Store viral stocks at −80°C.
3.4. Titering by ELISA
Titering the LV stock is helpful in producing consistent transduction results by using a consistent amount of active virus. Multiplicity of infection (MOI) is a measure of viral infectivity in a population of target cells. With lentivirus, the MOI is the ratio of transfer viral transducing particles to the target cells. A MOI of 5 indicates that there are five transducing units for every cell contained within a well. It is important to note that different cell types may require different MOIs for successful transduction. The following section will detail how to translate ELISA determined spectrophotometric readings to pg/mL p24 concentration and subsequently to transducing units (TU)/mL that are used to calculate the volume of LV required for a given MOI.
While it may not be necessary to determine the MOI for every application, it is necessary when predictably producing iPSCs for the following reasons:
Confirmation of the viability of the viral stock.
Determination of the maximum number of target cells that can be transduced by a given amount of virus.
Determination of the MOI-to-response ratio that produces the optimal induction profile.
Viral titer may be affected by any and all steps preceding viral titration. For instance, each freeze–thaw can reduce the functional titer (actual infection rate) by up to two- to four-fold. Likewise, low-quality cultures of 293FT will almost certainly lower the viral titer. Hence, titering the lentiviral stock serves as a useful quality control method. To more accurately gauge the performance of the lentivirus titer, we recommend freezing the viral stock before titering and/or titering your viral stock before each transduction.
The plasmid constructs used in this protocol do not contain a selectable marker gene (e.g., puromycin). Therefore, it is not possible to perform a colony-forming unit assay following LV transduction and drug selection.
We find it most convenient to titer lentiviral stocks by ELISA, using the HIV-1 p24 Antigen Capture Assay (Advanced BioScience Laboratories, 5421). The assay procedure is a double antibody sandwich enzyme immunoassay that is used to calculate the concentration of the amount of the HIV-1 p24 core antigen present in tissue culture samples. The assay’s linear range is between 3.1 and 100 pg/mL. Since p24 is highly conserved among various HIV-1 isolates, this assay detects p24 from various isolates with comparable sensitivity. In addition to allowing the user to determine the titer of LV produced above, this assay also allows the user to screen research cell lines for HIV-1 contamination prior to LV transduction and iPSC generation and thus help eliminate the risk of generating RCR. Demonstration of p24-negativity in transduced cells is also necessary prior to downgrading the cells from BSL2+ to BSL2 status.
-
Before performing the p24 ELISA assay, make 10−6, 10−7, and 10−8 serial dilutions from 10 μL of your concentrated LV stocks. Make at least 250 μL per dilution for the assay. If you did not concentrate the LV, do not dilute the LV stock (see Note 3).
Note: From experience, this range of dilutions gives data that consistently fits the best-fit line of the p24 standards.
Follow the directions included in the HIV-1 p24 Antigen Capture Assay, using the serially diluted stocks of virus as the test samples.
After reading absorbance at 450 nm, ensure that the test is valid by checking absorbance values of the negative control and 100 pg/mL p24 standard. Two or more negative control absorbance values over 0.120 will invalidate the assay. Absorbance values of the 100 pg/mL p24 standard should be >1.200 and <2.200. Likewise, absorbance values outside of this range will invalidate the assay.
Calculate the mean absorbance for each test sample dilution, negative control, and p24 standard dilution. Subtract the mean of the negative control (background absorbance) from the means of each p24 standard dilution. Likewise, subtract the mean of the negative control (background absorbance) from the means of each test sample.
Determine the p24 concentration of each test sample dilution by extrapolating from a standard curve or by using linear regression analysis.
Find the mean p24 antigen concentration in pg/mL of each test sample: Multiply the mean of each test sample dilution (from step 4) by the reciprocal of its dilution. For instance, multiply your determined 10−6 value from step 4 by 1/10−6 (or 106).
Find the mean p24 antigen concentration in pg/mL of your LV Stocks: Calculate mean p24 antigen concentrations of your 10−6, 10−7, and 10−8 dilutions (from step 6).
Convert to TU/mL: 10 TU corresponds to approximately 1 pg of p24 (see Note 3). Multiply your mean concentration (from step 7) by the ratio 10 TU/pg.
- Calculate the volume of virus needed for transduction. We recommend starting with an MOI of 5. Use the following equation to determine the volume of virus needed:
3.5. Transduction (Reprogramming) of HDFs
For optimal reprogramming of HDFs into iPSCs, fibroblast cultures should be healthy, relatively low passage (passage 6 or earlier), and rapidly dividing. For context, “rapidly dividing” means the following: If 2 × 105 fibroblasts are plated in a 35-mm dish, the culture should reach 70% confluence overnight. Such cultures are “rapidly dividing.” Neonatal fibroblasts will generally fulfill this requirement. Adult fibroblasts usually do not. Addition of 4 ng/ mL human bFGF will increase the cell division rate of adult fibro-blast cultures and greatly improve their transduction efficiencies.
The following protocol describes the transduction of a HDF cell line.
Day 0: Coat two wells of a 6-well plate with 1 mL/well 0.1% (w:v) gelatin solution. One well will be infected with LV. The other will be mock-infected and serve as a negative (morphology) control. Incubate at 37°C for at least 1 h.
Detach HDFs from their culture vessel by trypsinization. Pipette the cells through a 10-mL serological pipette a couple of times to ensure a good dispersion of cells. Count HDF, either by Coulter counter or with a hemacytometer.
Remove the gelatin solution from the 6-well plate in step 1.
Plate 2 × 105 HDFs/well in the gelatin-coated well with 2 mL HDF medium/well.
Incubate overnight at 37°C in a humidified 5% CO2 incubator.
-
Day 1 (Day of transduction): Prepare the transduction medium in plain, sterile DMEM. In a sterile tube, add the lentiviral stock(s) containing OCT4/SOX2, KLF4/c-MYC, and NANOG/LIN28 gene inserts. Bring the final volume up with DMEM to 600 μL/well/transduction. Include 4 μg/ mL polybrene in transduction medium. (The control medium contains only DMEM and Polybrene.)
Note: Polybrene is a polycation that reduces charge repulsion between the virus and the cellular membrane. Excessive exposure to polybrene (>24 h) can be toxic to cells. The optimum final concentration of polybrene may need to be determined empirically but generally falls within a range of 2–12 μg/mL.
Wash each well of HDFs twice with DPBS. Aspirate the final wash, and add the transduction medium, tilting the plate back-and-forth and side-to-side to disperse the virus.
Incubate at 37°C in a humidified 5% CO2 incubator over-night. For the first hours after transduction, it is beneficial to rock the plate back-and-forth and side-to-side every 10–15 min to further disperse the virus.
Day 2: Remove virus-containing and control medium, wash each well three times with DPBS, and add 2 mL fresh HDF medium. Incubate at 37°C in a humidified 5% CO2 incubator.
Day 3: Prepare 2 gelatin-coated 6-well plates: Coat all wells of a 6-well plate with 1 mL/well 0.1% (w:v) gelatin solution. Incubate at 37°C for at least 1 h.
After 1 h, thaw vials of irradiated MEFs (iMEFs), and plate iMEFs at a density of 2 × 104 cells/cm2. For each gelatin-coated 6-well plate, plate 1–2 × 106 cells/dish (the total area of a 6-well plate is approximately 60 cm2) in 2 mL MEF medium per well. Incubate at 37°C in a humidified 5% CO2 incubator overnight.
Day 4: Lift the transduced HDFs and control with trypsin separately. Once the cells have sufficiently detached, inactivate the trypsin with trypsin inhibitor. Centrifuge at 200 × g for 5 min. Aspirate the supernatant. Resuspend pellets in 12 mL HDF medium. Optional: We recommend supplementing this medium with 10 μM Y-27632 (ROCK inhibitor).
Aspirate the MEF medium from the 6-well plates containing iMEFs. Wash each well 1× with DPBS with Ca2+ and Mg2+ to clear out any debris.
Add 2 mL/well of resuspended transduced or control HDFs to the 6-well plate containing iMEFs. Incubate at 37°C in a humidified 5% CO2 incubator.
Day 5: Aspirate the spent HDF medium, and replace with 2.5 mL/well fresh PSC medium. Replace medium daily, thereafter.
3.6. Live-Cell Staining and Live-Cell Imaging
Transduced fibroblasts are usually reprogrammed very quickly, if successful. Potential iPSC colonies are usually present in abundance at approximately day 14 posttransduction (see Fig. 1). Additional colonies may appear later. Many colonies, however, will be only partially transduced, but will have very similar morphology to bona fide iPSCs. These colonies may represent a stage of the spectrum of evolution toward bona fide iPSCs. Therefore, it is important to characterize the cultures by live staining during iPSC generation. Colonies that stain positive for Tra-1-60 and Tra-1-81 and stain dim for Hoechst 33342 are reliably bona fide iPSC colonies and can be passaged immediately after staining (see Note 1 and Fig. 2).
Fig. 1.
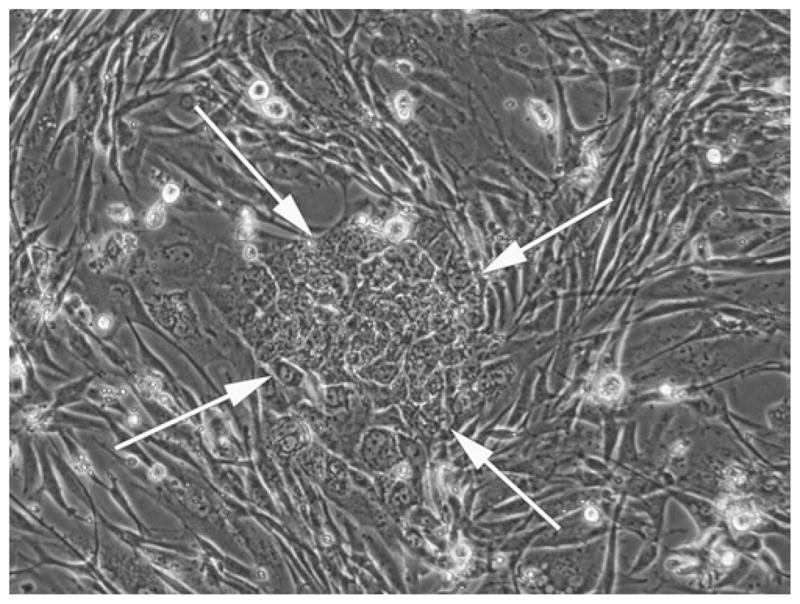
Early morphology of transduced human dermal fibroblasts. Picture taken Day 9–10 post-transduction. Early in reprogramming, transduced human fibroblasts begin forming colonies, become small and rounded compared to nontransduced fibroblasts surrounding the colonies, but are still hyper-cytoplasmic compared to embryo-derived pluripotent stem cells. 10× objective.
Fig. 2.
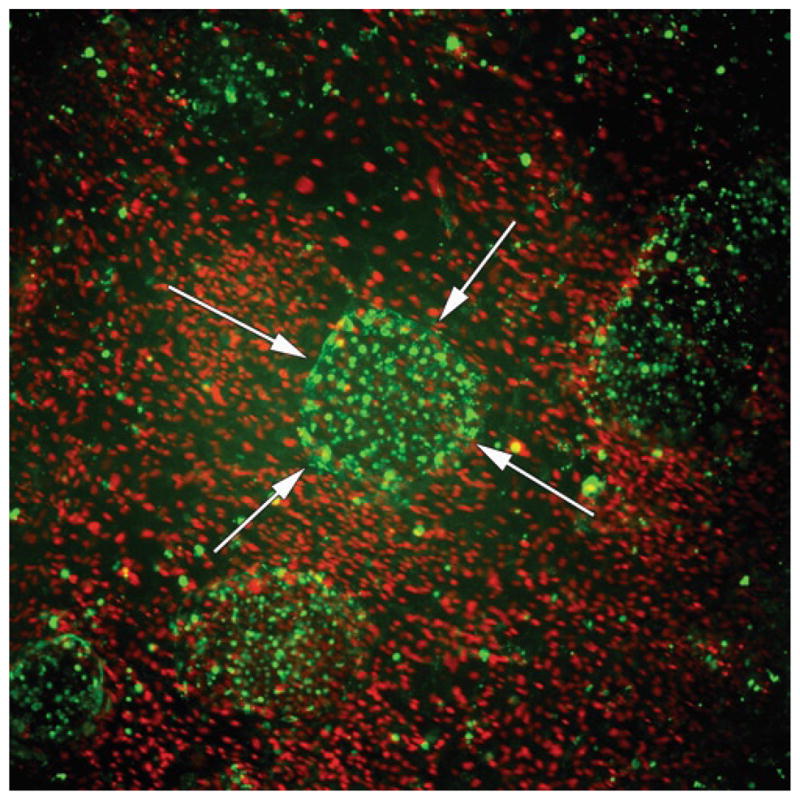
Live staining immunocytochemistry of human iPSCs derived following lentiviral reprogramming of human dermal fibroblasts. Reprogrammed iPSCs stain positive for the pluripotent marker TRA-1-60 (green, colony marked by arrows). The surrounding iMEFs and undifferentiated HDFs brightly stain for Hoechst 33342 (red), while human iPSC colonies stain dim for Hoechst 33342. Of note is the discriminate Hoechst staining of the cells surrounding the iPSC colonies which stain brighter. 4× objective.
Make 1:100 dilutions of the conjugated antibodies in a sterile tube. For each well of a 6-well plate, add 6 μL of Tra-1-60-Alexa Fluor 647 and 6 μL of Tra-1-81-Alexa Fluor 488 to 600 μL of PSC medium.
Aspirate the medium from the well(s) to be stained. Wash each well three times with DPBS with Ca2+ and Mg2+ before adding the antibody-medium mixture.
After the final wash, apply the antibody-medium mixture (from step 1) to the well. Briefly, rock the plate back-and-forth and side-to-side gently to adequately disperse the antibody throughout the well.
Incubate at 37°C in a humidified 5% CO2 incubator for 90 min.
In the meantime, prepare an excess of 2× solution of Hoechst 33342 in PSC medium (1.0 μg/mL). Prepare 1,000 μL for each well being stained. Filter the 2× solution using a 0.22-μm syringe filter.
After the 90 min incubation, add 600 μL of 2× Hoechst 33342 directly to the well. The final concentration of Hoechst is 0.5 μg/mL.
Incubate at 37°C in a humidified 5% CO2 incubator for 30 min.
Wash each well three times with DMEM/F12 without phenol red. The subtraction of phenol red allows for better fluorescent visualization of the live-stained cultures.
Add 1 mL PSC medium without phenol red.
Visualize live-stained cultures under a fluorescence microscope. Take note of the morphology and staining patterns of iPSC colonies. The Hoechst is used primarily to train the eye (see Note 1).
Proceed to Subheading 3.7 on picking and passaging colonies.
3.7. Picking and Passaging iPSC Colonies
Colonies should be expanded depending on their number and size. As a point of reference, 2–4 colonies can be split into one well (~10 cm2) of a 6-well plate but it is preferable to split only 1 colony per well to maintain clonality. Scale the reagents and tissue culture vessels appropriately. Once a colony is chosen for manual passaging, turn the objective wheel to the object marker (see Fig. 3), and mark the site of the colony on the underside of the plate. This product is relatively inexpensive and saves valuable time in picking the best colonies.
Fig. 3.

Use of an object marker greatly simplifies identification of early iPSC colonies for picking and passaging. The marker (shown with the arrow in the left panel) simply takes the place of an objective. When a colony is identified (usually by using the 10× objective), the marker is rotated into place and the position of the colony is marked on the bottom of the plate (marked colonies are shown in the right panel). The plate can then be transferred to a BSC for picking and passaging of the marked colonies.
The following protocol can be used for both unstained and live-stained iPSC cultures.
Day 0: For each colony to be passed, prepare 1 well of a 6-well tissue culture plate with gelatin. Add 1 mL of 0.1% (w:v) gelatin solution per well. Incubate at 37°C for at least 1 h.
After 1 h, thaw vials of irradiated MEFs (iMEFs), and plate iMEFs at a density of 2 × 104 cells/cm2 in 2 mL MEF medium per well. Incubate at 37°C in a humidified 5% CO2 incubator overnight.
Day 1: Aspirate MEF medium and wash plates twice with DPBS with Ca2+ and Mg2+. Replace medium with 1 mL/well PSC medium. Optional: Supplement media with Y-27632 (ROCK inhibitor), of 10 μM at a final concentration.
Exchange the medium of iPSC cultures with 1 mL/well of fresh PSC medium.
Under a microscope equipped with a Microscope Object Marker, locate bona fide iPSC colonies (Tra-1-60/Tra-1-81-positive). Mark these colonies on the underside of the well.
In a Class II BSC, manually detach the marked iPSC colony with the end of a sterile P10 micropipette tip. Try to break this marked colony into the smallest pieces possible.
Pool together the manually detached iPSC colony pieces in a sterile tube.
Wash the well of the previously detached colony pieces with 1 mL/well of PSC medium. Pool this wash with the iPSC colony pieces. Repeat steps 4–8 for each colony to be passaged.
Evenly distribute the detached iPSC colony pieces over the 6-well plate containing iMEFs in PSC medium. Gently rock the plate back-and-forth and side-to-side to disperse the colony pieces evenly.
Incubate at 37°C in a humidified 5% CO2 incubator. Leave newly passaged cultures undisturbed for 2–4 days. Feed daily thereafter. Passage as required. Cultures may be examined for the presence of markers of pluripotency by immunocytochemistry (see Chapter 15) after passage 10 (Fig. 4).
Fig. 4.
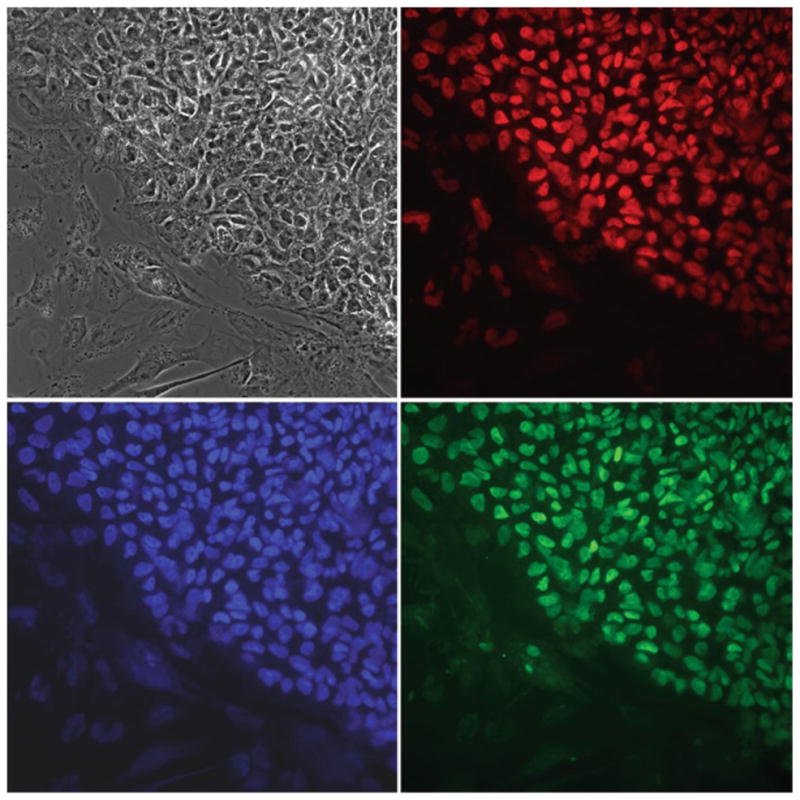
Immunocytochemistry of lentivirus-derived human iPSC colonies propagated on iMEFs. iPSCs stain uniformly positive for the pluripotent markers OCT4 (red), SOX2 (green), and NANOG (blue). The phase contrast image illustrates the morphological similarity of the iPSC colony to classical human ESC colonies. 40× objective. Photos taken by Alexander Stover.
Acknowledgments
This work has been funded by the National Institutes of Health (T15HL074286, R21MH087925, R01HD059967). The NCI Preclinical Repository supplied FGF-2. We also recognize Richard Pepple for proof-reading this chapter.
Footnotes
Hoechst 33342.
The basis for the presence or absence of Hoechst staining after loading the cells with Hoechst is the presence or absence of the multidrug resistance transporter ABCG2. In the presence of ABCG2, which appears during the early phases of transduction, Hoechst is actively transported out of the cells, leading to cells that are Hoechst-dim compared to surrounding nontransduced cells that are Hoechst-bright (7). As the iPSCs mature, the ABCG2 transporter is downregulated and the cells remain Hoechst-bright after loading so it is of little use then. Since Tra-1-60 staining appears early in the transduction process and is not seen in nontransduced cells, the appearance of Tra-1-6-bright/Hoechst-dim colonies allows a nice, early indication of an iPSC colony that can be picked out of a mixed colony using fluorescence as your guide. Unfortunately, Hoechst is also mutagenic and thus cannot reasonably be used for routine picking of iPSCs. It is, therefore, omitted from routine use and is used solely to train the eye of the novice iPSC-deriver as to what a bona fide early iPSC colony looks like both by phase microscopy and by fluorescence with Tra-1-60 (with or without additional Tra-1-81).
High-copy versus low-copy plasmids.
Plasmid copy number is perhaps the most important factor affecting plasmid DNA yield. Plasmids vary widely in their copy number depending on the origin of replication they contain, which determines whether they are under relaxed or stringent control, as well as the size of the plasmid and its associated insert. Some plasmids also have mutations which allow them to reach very high-copy numbers within the bacterial cell. Low-copy plasmids have approximately 10 copies/ cell, while high-copy plasmids have up to 100’s of copies/cell. Therefore, it is necessary to scale up the LB culture medium volume to obtain higher plasmid yield. It is best to use the suggested culture volumes. Using larger culture volumes will lead to an increase in biomass and can affect the efficiency of alkaline lysis during plasmid purification, leading to reduced yield and purity of the preparation.
The relationship between TU and pg of p24 is approximately 2,000 molecules of p24 per physical particle (PP) of HIV, which indicates there are approximately 104 PP/pg of p24 detected. It is also important to note that while freeze–thaw of the LV produced may not affect the quantity of p24 detected, this quantity is still used to estimate the MOI, and thus infectivity will likely be reduced. Thus, it is important to choose aliquot size carefully and, ideally, freeze–thaw no more than once.
References
- 1.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 3.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 4.Belmonte JC, Ellis J, Hochedlinger K, Yamanaka S. Induced pluripotent stem cells and reprogramming: seeing the science through the hype. Nat Rev Genet. 2009;10:878–883. doi: 10.1038/nrg2700. [DOI] [PubMed] [Google Scholar]
- 5.Cowan CA, Atienza J, Melton DA, Eggan K. Nuclear reprogramming of somatic cells after fusion with human embryonic stem cells. Science. 2005;309:1369–1373. doi: 10.1126/science.1116447. [DOI] [PubMed] [Google Scholar]
- 6.Stojkovic M, et al. Derivation of a human blastocyst after heterologous nuclear transfer to donated oocytes. Reprod Biomed Online. 2005;11:226–231. doi: 10.1016/s1472-6483(10)60962-5. [DOI] [PubMed] [Google Scholar]
- 7.Chan EM, Ratanasirintrawoot S, Park IH, Manos PD, Loh YH, Huo H, Miller JD, Hartung O, Rho J, Ince TA, Daley GQ, Schlaeger TM. Live cell imaging distinguishes bona fide human iPS cells from partially reprogrammed cells. Nat Biotechnol Nov. 2009;27(11):1033–7. doi: 10.1038/nbt.1580. [DOI] [PubMed] [Google Scholar]