

Abstract
Culturing human embryonic stem cells (hESCs) requires a significant commitment of time and resources. It takes weeks to establish a culture, and the cultures require daily attention. Once hESC cultures are established, they can, with skill and the methods described, be kept in continuous culture for many years.
hESC lines were originally derived using very similar culture medium and conditions as those developed for the derivation and culture of mouse ESC lines. However, these methods were suboptimal for hESCs and have evolved considerably in the years since the first hESC lines were derived. Compared with mouse ESCs, hESCs are very difficult to culture – they grow slowly, and most importantly, since we have no equivalent assays for germline competence, we cannot assume that the cells that we have in our culture dishes are either stable or pluripotent. This makes it far more critical to assay the cells frequently using the characterization methods, such as karyotyping, immunocytochemistry, gene expression analysis, and flow cytometry, provided in this manual.
Keywords: ESC, iPSC, PSC hESC culture, feeder cells, mechanical passaging, human pluripotent stem cells
1. Introduction
Following fertilization, the cells of the human embryo undergo several rounds of cell division and rearrange to form a hollow sphere of cells termed the blastocyst. The cells of the blastocyst segregate into an outer layer called the trophectoderm and an inner cell mass (ICM). The trophectoderm becomes the fetal contribution to the placenta while the cells of the ICM give rise to the embryo proper. The isolation and culture of a unique population of cells, embryonic stem cells (hESCs), from the ICM of human blastocysts was first accomplished in 1994 by Ariff Bongso’s group (1). In 1998, James Thomson and colleagues described the derivation of the first hESC lines that were propagateable and cryopreservable cell cultures (2). Although hESCs are typically produced from the ICM, they can also be produced from earlier stage embryos, including the morula and cleavage stages.
hESCs display two unique properties: the ability to self-renew indefinitely and the potential to give rise to all cell types of the human body (2). Thus, hESCs are pluripotent stem cells (PSCs) because they are able to form cell lineages of all of the three germ layers – endoderm, ectoderm, and mesoderm. These features of hESCs have made them extremely attractive tools not only for the study of development and cancer but also for their potential use in regenerative medicine strategies to repair or replace damaged tissues.
hESCs in culture (under the conditions described in this chapter) grow as tightly compact colonies of cells with high nucleus-to-cytoplasm ratios. On a molecular level, hESCs in culture express characteristic and specific (1) surface antigens such as the stage-specific embryonic antigen SSEA-4 and the teratocarcinoma recognition antigens TRA-1-60 and TRA-1-81 and (2) pluripotency-specific transcription factors such as Oct-4 and Nanog (3).
In this chapter, we describe basic culture methods for hESC (PSC) culture. These methods may also be used for the propagation of PSCs produced by cellular reprogramming (induced PSCs or iPSCs as described in Chapters 5–7). While these methods have been used successfully for years by many laboratories, and many publications cite them, it should be noted that more modern methods exist, and these are covered in Chapters 9 and 10. In addition, we also cover cryopreservation methods. Importantly, since PSC cultures are often kept in continuous culture for months, even years, it is critical that genetic and developmental drift (4) be monitored in the cultures (see Notes 1 and 2). The best way to control for drift is to generate a large bank of frozen cells as soon as possible after the cultures are first expanded as described in Chapter 3. The importance of this cannot be overemphasized – the value of discoveries based on PSCs depends on the reproducibility of results.
2. Materials
2.1. Reagents
Dulbecco’s modified Eagle medium: Ham’s F12 (DMEM/ F12 + GlutaMAX. Invitrogen, #10565).
Dulbecco’s phosphate buffered saline (DPBS−−) without Mg2+ and Ca2+.
Dulbecco’s phosphate buffered saline (DPBS++) with Mg2+ and Ca2+.
KnockOut™ Serum Replacement (KSR) (Invitrogen, #108280-028).
2-Mercaptoethanol, 55 mM (1,000×) in PBS (such as Invitrogen, #21985-023).
GlutaMax (100×) (Invitrogen, #35050).
Human basic fibroblast growth factor (bFGF) (Stemgent, #03-0002).
MEM-Nonessential amino acids (NEAA) 100× (10 mM) (Hyclone, #SH30238.01).
Hybri-Max dimethyl sulfoxide (Sigma-Aldrich, #D2650).
Water for embryo transfer (Sigma, #W1503, see Note 3).
Fetal bovine serum (FBS) (Hyclone, #SH30070.03).
Pen-strep 100× (optional) (Invitrogen, #15070-063).
TrypeLE-Express (Invitrogen, #12604).
Collagenase IV (20,000 U, Invitrogen, #17104-019).
CF-1 mouse embryonic fibroblasts (ATCC, #SCRC-1040).
Nikon Object Marker, catalog # MBW10020 (optional).
6-Well vacuum gas plasma-treated tissue culture dishes (such as BD Falcon, #353046).
Sterile nylon membrane syringe filter (Pall Life Sciences, #PN 4433).
Nalgene freezing container (containing isopropanol).
20 μL pipette tips (Eppendorf and others).
150 mm Tissue culture dishes (TPP, #93150).
2.2. Media and Stock Solutions
2.2.1. Human Basic FGF (bFGF) (10 μg/mL, 1 mL, See Note 4)
Dissolve 10 μg of human bFGF in 1 mL KSR.
Aliquot in 50 μL samples.
Store thawed aliquots at 4°C for up to 2 weeks.
Store frozen aliquots at −20°C or −80°C for 6 months.
2.2.2. Collagenase IV (200 U/mL, 100 mL, See Note 5)
Dissolve 20,000 U of collagenase IV in 100 mL of DMEM/F12 + GlutaMax. This is usually ~1 mg/mL final concentration.
Add to a 250 mL 0.2 μm filter unit and filter sterilize.
Aliquot in 5–10 mL in sterile tubes and store at −20°C until use.
2.2.3. KnockOut™ Serum Replacement
Aliquot as follows:
Thaw 500 mL bottle at 4°C and aliquot into sterile 50 mL tubes and store at −20°C.
Mix thoroughly when thawed, both for the initial aliquotting and when for use in media preparation.
2.2.4. MEF Medium (500 mL)
Combine 440 mL DMEM/F12 + GultaMax, 50 mL FBS, 5 mL GlutaMax, 5 mL NEAA.
Sterile filter 2 μm.
Store at 4°C.
Warm to room temperature in the hood before use, discard unused medium after 2 weeks.
2.2.5. Human PSC Medium (100 mL)
Combine, in order, 78.8 mL DMEM/F12 + GlutaMax with 20 mL KSR, 1.0 mL 100× NEAA, 100 μL bFGF stock solution, and 100 μL of 1,000× 2-mercaptoethanol.
Filter using 2 μm PES filter.
Store at 4°C when not in use and discard any unused medium after 2 weeks.
2.2.6. Human PSC Cryopreservation Medium (10 mL, 2×)
Combine 2 mL of human PSC medium, 6 mL of FBS, and 2 mL of DMSO.
Mix thoroughly.
Sterile filter using a syringe filter approved for use with DMSO (e.g., nylon membrane).
Keep cold and use immediately. This is a 2× solution.
3. Methods
These methods assume that all PSC culture is carried out in 6-well plates (see Notes 6–8 for helpful general suggestions).
3.1. Preparation of Feeder Cell Stocks
The traditional feeder cells are mitotically inactivated, low-passage mouse embryonic fibroblasts, usually from CF-1 strain mice (5). These MEFs are seeded at a wide range of densities depending on the different PSC cell line being grown. For example, the original “H” series lines from WiCell were grown in the presence of MEFs seeded at 75,000 cells/cm2. However, these lines and others have also been successfully grown on denser feeder layers, as have many others. It will take some trial and error in your laboratory to determine the optimum density. Human-derived fibroblasts of various origins have also been successfully used as a feeder cell layer. Any distinct advantages of human versus mouse feeder cells have not been agreed upon. As with MEFs, if you choose to use human feeders, it will require some trial and error to determine the best concentration.
Thaw a cryopreserved vial (such as the ATCC product cited) of mouse embryonic fibroblasts quickly in a 37°C water bath (without submerging the cap), and wash with 70% alcohol before moving it to the tissue culture hood. Carefully move the contents into a 15 mL conical tube. Slowly and dropwise, add 10 mL of warm MEF medium, while gently shaking the tube.
Centrifuge at 200 × g for 5 min, aspirate supernatant, and resuspend the pellet in 5 mL of MEF medium.
Seed onto a 0.1% gelatin-coated 150 mm TPP tissue culture dish and add an additional 15 mL of MEF medium. Place in incubator and gently move the plate back-and-forth and then side-to-side, so as to evenly distribute the cells.
Monitor the cells daily. They should divide extremely quickly but not require daily feeding before reaching confluence. This often takes only 24 h.
When the cells become confluent, split at a 1:2 ratio using TrypLE-Express. Aspirate the medium, rinse the plate with 5 mL DPBS−−, and add 10 mL of RT or 37°C TrypLE-Express. When the cells start to lift off the plate, inactivate the enzyme by adding 10 mL of warm MEF medium. Collect the cells in a sterile conical tube and centrifuge them at 200 × g for 5 min. Aspirate the supernatant, resuspend in a 40 mL of medium, and reseed into 2–150 mm dishes. This is considered passage 1.
Continue monitoring and splitting the MEFs until passage 5 is reached. At this point, lift the cells, and irradiate them with 3,000 rads to inactivate them (see Note 9).
Freeze the cells in DMEM with 30% FBS and 10% DMSO. The freezing density will depend on the PSC line with which you are working. We recommend that you freeze enough feeder cells in one vial (~3.5 × 106/vial) to seed an entire 6-well plate.
3.2. Preparation of a Feeder Layer
24 h before a plate of feeder cells is needed for PSC culture, coat the plate with 0.1% gelatin for 2–24 h before plating MEFs to help the PSCs attach.
Thaw a vial of prepared irradiated/inactivated MEFs as described above, and seed onto the plate in MEF medium.
After the MEFs have attached overnight, aspirate the MEF medium, rinse with DPBS++, and add 1 mL/well PSC medium. Allow the MEFs to condition this medium for at least an hour before seeding the PSCs (1 mL/well, as described in Subheading 3.4.1, step 9).
3.3. Thawing of Cryopreserved PSCs
Quite frequently, there is a growth lag after thawing and plating PSCs – it may take several days to see colonies (6). It is advisable to observe the cultures under 4× magnification 24 h after thawing, but not to exchange the medium for at least 48 h. There may be considerable floating debris and dead cells upon thawing the cells – this is normal.
Gently but quickly thaw the vial of cells by shaking it in a 37°C water bath until the last sliver of ice has melted (about 60 s). Spray the tube with 70% alcohol and dry with a Kimwipe.
In the biosafety cabinet, aseptically remove the vial contents and place them in 15 mL conical tube. Slowly, with gentle tapping, add 10 mL of room temperature PSC medium.
Spin at 200 × g for 5 min.
Aspirate the supernatant.
Add 3 mL of PSC medium to the tube, triturate gently, and transfer the contents to one well of a 6-well dish that has been prepared with inactivated MEFs as described in Subheading 3.2.
Place plate into the incubator.
Allow 3–7 days for the cells to attach. During this time, replace half of the medium every other day being careful not to aspirate the cells.
The medium should be replaced daily starting 4–7 days after thawing the cells, or when the cells appear to have attached (Fig. 1 shows the appearance of traditionally cultured PSCs).
Fig. 1.
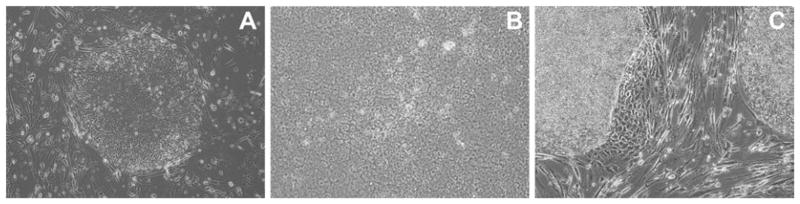
Phase-contrast images of human PSC colonies in culture. (a) A PSC colony shown at 20× a few days after passaging. Note the distinct border between the PSCs and the feeder layer and the apparent heterogeneity of the PSCs, comparing the outside of the colony to the inside. This heterogeneity is typical in a recently passaged culture. (b) A closeup (40×) of a larger colony, indicating the classic cell morphology of PSCs. Note the high nucleus-to-cytoplasm ratio as well as the very prominent nucleoli. (c) A more mature culture at 10× showing both the relative homogeneity of cell morphology throughout the colony as well as an area of very noticeable differentiation (the area of larger phase-dark cells) on the lower-right border of an otherwise undifferentiated colony. Notice the difference between these differentiated phase-dark cells and the loose undifferentiated cells near the periphery of the early stage colony in (a).
3.4. Passaging PSCs
Human PSCs have traditionally not survived well when dissociated to single cells. Thus, the most reliable method for passaging undifferentiated PSC cultures has been manual dissection of the colonies. This method may seem tedious but it is virtually foolproof and we recommend that novices use this method until they have familiarity with the cells and can easily recognize differentiation in the cultures. We also recommend manual passaging for producing cell banks of low-passage PSCs (see Note 10).
3.4.1. Mechanical Dissociation
The choice of tool for mechanical passaging is an individual preference, but we have found that needles and pipette tips are the most common choice. They are inexpensive to obtain and provide consistency (see Note 11).
Evaluate the culture daily under 4× or 10× phase-contrast optics.
The cells can be split among 3–6 plates of the same size as the original culture, depending on the density of the original culture. If you wish to put the cells in different-sized plates or dishes, calculate the volume to add based on the surface area.
Mark (or remove) overtly differentiated colonies so as not to disturb these during the dissociation process.
Remove the medium from the dish and replace with fresh PSC medium.
Dissect the colonies by hand, either under a low-power dissecting microscope (in a horizontal flow hood) or without a microscope in the tissue culture hood (see Fig. 2).
Break up each colony and move it into suspension by moving the tip around and across each colony in a crosshatch or a spiral motion. Pipette tips are a much better tool for this than needles due to their larger bore. Since the colonies are large at the time of passage, it is relatively easy to see individual colonies on the plate and, with practice, one can quickly passage an entire plate in less than 20 min (see Note 12).
After all of the colonies are dissected (from an entire 6-well plate, for example), use a 5 mL pipette to transfer the culture medium containing the dissected colonies to a 15 or 50 mL conical tube. Rinse the plate with 1 mL PSC medium, moving the medium sequentially from well to well before adding it to the same 15 mL tube.
Using PSC medium, bring the volume of medium and cells in the tube to the appropriate amount for seeding new plates. For example, if you have just passaged one well of a 6-well plate, and are passaging 1:6, you should bring the final volume to 12 or 2 mL for each new well that will be seeded (but see the slightly different procedure if you are cryopreserving the cells, Subheading 3.5).
Gently triturate the cell clumps using a sterile 10 mL pipette and divide the cell suspension into the prepared culture dishes on feeder layer. Do not overtriturate; triturate gently, trying to achieve a relatively uniform distribution of the cell clumps without creating single cells.
Place the newly seeded plates in the incubator. Briskly move the plate(s) back-and-forth, side-to-side, and forwards-and-backwards to ensure even dispersion, while being careful not to splash any medium onto the cover of the culture dish.
Fig. 2.
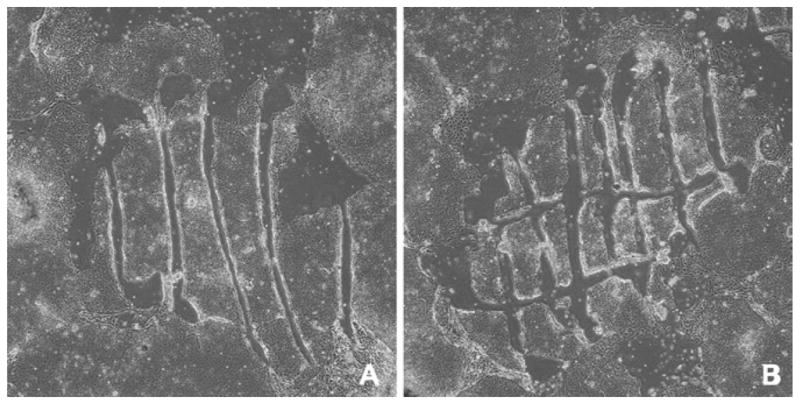
Manual passaging of human PSCs in culture showing the sterile needle or pipette method used for slicing the colonies into about 100 pieces. The colony is cut into strips (a), and then into squares (b). Each piece of the colony has a few hundred cells (4× phase-contrast).
3.4.2. Collagenase Dissociation
Enzymatic dissociation methods vary widely, and the exact conditions need to be developed for each laboratory. Most importantly, cultures that have been maintained by manual passaging cannot be passaged by enzymatic dissociation unless exceptional care is taken to adapt the cells to this new set of conditions. When done properly though, enzymatic passaging can provide the investigator with a convenient and efficient way of maintaining PSC culture stocks (see Note 5).
Remove the culture medium.
Rinse the culture with DPBS++.
Treat with 2 mL/well 200 U/mL of collagenase IV solution for 5–10 min at 37°C or until the edges of the colonies start to curl up. Observe the culture under the microscope.
Remove the collagenase and replace with 2 mL/well of PSC medium.
Using a 5 mL pipette, gently dislodge the “good” colonies from the plate and transfer them to a 15 mL conical tube. Alternatively, one could remove the differentiated colonies prior to treating the culture dish with collagenase.
Gently triturate the cell clumps using a sterile 10 mL pipette and plate on a feeder layer of MEFs. Do not make a single-cell suspension but try to achieve a relatively uniform suspension of cell clumps containing several hundred cells each.
The cells can be divided among 3–6 dishes of the same size as the original culture, depending on density of the original culture. If you wish to put the cells in different-sized dishes, calculate the dilution based on surface area.
3.5. Cryopreservation
Cryopreservation is used to stabilize cultures with specific genetic characteristics at specific points in time. Without the ability to cryopreserve our cell lines, we are forced to continuously subculture them, during which time the cells may accumulate genetic changes and become heterogeneous. Using validated stock vials to initiate new experiments maximizes the long-term usefulness of a cell line and minimizes experimental variation.
For many years, the traditional method of cryopreserving PSCs has involved freezing the cells in large clusters with a medium containing FBS and DMSO. PSCs have very poor survival with this cryopreservation method, however (6). As a result, the time from thawing the vial to having cultures suitable for experimentation can be weeks to months. Vastly more efficient techniques have been recently developed in conjunction with alternative culture systems, and these are outlined in Chapters 9 and 10. Nonetheless, for researchers interested in pursuing traditional PSC culture, the historical method is presented in this chapter.
Prepare cells for cryopreservation when they have reached the same stage at which you would normally passage them.
Change the culture medium just before harvesting the cells.
Label 1.8 mL cryogenic vials with cell line name, date, and passage number.
Prepare 2× stock cryopreservation medium (see Subheading 2.2) and keep on ice.
Dislodge the colonies from the plate, mechanically, using a sterile pipette tip or treat with 2 mL/well 200 U/mL of collagenase IV in DMEM/F12 + Glutamax for 5–10 min at 37°C.
Remove collagenase and replace with PSC medium (3 mL for each well of a 6-well dish).
For each well of a 6-well dish, collect the cells in 3 mL of PSC medium and transfer to a 15 mL conical tube.
Centrifuge 5 min at 200 × g. Aspirate supernatant, leaving a small amount of medium covering the pellet.
Gently resuspend the pellet in conditioned PSC medium (usually 1.5 mL for each well of a 6-well dish or one half of the final freezing volume). Use a 5 mL pipette to gently triturate the clumps.
Dropwise, add an equivalent volume of ice-cold 2× cryopreservation medium, mixing constantly by tapping the tube (see Note 13).
Place 1.0 mL of cell mixture in each cryogenic vial (i.e., about three vials per well).
Rapidly transfer the vials to a precooled (4°C) Nalgene freezing container (containing isopropanol), and place immediately in a freezer at −70°C to −80°C. The next day, transfer cells to liquid nitrogen for long-term storage.
Fig. 3.
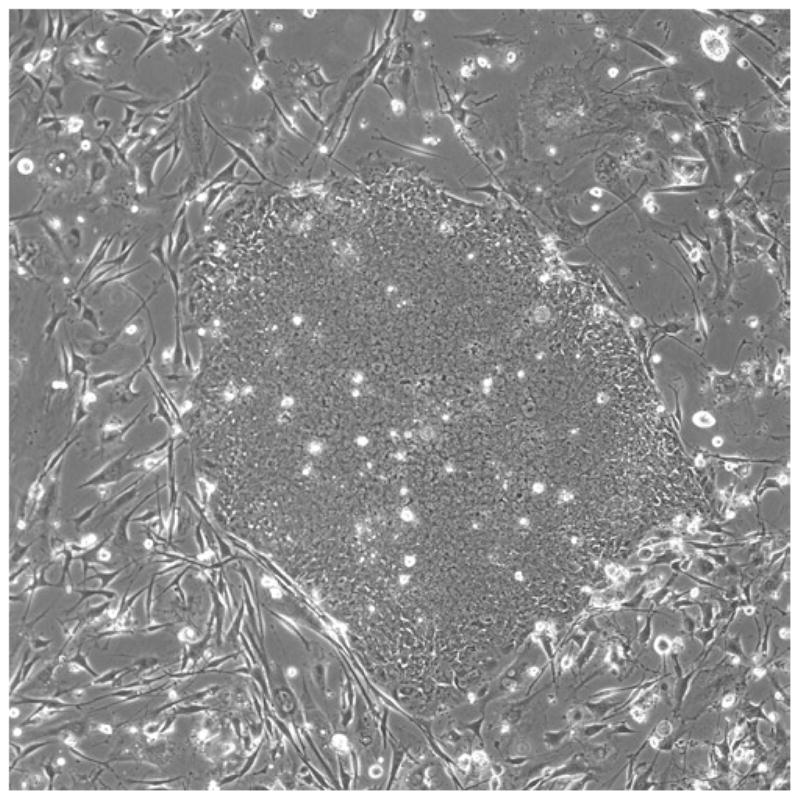
A colony about half the diameter of the 10× field contains about 4,400 cells; a colony filling the field contains about 15,000 cells.
Fig. 4.
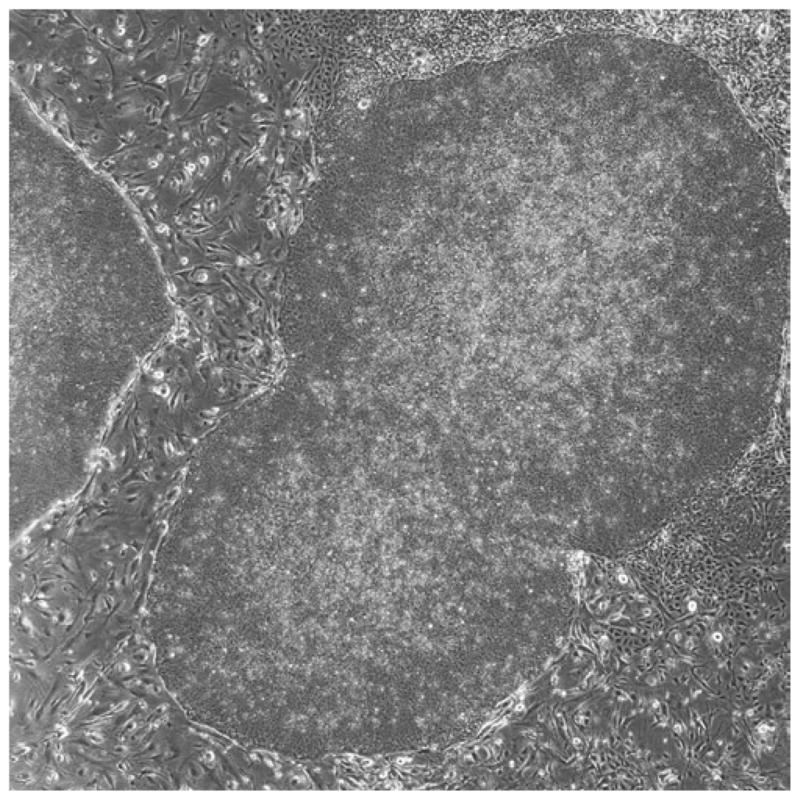
Example of a high-density culture on mouse embryonic fibroblasts. Note how two large colonies have merged together and there is some noticeable differentiation around the edges of the colonies (4× phase-contrast). This culture would need to be passaged right away.
Acknowledgments
This work has been funded by the National Institutes of Health (T15HL074286, R21MH087925, R01HD059967). The NCI Preclinical Repository supplied FGF-2. We would also like to recognize the contributions of Chris Stubban, Rodolfo Gonzalez, Richard Pepple, Heather Maxwell, and Nicole Sheridan in the production of these protocols.
Footnotes
We know that PSCs acquire chromosomal abnormalities over long periods of culture, so karyotyping or other genetic analysis methods must be performed on a regular basis. For detailed information about how to monitor genetic drift, see Chapters 13 and 14. Keep in mind that changes during the time the cells are cultured in your laboratory can only be detected if you first analyze the cells very soon after you obtain them.
PSCs can also drift toward a more differentiated state over periods of extended culture. Since there is no assay for pluripotence equivalent to the germline transmission assay for mouse PSCs, surrogate markers, such as antibody markers, should be routinely checked, especially if the morphology of the cells seems to be different from the earlier cultures. The gold standard for measuring the pluripotency of a PSC line is to transplant it to an immune-deficient mouse to form a teratoma (Chapter 17). Keep in mind that it will require histological expertise to identify cell types and tissues in the tumors. In vitro differentiation of PSCs using embryoid body culture (Chapter 28) will allow at least a cursory analysis of PSC differentiation potential. However, embryoid bodies never achieve the maturity of cells that develop in teratomas, and since the methods used to assess differentiation in vitro usually involve a small number of markers assayed by immunocytochemistry (Chapter 15), it is more difficult to judge the full range of pluripotence using this method. The best approach to monitoring developmental drift is to pick a particular method and differentiated cell type to check periodically (see Chapter 30 on embryoid body and neuroepithelial differentiation, as well as the specific chapters on neuronal, cardiac, and hematopoietic cells, Chapters 29–33).
An important consideration for all cell cultures, but most especially for PSC culture, is that all reagents, including the water used to make them up, be qualified for use in the particular culture at hand. Water quality, even that purified by double distillation and/or “MilliQ” water, can vary significantly from geographic area to geographic area and from season to season. With this in mind, our laboratory tends not to buy media powders that we must ourselves reconstitute but rather the fully diluted media. In addition, for all dilutions of reagents that will come into contact with live cells, directly or indirectly, we use water that has been qualified for embryo culture, such as the Sigma product we cite. Although one might save pennies doing everything oneself, the likelihood, in this research area, of losing dollars by doing so is too high to justify it.
For all stock or small quantity growth factor solutions, prewet all pipette tips, tubes, and filters with DPBS with 0.2% BSA to lessen the loss of the growth factor.
The type of enzyme used for dissociation is critical. For example, passaging with trypsin appears to put more selective pressure on the cultures than other methods, resulting in a higher incidence of drift of PSC lines toward aneuploidy. But some PSC lines have been derived using trypsin from the outset; thus some lines can be routinely passaged using whatever enzymatic technique is used by the supplier. Microbial collagenase is preferred by many laboratories, perhaps because of the way in which it is used. Collagenase is used to loosen the PSC colonies from the dishes, not to dissociate them to single cells, but the cell clumps have to be further dissociated by trituration. Collagenase is isolated from Clostridium histolyticum. Type IV is selected because of its low tryptic activity, and is recommended for isolation of pancreatic islets. This is a crude product, so expect a wide lot-to-lot variation. EDTA inhibits this enzyme’s activity. Accutase is also becoming more popular because of its unique ability to dissociate PSCs into single cells while maintaining viability (7); however, this requires the use of defined medium and a Matrigel substrate (see Chapter 10). The potent apoptosis-blocking ROCK Inhibitor Y27632 is frequently used alongside Accutase and other enzymatic and mechanical passaging techniques to further help maintain viability (8). Another enzymatic reagent which has been used is Invitrogen’s TrypLE-Express. TrypLE is a fungally derived Trypsin-like enzyme that dissociates cells extremely quickly. It has not been widely used for PSC passaging, but may be useful for breaking apart colonies for use in flow cytometry or cytogenetics. Nonenzymatic cell dissociation buffers are also available. The latter are Ca- and Mg-free saline solutions containing EDTA or EGTA. They have not been as widely used for PSC dissociation as the methods described above, however, they should offer advantages for assays that require intact cell surface proteins such as flow cytometry. Commercial formulations are available, such as Cell Dissociation Buffer (Invitrogen catalog no. 13150016) which contains glycerol as well as a proprietary mixture of salts and chelators. Keep in mind that enzymes are not highly purified recombinant products, and they may contain animal products. Trypsin is generally prepared from porcine tissue, and collagenase is a crude microbial product.
PSCs are usually cultured without antibiotics; with good culture technique, bacterial and fungal contamination should not be a problem. However, we recommend that antibiotics be used while new investigators are being trained in the techniques. Antibiotics such as amphotericin, penicillin, and streptomycin, however, do not have any effect on mycoplasma. Mycoplasma is highly infectious and commonly occurs when new cells are introduced into laboratories. The ATCC (American Type Culture Collection) estimates that 16% of cell cultures are contaminated by mycoplasma. This bacterium can also come from tissue culture reagents such as serum and media supplements as well as from laboratory staff. Mycoplasma is a serious problem in laboratories that culture multiple cell lines or have inadequately trained personnel. Cultures must be monitored for mycoplasma on a regular basis, and contaminated cultures must be destroyed. The best defense against mycoplasma contamination is good aseptic technique; the laboratory should not allow inexperienced or careless workers to share cell lines, solutions, or tissue culture equipment. As a precaution, all cultured cell lines should be tested at least four times a year. Testing for mycoplasma can be done by enzymatic, polymerase chain reaction (PCR), fluorescent staining, or culture methods.
It is important to keep in mind the actual source of the materials and reagents used in the culture and maintenance of PSCs. Since many reagents are derived from animal sources, there is inherent lot-to-lot variability. While vendors make every effort to control the variability by setting production specifications, these are usually ranges and as long as the product falls within the approved range, the product passes inspection and is distributed. Ideally, you should have your own quality control methods to test new lots of products. At the very least, record the lot numbers of reagents used; if an experimental result cannot be replicated, or a cell line fails to thrive, you will save considerable time if the problem is traceable to a suboptimal reagent.
The cells should be passaged at about 1:3–1:6 every 5–7 days. Prepare the feeder layer or extracellular matrix (ECM) substrata the day before passaging. Depending on the cell line, passaging on a Friday afternoon may be a good routine. The cells are usually left undisturbed for 2–3 days following passaging, which allows them to settle down on the substratum, attach and begin dividing before the medium is changed. There will be considerable variation in the size of colonies in a single dish. Human PSCs do not substantially pile up on each other, and their colonies can grow to a large diameter while remaining undifferentiated. Culture conditions affect the flatness of the colonies, but as an approximation, they are ready to split when the diameter fills the 10× field when observed under the microscope. As shown in Fig. 3, a colony about half the diameter of the 10× field contains about 4,400 cells. A colony filling the field would contain about 15,000 cells. When the colonies grow very large and start to merge into one another (see Fig. 4), they must be passaged to avoid differentiation and/or starvation of the culture.
When passaging by most methods, do not make a single-cell suspension; dissociate the colonies into smaller colonies of a few hundred cells. Examine the culture daily for colony morphology under the phase-contrast or dissecting microscope. With experience, one can get a good overview of colony morphology by holding the dish up to a light and looking at the bottom of the dish. The differentiated colonies will have ragged edges and hollow or dark centers. On the bottom of the dish, mark colonies that are badly differentiated or parts of the colony that you do not wish to transfer to a new culture dish. This can be easily accomplished with a special microscope attachment sold by Nikon (see Subheading 2.1 “Reagents”). To be certain that the colonies selected are undifferentiated, it is advisable to dissect the colonies while viewing the dish under a dissecting microscope with illumination from the base. But this is not absolutely necessary, and some prefer passaging cells without magnification.
The single most important skill in successful culturing of PSCs may be the ability to recognize the morphology of undifferentiated cells under a variety of conditions (see Fig. 1).
- Feed cells every day, except for 1 or 2 days following passage.
- Examine the cultures every day under 4× and 10× phase-contrast. This will allow you to become familiar with the morphologies of undifferentiated and differentiated cells and colonies.
- When they are cultured on feeder layers, PSCs tend to undergo spontaneous differentiation in the centers of the colonies. When passaging, take care to avoid passaging these differentiated “centers” to the new culture.
- Most PSC lines double every 31–35 h.
- Store medium at 4°C, protect from light, and discard any unused medium after 10 days. Best results are achieved when medium is prepared in small batches once a week.
Mitomycin C can be used in the place of irradiation for inactivation but it is a cytotoxic antitumor agent and must be handled carefully; it works by cross-linking the DNA, which blocks cell division. Follow your institution’s rules for safe handling and disposal. Handlers should wear latex or nitrile protective gloves and work in a biological safety or fume hood. One effective method is to inactivate the mitomycin C with an equal volume of household bleach. Inactivation is rapid.
- Remove the feeder cell medium.
- Add 10 mL/75 cm2 of mitomycin C medium. Make sure the entire flask is covered with mitomycin C medium so that the inactivation is complete and all cells are exposed for the entire incubation time.
- Incubate for 3 h at 37°C in 5% CO2.
- Remove mitomycin C, neutralizing it with bleach or other recommended procedure.
- Wash inactivated feeder layer three times with 10 mL each of DPBS++.
- Trypsinize the cells to remove them from the dish.
- Use the cells immediately for plating and/or cryopreserve them for later use.
An unfortunate historical accident has been using the number of passages as a measure of the age (or of the number of cell divisions) of a PSC line. Because of the inconsistencies in PSC culture procedures in different labs, cells are passaged at different time intervals, ranging from 4 to 7 days. Therefore, the number of passages for one line might not be representative of another, even though the cells have been in culture for exactly the same amount of time. Passage ratios vary from 1:2 to 1:80 among different laboratories. A better measure would be the number of doublings, but to count the number of cells in a culture is difficult since the cells form tight clusters and are not passaged as single cells, but as clumps.
Fire-drawn glass pasteur pipettes were the choice for early PSC labs, but they are labor-intensive to prepare, and no two are alike. Needles have the advantage of greater precision; however, for the novice user, they have a tendency to scrape ribbons of plastic off of the plate and introduce them into the medium. Needles, however, are very useful in instances where one may need to isolate a very small colony or where there is a small patch of undifferentiated cells surrounded by areas of spontaneous differentiation. Pipette tips, on the other hand, have the advantage of greater efficiency for more confluent plates with larger, less-differentiated colonies. This is due to their larger bore, and the ability to use them like a scoop to shovel colony chunks into suspension. At the same time, relatively precise cuts can be made using the edge of the tip. We recommend individually wrapped 20 μL pipette tips from Eppendorf since this eliminates the possibility of another technician accidentally contaminating a shared box of autoclaved pipette tips.
As the basal media used for most common human cell cultures is bicarbonate-based, do not keep a culture outside the incubator for more than about 15 min at a time. Any time the medium is outside the CO2 environment, it loses CO2 and its pH rises, going above pH 8 in about 30 min. Thus, if your dissections take longer than about 20 min, put the cultures back into the incubator for about 10 min before continuing.
Do not leave the cells in DMSO at room temperature for long periods of time as DMSO is toxic to the cells and is, under a variety of conditions, also known to induce differentiation.
References
- 1.Bongso A, Fong CY, Ng SC, Ratnam S. Isolation and culture of inner cell mass cells from human blastocysts. Hum Reprod. 1994;9:2110–7. doi: 10.1093/oxfordjournals.humrep.a138401. [DOI] [PubMed] [Google Scholar]
- 2.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–7. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 3.Müller FJ, Laurent LC, Kostka D, Ulitsky I, Williams R, Lu C, Park IH, Rao MS, Shamir R, Schwartz PH, Schmidt NO, Loring JF. Regulatory networks define phenotypic classes of human stem cell lines. Nature. 2008;455:401–5. doi: 10.1038/nature07213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitalipova MM, Rao RR, Hoyer D, Jones K, Johnson J, Dalton S, Meisner L, Stice SL. Preserving the genetic integrity of human embryonic stem cell lines. Nature Biotechnology. 2005;23:19–20. doi: 10.1038/nbt0105-19. [DOI] [PubMed] [Google Scholar]
- 5.Amit M, Margulets V, Segev H, Shariki K, Laevsky I, Coleman R, Itskovitz-Eldor J. Human Feeder Layers for Human Embryonic Stem Cells. Biology of Reproduction. 2003;68:2150–6. doi: 10.1095/biolreprod.102.012583. [DOI] [PubMed] [Google Scholar]
- 6.Heng BC, Ye CP, Liu H, Toh WS, Rufaihah AJ, Yang Z, Bay BH, Ge Z, Ouyang HW, Lee EH, Cao T. Loss of viability during freeze-thaw of intact and adherent human embryonic stem cells with conventional slow-cooling protocols is predominantly due to apoptosis rather than cellular necrosis. Journal of Biomedical Science. 2006;13:433–45. doi: 10.1007/s11373-005-9051-9. [DOI] [PubMed] [Google Scholar]
- 7.Bajpai R, Lesperance J, Kim M, Terskikh AV. Efficient propagation of single cells accutase-dissociated human embryonic stem cells. Molecular Reproduction and Development. 2007;75:818–27. doi: 10.1002/mrd.20809. [DOI] [PubMed] [Google Scholar]
- 8.Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, Takahashi JB, Nishikawa S, Nishikawa S, Muguruma K, Sasai Y. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nature Biotechnology. 2007;25:681–6. doi: 10.1038/nbt1310. [DOI] [PubMed] [Google Scholar]