

Abstract
Formation of new neurons in the adult brain takes place in the subventricular zone and in the subgranule layer of the dentate gyrus throughout life. Neurogenesis is thought to play a role in hippocampus- and olfaction-dependent learning and memory. However, whether impairments in neurogenesis take place in learning and memory disorders, such as Alzheimer’s disease, is yet to be established. More importantly, it remains to be elucidated whether neurogenic impairments play a role in the course of the disease or are the result of extensive neuropathology. We now report that transgenic mice harboring Familial Alzheimer’s disease-linked mutant APPswe/PS1ΔE9 exhibit severe impairments in neurogenesis that are evident as early as two months of age. These mice exhibit a significant reduction in the proliferation of neural progenitor cells and their neuronal differentiation. Interestingly, levels of hyperphosphorylated tau, the cytotoxic precursor of the Alzheimer’s disease hallmark neurofibrillary tangles, are particularly high in the neurogenic niches. Isolation of neural progenitor cells in culture reveals that APPswe/PS1ΔE9-expressing neurospheres exhibit impaired proliferation and tau hyperphosphorylation compared to wild type neurospheres isolated from nontransgenic littermates. This study suggests that impaired neurogenesis is an early critical event in the course of Alzheimer’s disease that may underlie memory impairments, at least in part, and exacerbate neuronal vulnerability in the hippocampal formation and olfaction circuits. Furthermore, impaired neurogenesis is the result of both intrinsic pathology in neural progenitor cells and extrinsic neuropathology in the neurogenic niches. Finally, hyperphosphorylation of the microtubule-associated protein tau, a critical player in cell proliferation, neuronal maturation and axonal transport is a major contributor to impaired neurogenesis in Alzheimer’s disease.
Keywords: Neurogenesis, Alzheimer’s disease, tau, amyloid, stem cells
Introduction
Neurogenesis in the adult brain takes place in discrete microenvironments, namely, the subventricular zone (SVZ) and the subgranule layer (SGL) of the dentate gyrus (DG), throughout life. Newly formed neurons integrating in the olfactory bulb and granule layer of the DG, respectively, are thought to play a role in olfaction- and hippocampus-dependent forms of learning and memory [for example (Imayoshi et al. 2008; Paton and Nottebohm 1984; Thuret et al. 2009; van Praag et al. 2002), for review see (Aimone et al. 2006; Zhao et al. 2008)]. In addition, neurogenesis is thought to modulate brain plasticity and repair [For review see (Zhao et al. 2008)], by providing neurotrophic support for neurons and a pool of neural progenitor cells (NPC) with the capacity to differentiate into neurons, astrocytes and oligodendrocytes [For review see (Jagasia et al. 2006; Peterson 2002)]. While it is well established that extent of neurogenesis declines with aging, whether impaired neurogenesis may play a role in the aging-linked learning and memory disorder Alzheimer’s disease (AD) is yet to be determined.
Loss of hippocampus-dependent learning and memory has been extensively described in humans affected with AD [For review see (Price et al. 1998)] as well as in FAD-linked transgenic mice [For review see (Ashe 2001)]. These mice express genes mutated in human pedigrees affected with early onset AD (FAD), i.e., amyloid precursor protein (APP), presenilin-1 (PS1) and presenilin-2 (PS2). Loss of olfaction function appears to be one of the earliest markers of possible AD (Albers et al. 2006). Deficits in olfactory sensitivity, odor discrimination, and odor identification are common and appear early in disease progression (Bacon et al. 1998; Kesslak et al. 1988; Serby 1987; Warner et al. 1986). Loss of neurons in the hippocampus and cortex starts early in the disease and progresses with time. The hallmark lesions amyloid deposition and neurfibrillary tangles accumulate in these brain areas. These lesions composed of aggregated β-amyloid (Aβ) peptides and paired helical filaments of hyperphosphorylated tau, respectively.
Located in the most affected areas of AD neuropathology, both the SGL and the SVZ/olfactory bulb, may be heavily distressed, leading to impaired or altered neurogenesis, that in turn may exacerbate neuronal vulnerability and neuropathology and contribute to memory impairments. Several studies examined the fate of neurogenesis in AD in transgenic animals that exhibit amyloid deposition, in an attempt to associate amyloid pathology with alterations in neurogenesis. Some of these studies suggest a correlation between extensive amyloidosis, amyloid deposition later in life and neurogenic deficits (Donovan et al. 2006; Ermini et al. 2008; Haughey et al. 2002a; Haughey et al. 2002b; Niidome et al. 2008; Taniuchi et al. 2007; Verret et al. 2007; Zhang et al. 2007). However, several other studies observed an increase in extent of hippocampal neurogenesis (Gan et al. 2008; Jin et al. 2004b; Kolecki et al. 2008; Lopez-Toledano and Shelanski 2007; Mirochnic et al. 2009). These conflicting observations may be due to two major reasons. First, several FAD-linked players, such as PS1 and metabolites of APP regulate neurogenic signaling pathways differentially (Lazarov 2009). Second, some of these animal models exhibit different degrees of pathology that may have a differential effect on neurogenesis. Gaining an insight into the mechanism underlying FAD-induced alterations in neurogenesis may reconcile these discrepancies.
Importantly, there have been no studies examining the fate of neurogenesis in these mice prior to extensive brain pathology. Thus, whether neurogenesis is impaired early in the disease, contributing to its progression, or a side effect of massive pathology is not known.
To address these issues, we examined the fate of NPC in both the SGL and the SVZ of young transgenic mice harboring FAD-linked APPswe/PS1ΔE9. Here we show that proliferation and differentiation of NPC is severely impaired in the SVZ and in the SGL of these mice as early as two months of age, preceding onset of amyloid deposition. This suggests that neurogenic impairments are an early event in the disease rather than a secondary effect. In addition, neurogenic impairments precede onset of memory impairments in these mice (Jankowsky et al. 2005), suggesting that impaired neurogenesis may underlie, at least in part, memory deficits. Furthermore, we show a dramatic increase in steady state levels of Aβ and tau phosphorylation in the neurogenic niches in the brains of APPswe/PS1ΔE9 mice. Increase in tau phosphorylation was detected by PHF-1 and AT-8 antibodies, raised against epitopes hyperphosphorylated in AD (Goedert et al. 1995; Otvos et al. 1994). Importantly, increases in tau phosphorylation take place in neurogenic areas where tau is expressed in NPC and neuroblasts in particular. Using neurosphere culture we further show that APPswe/PS1ΔE9-expressing neurospheres exhibit reduced extent of proliferation and increased levels of tau phosphorylation, suggesting that FAD-linked APPswe/PS1ΔE9 mutations have an intrinsic effect on NPC. Taken together, the results of this study suggest that NPC are affected early in AD in both neurogenic areas of the adult brain and may contribute to deficits in hippocampus- and olfaction-dependent memory.
Materials and Methods
Transgenic Animals
Mice co-expressing FAD mutant human PS1ΔE9 and a chimeric mouse–human APP695 harboring a human Aβ domain and mutations (K595N, M596L) linked to Swedish FAD pedigrees (APPswe) were previously described (Jankowsky et al. 2001). These mice were generated by coinjection of two transgene constructs into one founder (Jankowsky et al. 2001). Transgene constructs have been described (Borchelt et al. 1996; Lee et al. 1997). Nontransgenic wild type littermates of the APPswe /PS1ΔE9 mice served as controls. Mice were group housed under a 14:10 light:dark cycle with free access to food and water. All mice used for experiments were male. Transgenic mice harboring FAD-linked presenilin human wild type (PS1HWT) and PS1ΔE9 mutant were included in biochemical analyses for PS1 human transgene expression control. These mice have been described previously (Borchelt et al. 1996; Lazarov et al. 2005; Lee et al. 1997).
Antibodies
The following primary antibodies were used in this study: mouse anti-BrdU monoclonal (1:300 Novocastra, Newcastle, UK), early neuronal differentiation marker goat anti-doublecortin polyclonal (DCX, 1:400 Santa Cruz, Santa Cruz, California), rabbit anti-PS1NTF polyclonal (1:5000, a generous gift from Dr. Gopal Thinakaran), 6E10 mouse anti- human Aβ monoclonal antibodies (1:1000, Chemicon, Billerica, MA), 369 mouse anti-APP (1:1000, generous gift from Dr. Sangram S. Sisodia), PHF-1 mouse anti-phosphorylated tau [Ser396 and Ser404 PHF-1 (Otvos et al. 1994) 1:2500], Tau-5 mouse anti-Tau monoclonal (1:1,000, Chemicon), R1 rabbit anti-tau polyclonal (1:1000, a generous gift from Dr. Lester Binder), rabbit anti-glial fibrillary acidic protein polyclonal (GFAP; 1:400, Dako, Glostrap, Denmark), mouse anti-NeuN monoclonal (1:400, Chemicon), mouse anti-actin monoclonal (1:5000, Chemicon), mouse anti-TUJ-1 monoclonal (1:5000, Promega, Madison, WI). Secondary antibodies were donkey anti-goat Cy5 (1:250, Jackson ImmunoResearch, West Grove, PA), donkey anti-mouse Cy3 (1:500, Jackson ImmunoResearch), donkey anti-rabbit Cy5 (1:250, Jackson ImmunoResearch), donkey anti-rabbit biotin (1:250, Jackson ImmunoResearch), donkey anti-mouse biotin (1:250, Jackson ImmunoResearch), Cy2 Streptavadin (1:250, Jackson ImmunoResearch). Secondary antibodies used for Western blot were rabbit anti-mouse horseradish peroxidase (1:5000) and protein A-peroxidase (1:1000, Pierce, Rockford, IL).
Neurosphere Culture
Neural stem cells (NSC) were isolated from the SVZ of mice at 2 months of age and cultured as previously described (Bez et al. 2003; Lois and Alvarez-Buylla 1993; Reynolds and Weiss 1992; Vescovi et al. 1993). Briefly, a 2mm thick coronal section is cut starting ventrally 1mm behind the olfactory bulbs and subsequently removing the section by cutting 2mm dorsal to the initial section. Using a dissecting microscope, the lateral and anterior wall of the ventricles is dissected, containing the SVZ. The crude SVZ extract is chopped briefly with a sterile scalpel and the pieces collected in Hank’s Balanced Salt solution containing .005% Papain (w/v), .001% Cystine (w/v), .001% DNAse (v/v) and .001% EDTA (w/v) and incubated at 37°C for 45 minutes. Following incubation, tissue pieces are centrifuged at 1000 × g for 5 minutes and all but 300μl of supernatant is aspirated off. The tissue is then dissociated by repeated pipetting using a P200 pipette in the remaining media. Primary cultures were incubated (37°C, 5% CO2) for 10 days in complete medium [Water, DMEM-F12 (Gibco, Carlsbad, CA), glucose (Sigma, St. Louis, MO), NaHCO3 (Sigma), HEPES (Sigma), L-Glutamine (Gibco), Penicillin/Streptomycin (Gibco), Putrescine (Sigma), Apo-Transferrin (Sigma), Insulin (Roche, Indianapolis, IN), Selenium (Sigma), Progesterone (Sigma), BSA (Sigma), Heparin (Sigma), EGF (20ng/ml, Peprotech, Rocky Hill, NJ), bFGF (10ng/ml, Peprotech)] before passage. To passage, NSCs were collected in conical tubes and pelleted by centrifugation at 1000 × g for 10 minutes. Old medium was removed and cells were mechanically dissociated by 60–70 passages through a 200μl pipette. Singly dissociated cells were then plated in new medium at a density of ~20,000 cells/ml and placed back in incubation. This cell-passage procedure was repeated every 5–7 days. Cells were used for experiments once most of the debris from the primary preparation had cleared (3–4 passages) and a substantial population of cells was acquired.
Neural progenitor cell BrdU Proliferation Assay
NPC were dissociated into single cells and were plated into a 96-well plate (15,000 cells/well). Each well was treated with 5 μM BrdU and incubated for 48 hours at 37°C, 5% CO2. Cells were fixed with 70% ethanol/0.1N HCl for 30 minutes at room temperature and incubated in mouse mAb BrdU (1:300, Novocastra, Newcastle, UK) for 1 hour at room temperature. Cells were rinsed in 1XTBS three times and incubated in secondary antibody rabbit anti-mouse HRP (1:5000, Pierce) for 30 minutes at room temperature, followed by the addition of tetramethylbenzidine (TMB) substrate solution (Invitrogen) in the dark for 15 minutes. The reaction was terminated by adding 2.5N sulfuric acid into each well. Absorbance was measured using spectrophotometric plate reader at dual wavelength of 450–595 nm. Each experimental group includes 5 replicates (n=5).
BrdU injection
Animals received intraperitoneal injections of 5′-bromo-2′-deoxyuridine (BrdU, Sigma) every 12 hours for 3 days at a dose of 100 mg/kg body weight in 0.7% NaCl. All mice were anesthetized with a mixture of ketamine and xylazine and transcardially perfused with 250 ml of ice cold phosphate buffer saline followed by 4% paraformaldehyde. The brains were then removed and placed into 4% paraformaldehyde and kept at 4°C.
Immunohistochemistry
Brains from perfused mice were post-fixed in 4% paraformaldehyde for 3 days and stored in 30% sucrose at 4°C. The brains were then sectioned sagitally at 50μm using a microtome and placed into cryoprotectant (23.8% glycerol, 28.5% ethylene glycol, 47.6% PBS v/v). Brain sections were blocked using 1X PBS containing 5% normal donkey serum and .05% Triton X-100 (Sigma). Sections were then incubated for 72 hours in primary antibodies followed by 2 hours of blocking and 2 hours of incubation in secondary antibodies. Slide mounting was performed using PVA/DABCO. Negative controls employing secondary antibodies only were used to assess antibody specificity. Every sixth section was taken for immunohistological treatment and/or stereological analysis.
BrdU Pretreatment- Brain sections were incubated in deionized formamide/SSC solution (50% Deionized formamide, 10% 5x SSC v/v in water) for 2 hours at 65°C. Sections were then incubated in 2N HCl at 37°C for 30 minutes and subsequently washed with 0.1M borate buffer at room temperature for 10 minutes.
Western Blot analysis
Protein extraction from brain tissue was performed in 1X TNE lysis buffer (50mM Tris, 150mM NaCl, 5mM EDTA, Protease inhibitor cocktail (Sigma) and 100mM PMSF). Quantification of protein was performed using the BCA-method (Pierce) and equal amounts of protein were subjected to direct immunoblotting using Tris-glycine gels and transferred onto nitrocellulose membranes. For phosphorylation specific antibodies ROLB buffer [10mM HEPES, pH7.4, 0.5% Triton X-100, 80mM βglycerophosphate, 50mM sodium fluoride, 2mM sodium orthovanadate, 100nM staurosporine, 100nM K252a, 50nM okadaic acid, 50nM microcystin, mammalian protease inhibitor cocktail (Sigma), Phosphatase inhibitor cocktail II (Calbiochem)] was used.
Stereological Analysis
The number of BrdU+ and BrdU+DCX+ cells in sagittal brain sections was quantified using design-based stereology (StereoInvestigator version 7, MBF Bioscience, Williston, VT, USA). For the analysis, every sixth section of brain tissue was quantified by applying Nv × VRef method(Peterson 1999). The following parameters were used; for SVZ, sections were traced using a Zeiss AX10 microscope (Carl Zeiss Ltd., Hertfordshire, England) in low magnification (5X) and counting was performed at high magnification (63X), counting frame= 100μm×100μm, grid size 200μm×300μm and all sections were counted using 15μm top and bottom guard zones. For the dentate gyrus the counting frame was set equal to the grid size (140μm × 140μm) in order to count the entirety of the DG due to the relative paucity of cells. All other parameters remained the same.
Statistical Analysis
All error bars represent standard errors of the mean value. For statistical analysis of both the stereological data and the proliferation assay a student’s t-test was used and values that are statistically significant (P<0.05) are clearly indicated with an asterisk (*) and, where necessary, lines demarcating which two values reach significance.
Results
Proliferation of cells in the SVZ and hippocampus is significantly reduced in transgenic mice harboring FAD-linked APPswe/PS1ΔE9 as early as the age of 2 months
To determine the effect of expression of FAD-linked mutant APPswe/PS1ΔE9 on neurogenesis we first examined the extent of proliferation of NPC in the neurogenic regions, i.e., SVZ and SGL, of transgenic mice co-expressing APPswe/PS1ΔE9 mutations. For this purpose FAD-linked transgenic mice harboring APPswe/PS1ΔE9 were injected with the thymidine analog 5′-bromo-2′-deoxyuridine (BrdU) at 2 months of age, 3 months prior to onset of amyloid deposition in APPswe/PS1ΔE9 mice. Control groups of nontransgenic littermate mice and of mice harboring human wild type presenilin-1 (PS1HWT) were injected using the same regimen as transgenic mutant mice. We then quantified the number of cells immunoreactive for BrdU in brain sections of these mice. Stereological analysis of immunolabeled sections revealed that proliferation of BrdU+ cells in both the SVZ (Figure 1A) and SGL (Figure 2A) was drastically reduced in the brains of APPswe/PS1ΔE9 mice when compared with PS1HWT and nontransgenic controls. These results suggest that either the mutations intrinsically affect the proliferation of NPC or that alterations in the neurogenic niche of APPswe/PS1ΔE9 take place long before amyloid plaque formation.
Figure 1. Reduced proliferation and neuronal differentiation is an early life primary neuropathological event in FAD-linked mice co-expressing APPswe and PS1ΔE9 mutations: the subventricular zone neurogenic microenvironemnt.

(A) The number of BrdU immunoreactive cells is reduced by 36.09% in the subventricular zone of mice co-expressing APPswe/PS1ΔE9 mutations when compared with PS1HWT or non-transgenic littermates. (B) The number of BrdU immunoreactive cells that are co-labeled with the immature neuronal marker doublecortin (DCX) is dramatically reduced in APPswe/PS1ΔE9 mice when compared with PS1HWT or nontransgenic controls. (C) Representative images of BrdU immunoreactivity (red) in the subventricular zone of nontransgenic (top) and APPswe/PS1ΔE9 (bottom) mice (*p ≤ 0.005, students t-test). Scale bar= 100μm.
Figure 2. Reduced proliferation and neuronal differentiation is an early life primary neuropathological event in FAD-linked mice co-expressing APPswe and PS1ΔE9 mutations: the dentate gyrus neurogenic microenvironemnt.

(A) The number of BrdU immunoreactive cells in the dentate gyrus of APPswe/PS1ΔE9 mice is greatly reduced when compared with PS1HWT or non-transgenic littermates. (B) As in the SVZ, the number of BrdU immunoreactive cells co-expressing doublecortin is also vastly reduced in the FAD-linked APPswe/PS1ΔE9 mice. (C) Representative images of BrdU immunoreactive cells in the dentate gyrus of nontransgenic (top) and APPswe/PS1ΔE9 (bottom) mice (*p ≤ 0.05, students t-test). Scale bar= 100μm.
Formation of new neurons is impaired in APPswe/PS1ΔE9 transgenic mice at 2 months of age
To examine whether reduced extent of proliferation of NPC in the brains of mice harboring FAD- linked mutant APPswe/PS1ΔE9 manifests in reduced number of new neurons, brain sections were immunolabeled with antibodies raised against BrdU and with antibodies raised against the early neuronal differentiation marker doublecortin (DCX). Newly differentiating neurons expressing BrdU+DCX+ were quantified in the SGL and SVZ of PS1HWT, APPswe/PS1ΔE9 and nontransgenic mice using unbiased stereology. We observed a drastic reduction in the number of BrdU+/DCX+ cells in both the SVZ (Figure 1B) and SGL (Figure 2B), suggesting that extent of neuronal differentiation is decreased in these mice. Taken together, these results suggest that proliferation of NPC and their differentiation into neurons is impaired in both the SVZ and SGL of APPswe/PS1ΔE9 transgenic mice, and that these impairments take place very early in life.
APP misprocessing and amyloid pathology in the neurogenic areas of FAD-linked transgenic mice harboring APPswe/PS1ΔE9 mutations
To examine the mechanism underlying impaired neurogenesis we addressed the possibility that alterations in neurogenesis are the result of FAD-induced APP misprocessing (Borchelt et al. 1996; Thinakaran et al. 1996) in the neurogenic microenvironments. For this purpose, expression of APP metabolites was examined in protein extracts prepared from the neurogenic (i.e. SVZ and hippocampus) and non-neurogenic (i.e., cortex) areas of APPswe/PS1ΔE9 mice using Western blot analysis, and compared these to expression levels in mice harboring FAD -linked PS1HWT and PS1ΔE9. As expected, steady state levels of full length APP (FL-APP) were 2–3 fold higher in all brain areas of APPswe/PS1ΔE9 mice compared to endogenous levels expressed in PS1HWT and PS1ΔE9 mice (Figure 3A, see FL-APP panel) (Thinakaran et al. 1996). In addition, levels of carboxyl-terminal fragments (APP-CTFs) were up regulated in protein extracts of APPswe/PS1ΔE9 brains (Figure 3A see APP-CTFs 369 panel). Significantly, levels of APP-CTFs compared to FL-APP were increased in APPswe/PS1ΔE9 mice in the neurogenic regions in particular (Figure 3D, top). To examine the nature of APP-CTFs that were increased in the neurogenic areas of APPswe/PS1ΔE9 mice, we reprobed the membrane with 6E10 antibodies recognizing amino acids 1–16 of Aβ. Levels of 6E10 revealed a dramatic increase in β-CTFs in APPswe/PS1ΔE9 mice (Figure 3A, see APP-CTFs 6E10 panel; Figure 3D bottom right), with particularly high levels in the SVZ of these mice (Figure 3A, see APP-CTFs 6E10 panel; Figure 3D bottom right), suggesting increased enzymatic activity of Bace-1 in the brain of APPswe/PS1ΔE9 mice (Thinakaran et al. 1996), and in the neurogenic SVZ in particular. Next we examined whether increased enzymatic activity of Bace-1 is manifested by increased Aβ levels. Western blot analysis revealed that Aβ levels are high in the hippocampus and cortex of APPswe/PS1ΔE9 mice, but significantly lower in the SVZ of these mice (Figure 3 C,D bottom left). Taken together, these results suggest that APP misprocessing is pronounced in the neurogenic niches in the brains of APPswe/PS1ΔE9 and manifested in increased levels of APP-CTFs, and β-CTFs in particular. This increased processing of APP suggests high activity levels of Bace-1 and dysfunction of γ-secretase. Intriguingly, in spite of apparently high levels of β-CTF production, Aβ levels are particularly low in the SVZ, suggesting either alterations in the half life of APP metabolites in this region, high activity level of Aβ-degrading enzymes or enhanced Aβ-clearance mechanisms in this neurogenic microenvironment. These results further suggest that while high Aβ levels play a role in altered neurogenesis in the hippocampus, they may not be a primary factor in the alterations in neurogenesis in the SVZ of APPswe/PS1ΔE9 mice.
Figure 3. Alterations in APP metabolites in the neurogenic microenvironments of APPswe/PS1ΔE9 mice.

(A) Steady state levels of APP metabolites and PS1 in protein extract prepared from the SVZ, hippocampus and cortex of FAD-linked transgenic mice. FL-APP panel: Comparable expression levels of full-length APP in brain samples of PS1HWT and PS1ΔE9. Note over-expression of APP in transgenic mice harboring FAD-linked APPswe/PS1ΔE9. APP-CTF-369 panel: Increase in APP-CTFs in the cortex and neurogenic areas of APPswe/PS1ΔE9 due to transgene expression. APP-CTF-6E10 panel: Increase in APPswe-derived β-CTFs, in the SVZ of APPswe/PS1ΔE9 mice. PS1 panel: levels of transgenic PS1HWT N-terminal fragments (PS1NTF) and PS1ΔE9 are comparable in all brain areas. (B) Schematic presentation epitope binding site of 6E10 and 369 antibodies to APP. (C) Western blot analysis of soluble Aβ in protein extract prepared from the SVZ, hippocampus and cortex of APPswe/PS1ΔE9 mice revealing high levels of soluble Aβ in the cortex and hippocampus but virtually undetectable levels in the SVZ. Levels of full length APP (FL-APP) were comparable in the different regions. (D) Quantification of protein expression level of APP-CTFs relative to FL-APP (upper panel); APPβ-CTFs relative to FL-APP (bottom left panel); Aβ relative to FL-APP (bottom right panel). Error bars represent S.E.M.
Hyperphosphorylation of tau in neurogenic microenvironments in FAD-linked APPswe/PS1ΔE9 transgenic mice
To examine the possibility that alterations in tau phosphorylation take place in the neurogenic niches of APPswe/PS1ΔE9 mice and may underlie impaired neurogenesis, we examined expression levels of tau in the brains of these mice. For this purpose, we prepared protein extracts of the SVZ, hippocampus and a non-neurogenic area (cortex or cerebellum), as before, and compared tau expression across FAD transgenic mice harboring PS1HWT, PS1ΔE9 and APPswe/PS1ΔE9 using phosphorylation-dependent antibodies. To examine alterations in tau phosphorylation we used AT8 antibodies that require tau protein to be phosphorylated at both serine 202 and threonine 205 (Goedert et al. 1995). These phosphorylation-dependent antibodies recognize hyperphosphorylated tau in paired helical filaments and neurofibrillary tangles (Biernat et al. 1992; Goedert et al. 1993; Mercken et al. 1992). Analysis of AT8 levels in protein extracts revealed a significant increase in tau phosphorylation in the SVZ, hippocampus and cortex of APPswe/PS1ΔE9 mice compared to equivalent brain regions of PS1ΔE9 and PS1HWT mice. Quantification of AT8 levels relative to total levels of tau revealed a dramatic increase in phosphorylated tau in the brains of APPswe/PS1ΔE9 mice (Figure 4A,B). These results strongly suggest that increased phosphorylation of tau is pronounced in the brains of these mice, and in the neurogenic niches in particular. To further investigate alterations in tau phosphorylation we used PHF-1 monoclonal antibodies that recognize phosphorylated epitopes at Ser-396/Ser-404 of tau (Otvos et al. 1994). The results show a dramatic increase in PHF-1 expression in the neurogenic areas and in the SVZ in particular in the mutant mice APPswe/PS1ΔE9 and PS1ΔE9 compared to PS1HWT (Figure 4C,D). Interestingly, in the cortex, thought to be a non-neurogenic area, PHF-1 levels in mice expressing APPswe/PS1ΔE9, PS1ΔE9 and PS1HWT are comparable (Figure 4C). Taken together, these results suggest that significant increases in tau phosphorylation in epitopes identified as clinically-relevant to Alzheimer’s disease, occur in the neurogenic areas in the brains of APPswe/PS1ΔE9 mice and may underlie impaired neurogenesis. These results further raise the possibility that increased levels of phosphorylated tau are the result of either altered regulation or function of kinases or phosphatases in the neurogenic microenvironments of APPswe/PS1ΔE9 mice.
Figure 4. Increased levels of phosphorylated tau in the neurogenic regions of APPswe/PS1ΔE9 mice.

(A) Expression levels of total tau and phosphorylated tau in protein lysates of SVZ, hippocampus and cerebellum of PS1HWT, PS1ΔE9 and APPswe/PS1ΔE9 mice, as detected by Western blot analysis using tau-5 and AT-8 antibodies, respectively. A dramatic increase in tau phosphorylated at Ser-202/Thr-205 was detected by AT8 antibodies in all regions tested. (B) Quantification of the amount of AT8/actin relative to tau/actin. Error bars represent S.E.M. (C) Levels of phosphorylated tau in protein lysates of SVZ, hippocampus and cortex of PS1HWT, PS1ΔE9 and APPswe/PS1ΔE9 mice as detected by PHF-1 antibodies. Western blot analysis shows a marked increase in tau phosphorylated at Ser-396/Ser-404 in the neurogenic regions but not in the cortex. (D) Quantification of the relative amount of PHF-1 tau to β-tubulin in arbitrary units (A.U.).
PHF-1 is expressed in newly generated cells in the SVZ and hippocampus of APPswe/PS1ΔE9 mice
To determine tau expression in NPC and its significance in their proliferation, development, migration and neuronal maturation we characterized tau expression in NPC specifically in the neurogenic niches. Triple-immunolabeling of brain sections of PS1HWT and APPswe/PS1ΔE9 mice with anti BrdU, anti- glial fibrillary acidic protein (GFAP) and anti tau-5 antibodies shows that in neurogenic areas tau co-localized with BrdU and GFAP, indicating that newly generated neural stem cells express tau (Figure 5C,D). In addition, in the rostral migratory stream (RMS), the path by which neuroblasts migrate to the olfactory bulb, we detected tau-5 immunostaining in proliferating BrdU+ cells expressing the early neuronal marker DCX (Figure 5A,B). Taken together, these results suggest that tau is expressed in neural stem cells, progenitor cells and neuroblasts, and imply that hyperphosphorylation of tau, as detected above in the neurogenic areas may have a direct impact on these cells. To determine this, we examined whether PHF-1 levels are pronounced in NPC present in APPswe/PS1ΔE9 mice by co- immunostaining brain sections of PS1HWT (Figure 6 C,D) and APPswe/PS1ΔE9 (Figure 6 A,B) mice with PHF-1 antibodies and DCX. In agreement with the Western blot analysis of PHF-1 expression in neurogenic areas (Figure 4A,B), this immunolabeling revealed that PHF-1 immunoreactivity was significantly higher in brain sections from APPswe/PS1ΔE9 mice compared to the PS1HWT in the neurogenic microenvironments in particular (Figure 6). In addition, colocalization of PHF-1 and DCX in APPswe/PS1ΔE9-expressing neuroblasts suggests that hyperphosphorylated tau is pronounced in newly differentiating neurons and migrating neuroblasts in these mice (Figure 6). These results imply that tau hyperphosphorylation may underlie impaired proliferation and neuronal maturation in APPswe/PS1ΔE9 mice.
Figure 5. Tau is expressed in neural stem cells, neural progenitor cells and migrating neuroblasts.
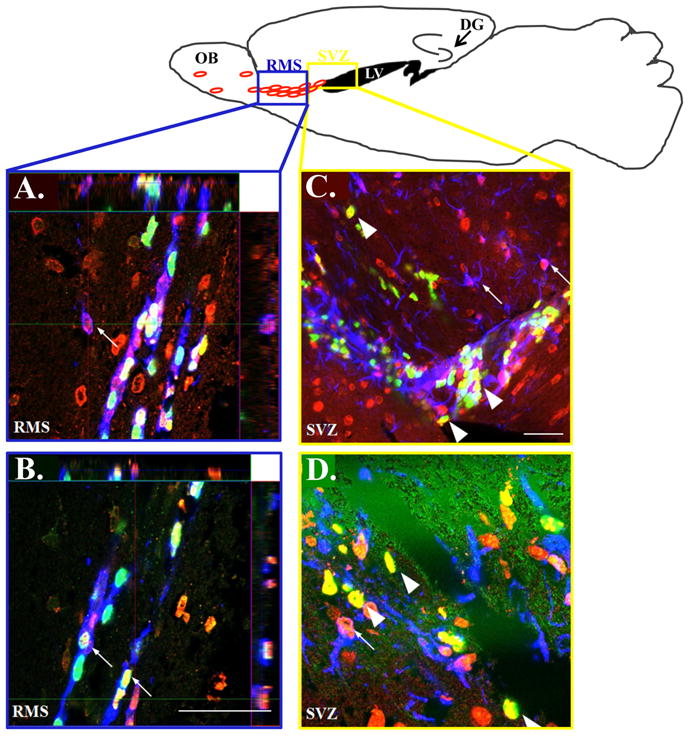
TOP: Schematic presentation showing the neurogenic niches in a sagittal slice through the mouse brain: the region outlined by the blue box is representative of the area from which confocal RMS (A,B) images were taken and the yellow box represents the region corresponding to the SVZ images (C,D). Confocal imaging of immunolabeled brain sections of APPswe/PS1ΔE9 mice shows that tau co-localizes with doublecortin (small arrows; A,B), BrdU (A–D big arrows in C,D) and GFAP (small arrows; C,D). Tau, red (A–D); BrdU, green (A–D); doublecortin, blue (A,B); GFAP, blue (C,D). Scale bar= 50μm
Figure 6. PHF-1 is expressed in neuroblasts in the subventricular zone of APPswe/PS1ΔE9 mice.

Upper Scheme: Schematic presentation of the area in the SVZ from which confocal images were taken. Left panel: PHF-1 expression is pronounced in DCX+ neuroblasts in the SVZ of APPswe/PS1ΔE9 (A) but hardly in PS1HWT (C) mice at 6 months of age. Right panel: High power orthogonal image showing strong co-localization of PHF-1 (green) and DCX (red) in the SVZ of APPswe/PS1ΔE9 (B) and no co-localization in PS1HWT mice (D). Blue arrows represent clear co-localization in the orthogonal view of APPSwe/PS1ΔE9 sections while the orange represent DCX+ cells in the PS1HWT SVZ that are clearly not PHF-1+. Scale bars= 50μm.
Neurospheres isolated from APPswe/PS1ΔE9 exhibit impaired proliferation and tau hyperphosphorylation
To address whether expression of FAD-linked APPswe/PS1ΔE9 mutations intrinsically affects neural progenitor cell proliferation, we isolated neurospheres from the SVZ of 2 month old APPswe/PS1ΔE9 and their nontransgenic littermates and examined the extent of proliferation by utilizing a BrdU incorporation assay [(Calbiochem); Figure 7A]. We observed a significant reduction in the extent of proliferation of neurospheres isolated from the SVZ of APPswe/PS1ΔE9 transgenic mice compared to neurospheres isolated from the SVZ of their nontransgenic littermates (Figure 7A), suggesting that expression of APPswe/PS1ΔE9 mutations intrinsically impairs proliferation of neural progenitor cells, independently of the neurogenic microenvironment. We next examined whether APPswe/PS1ΔE9-expressing neural progenitor cells isolated from this area exhibit tau hyperphosphorylation. We extracted protein from neurospheres isolated from the SVZ of APPswe/PS1ΔE9 mice and nontransgenic littermate controls and examined tau expression by Western blot analysis. Intriguingly, we observed that while total tau levels are comparable in APPswe/PS1ΔE9 and nontransgenic neurospheres, phosphorylated tau levels as detected by AT-8 antibodies are significantly higher in neurospheres derived from the APPswe/PS1ΔE9 mice (Figure 7B,C). These results suggest that APPswe/PS1ΔE9 mutant forms induce hyperphosphorylation of tau in NPC and reduce their proliferation.
Figure 7. Neurospheres derived from the SVZ of APPSwe/PS1ΔE9 mice exhibit impaired proliferation and increased tau phosphorylation.

(A) Proliferation assay examining BrdU incorporation in dissociated neurospheres shows a reduction in the proliferative capacity of neural progenitor cells derived from APPswe/PS1ΔE9 mice, compared to neural progenitor cells derived from nontransgenic littermates. (B) Western blot analysis of tau levels in protein extracts of neurospheres isolated from APPSwe/PS1ΔE9 and nontransgenic littermate mice. Total tau expression levels are comparable in neurospheres derived from APPSwe/PS1ΔE9 and nontransgenic mice compared to actin levels. However, a dramatic increase is detected in levels of phosphorylated tau in neurospehers derived APPSwe/PS1ΔE9 using AT-8 antibodies. Neurospheres isolated from APPSwe/PS1ΔE9 mice exhibit characteristic transgene expression pattern of APP (2–3 fold increase in FL-APP) and PS1 (lack of full length cleavage of PS1ΔE9). (C) Quantification of protein levels as detected in Western blot shows that the levels of AT8 are increased 3-fold relative to total tau (top) while tau levels are consistent when normalized to actin (bottom). Error bars represent S.E.M. (*p≤0.05, students t-test).
Discussion
This study shows for the first time that impaired neurogenesis is an early event in the development of AD pathology and may underlie functional and behavioral deficits characterizing AD, i.e., progressive loss of hippocampus- and olfaction-dependent learning and memory. In addition, this study reveals that tau hyperphosphorylation underlies, at least in part, impaired neurogenesis in the brains of mice harboring FAD-linked APPswe/PS1ΔE9. We show that tau is expressed in neural stem cells, neural progenitor cells and neuroblasts, and its hyperphosphorylated forms are pronounced in neural progenitor cells of APPswe/PS1ΔE9 mice.
The maintenance of pools of neural stem cells in the post-natal brain is increasingly recognized to play critical roles in learning and memory in both the olfaction system and the hippocampus, as newly-differentiating neurons incorporate in the olfactory bulb and granule layer of the dentate gyrus, respectively, and are thought to play a role in numerous aspects of learning and memory (Imayoshi et al. 2008; Zhao et al. 2008). Thus a deficiency in these processes at a very early stage of AD may cause or contribute to dysfunction of the olfaction system and hippocampus, leading to memory impairments. Very little is known about neurogenesis in humans affected with AD and in individuals with mild cognitive impairments (MCI). Numerous studies have suggested that the extent of neurogenesis in both the SVZ and DG declines with age, raising the possibility that reduced neurogenesis may account for, at least in part, impaired learning and memory and cognitive deterioration in the elderly (Kempermann et al. 1998; Kempermann et al. 2002; Kuhn et al. 1996; Seki and Arai 1995; Tropepe et al. 1997). Examination of neurogenesis in brain tissue of AD patients revealed increased expression of immature neuronal marker proteins(Jin et al. 2004b). However, these observations have been challenged recently (Boekhoorn et al. 2006). Other reports suggest that in the aged and AD brain, there is a significant decline in the extent of proliferation of progenitor cells and their numbers [For review see (Brinton and Wang 2006)].
Animal models are a critical and important tool in the examination of neurogenesis in the post-natal brain, as they enable a temporal and spatial analysis of the course of neurogenesis in relation to progression of neuropathology. This is particularly critical in AD, where neurogenesis may be altered in different ways during the progression of the disease, in response to events such as amyloid deposition (Faure et al. 2009; Jin et al. 2004a; Verret et al. 2007; Zhang et al. 2007), neurodegeneration and neuronal loss (Chen et al. 2008) and inflammation. Temporal proximity of alterations in neurogenesis to these neuropathological events raises the possibility that altered neurogenesis is a side effect, rather than a cause in AD. For example, three recent papers used similar transgenic animals to the APPswe/PS1ΔE9 mice we used in this study. Li and colleagues (2008) examined extent of cell proliferation in the SGL at age of 6 and 9 months (Li et al. 2008). While both time points are post amyloid deposition, they find a significantly reduced cell proliferation only at the 9 months group, suggesting that other factors aside from amyloid deposition may induce these alterations, and that neurogenesis changes as a function of age-specific neuropathology. Taniuchi and colleagues (Taniuchi et al. 2007) examined the number of proliferating and newly differentiating cells in the SGL of 5 and 9 month old APPswe/PS1ΔE9 mice. This study shows significant reductions in cell proliferation and early neuronal differentiation 9 months of age. Similarly to Li et al, both time points are post onset of amyloid deposition, and reflect changes in neurogenesis that take place at these specific time points. Another study of the same research group (Niidome et al. 2008) examined the number of proliferating cell nuclear antigen (PCNA)-positive cells in the SGL and SVZ of APPswe/PS1ΔE9 mice at age of 9 months and found no difference in the number of PCNA-positive cells in these areas in APPswe/PS1ΔE9 mice when compared with wild type mice. In contrast, they observed a reduction in the number of BrdU+ cells in the SGL but not SVZ of APPswe/PS1ΔE9 mice compared with wild type mice (Niidome et al. 2008). While the discrepancy between the observations using PCNA and BrdU as markers for cell proliferation is not fully addressed, this study, again, focuses on alterations in cell proliferation at 9 months of age, post amyloid deposition. In addition, both Niidome et al. and Li et al. examine cell proliferation solely, without any lineage-specific markers, and the relevance of these studies to alterations in neurogenesis is highly questionable. Finally, these studies fail to address whether neurogenesis is altered pre-onset of amyloid deposition, as a proof of concept. As APPswe and PS1ΔE9 mutant transgenes are expressed constitutively during development as well as post-natally, it is reasonable to assume that alterations in neurogenesis may take place early, prior to amyloid deposition.
Hence, we aimed at examining whether alterations in neurogenesis are an early event that plays a role in the neuropathology of the disease. As APPswe/PS1ΔE9 mice exhibit amyloid deposition in the neurogenic niche of the hippocampus 4–5 months of age, a process which, as discussed above, may alter neurogenesis (Verret et al. 2007; Zhang et al. 2007) we targeted neurogenesis at 2 months of age. The observation that proliferation and early differentiation of neural progenitor cells is severely impaired in the SVZ and in the SGL of the dentate gyrus of these mice as early as two months of age, long before amyloid deposition or memory impairments(Jankowsky et al. 2005; Lalonde et al. 2005), suggests that impaired neurogenesis is a contributing factor in Alzheimer’s pathology, rather than a consequence of neuronal dysfunction, and may underlie or exacerbate memory impairments and neuronal vulnerability in these brain areas.
While the molecular signaling pathways regulating neurogenesis in the post-natal brain are increasingly unraveled, the molecular mechanism underlying impaired neurogenesis in AD is poorly understood. To begin exploring this we first determined alterations of molecular determinants of AD, as they are pronounced in the neurogenic niches. We determined that while altered APP processing occurs in the SVZ and hippocampus, levels of soluble Aβ are relatively low in the SVZ, suggesting that Aβ levels and fibrillogenesis may differentially affect the neurogenic niche in the SVZ and the SGL of the dentate gyrus. Nevertheless, we found striking increases in levels of phosphorylated tau in the SVZ and the SGL of APPswe/PS1ΔE9 mice, suggesting that tau mis-phosphorylation may play a role in impaired neurogenesis in these mice. Hyperphosphorylation of tau in AD is thought to be the primary event inducing detachment of tau from microtubules (Bramblett et al. 1993), leading to filament formation and subsequently to accumulation of tau aggregates intracellularly [For review see (Binder et al. 2005)]. Notably, the density of tau inclusions correlates with cognitive decline in the disease(Arriagada et al. 1992; Ghoshal et al. 2002; Mitchell et al. 2002). Phosphorylation of tau occurs early in the pathogenesis of neurofibrillary tangle formation(Biernat et al. 1992). Several phosphorylation sites on tau, including Ser396, one of the epitopes recognized by PHF-1 antibodies (Otvos et al. 1994), modulate binding to microtubules (Bramblett et al. 1993). Hyperphosphorylation of these sites may decrease the affinity of tau for microtubules, leading to their depolymerization (Bramblett et al. 1993; Lindwall and Cole 1984). Functional implications of this process are far-reaching when it comes to developing neural progenitor cells, affecting mitosis, axonal transport, process elongation and neuronal maturation [For review see (Johnson and Stoothoff 2004)]. Previous studies observed hyperphosphorylation of tau, dystrophic neuritis and structures resembling paired helical filaments in the cortex and hippocampus of a similar animal model (Kurt et al. 2003). We observed that tau-5 immunoreactivity was colocalized with BrdU, GFAP and DCX, suggesting that alterations in tau phosphorylation may be detrimental to neural stem cells, neural progenitor cells and neuroblasts. Interestingly, recent evidence suggests that hyperphosphorylation of tau can lead to chromosome aberrations (Rossi et al. 2008) and subsequently obstructed mitosis. This may imply that reduced proliferation of progenitor cells, as was observed in vivo and in vitro, may be partially due to hyperphosphorylation of tau in these progenitors. Finally, several phosphatases and kinases have been proposed to play a role in tau phosphorylation [For review see (Trojanowski and Lee 1994; Trojanowski and Lee 1995)], such as Glycogen synthase kinase 3 (GSK3) and casein kinase 2 (CK2) (Morfini et al. 2002a; Wang et al. 1998). PHF-1 antibodies used in our preliminary studies recognize phosphorylated epitope at Ser-396/Ser-404 of tau (Hernandez et al. 2003; Seubert et al. 1995), thought to be preferentially phosphorylated by GSK3 (Godemann et al. 1999; Lee et al. 2003). Our data suggests that alterations in these enzymatic activities may take place in the neurogenic niches of APPswe/PS1ΔE9 mice and may underlie tau hyperphosphorylation in these brain areas. GSK3 is a component of the WNT signaling pathway, that plays a major role in adult neurogenesis (Lie et al. 2005), microtubule dynamics and fast axonal transport (Frame and Cohen 2001; Morfini et al. 2002b). GSK3 is inactivated by phosphorylation at serine 9 (Ser9) in its N′-terminus. Among GSK3’s numerous substrates are PS1, β-catenin, tau, and kinesin-I light chains (KLC) (Morfini et al. 2002b; Takashima et al. 1998; Tesco and Tanzi 2000) [For review see (Frame and Cohen 2001)]. Previous studies suggest that FAD-linked PS1 mutations affect GSK3 kinase activity in transfected cell lines (Takashima et al. 1998; Weihl et al. 1999a; Weihl et al. 1999b). Importantly, these kinases and phosphatases are known to inhibit fast axonal transport (Morfini et al. 2002a; Morfini et al. 2002b). In that regard, phosphorylation of KLC by GSK3 promotes the release of kinesin-I from membrane-bound organelles, leading to a reduction in fast anterograde axonal transport (Morfini et al. 2002a). Taken together, this evidence raises the possibility that axonal transport may be dysfunctional in newly formed neuroblasts, a hypothesis that is supported by our observation that PHF-1 immunoreactivity is pronounced in neuroblasts in the RMS of APPswe/PSΔE9 mice.
It has become increasingly clear that both APP and PS1 play major roles in neurogenesis in the post-natal brain, regulating proliferation, survival and differentiation of neural stem and progenitor cells. Hence, misregulation or dysfunction of these molecules, such as in AD, may compromise these processes or alter neurogenesis [For review see(Lazarov 2009a)]. PS1 has increasingly been considered an appealing signal in fundamental processes, such as Notch signaling, Wnt/β-catenin signaling, E-cadherin and ErbB-4-mediated signaling (De Strooper et al. 1999; Lie et al. 2005; Sardi et al. 2006; Tesco et al. 1998; Weihl et al. 1999a). ErbB-4 (HER4) belongs to the epidermal growth factor (EGF) family that comprises related receptor tyrosine kinases. Importantly, mice with genomic deletions of PSEN1 exhibit severely abnormal somitogenesis and neurogenic processes in the brain (Shen et al. 1997). Studies in transgenic mice harboring FAD-linked mutant PS1, reveal impaired hippocampal neurogenesis (Chevallier et al. 2005; Feng et al. 2001; Haughey et al. 2002a; Wang et al. 2004; Wen et al. 2004; Zhang et al. 2007). Interestingly, soluble APP (sAPP) has been shown to act as neurotrophic factor of EGF-responsive progenitor cells in the SVZ.(Caille et al. 2004; Ohsawa et al. 1999). By the examination of isolated neurospheres, in this study we determined that APPswe/PS1ΔE9 mutantations intrinsically impair the proliferation of neural progenitor cells in culture and induce tau hyperphosphorylation.
In summary, this study suggests that neural progenitor cells are affected, early and severely, in the APPswe/PS1ΔE9 animal model, in both the hippocampus and SVZ, leading to impaired neurogenesis. Expression of FAD mutations induces intrinsic alterations in NPC as well as alters their neurogenic microenvironments. We propose that these impairments in neurogenesis may contribute to hippocampal and olfactory dysfunction, as well as to neuronal vulnerability in AD.
Acknowledgments
This study was supported by NIH R01AG033570, Alzheimer’s Association Young Investigator Award and the Illinois Department of Public Health ADRF award (OL). We thank Dr. Ernesto R. Bongarzone (University of Illinois at Chicago) for his help and advice in establishing neurosphere cultures. We thank Dr. Sangram S. Sisodia (University of Chicago) for providing us with the transgenic animals and with 369 antibodies, Dr. Gopal Thinakaran for PS1NTF antibodies (University of Chicago), Dr. Lester Binder (Northwestern University) for the R1 antibodies and Drs. Gustavo Pigino and Scott Brady (University of Illinois at Chicago) for the PHF-1 antibodies.
References
- Aimone JB, Wiles J, Gage FH. Potential role for adult neurogenesis in the encoding of time in new memories. Nature neuroscience. 2006;9(6):723–727. doi: 10.1038/nn1707. [DOI] [PubMed] [Google Scholar]
- Albers MW, Tabert MH, Devanand DP. Olfactory dysfunction as a predictor of neurodegenerative disease. Curr Neurol Neurosci Rep. 2006;6(5):379–386. doi: 10.1007/s11910-996-0018-7. [DOI] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42(3 Pt 1):631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Ashe KH. Learning and memory in transgenic mice modeling Alzheimer’s disease. Learning & memory (Cold Spring Harbor, NY. 2001;8(6):301–308. doi: 10.1101/lm.43701. [DOI] [PubMed] [Google Scholar]
- Bacon AW, Bondi MW, Salmon DP, Murphy C. Very early changes in olfactory functioning due to Alzheimer’s disease and the role of apolipoprotein E in olfaction. Ann N Y Acad Sci. 1998;855:723–731. doi: 10.1111/j.1749-6632.1998.tb10651.x. [DOI] [PubMed] [Google Scholar]
- Bez A, Corsini E, Curti D, Biggiogera M, Colombo A, Nicosia RF, Pagano SF, Parati EA. Neurosphere and neurosphere-forming cells: morphological and ultrastructural characterization. Brain research. 2003;993(1–2):18–29. doi: 10.1016/j.brainres.2003.08.061. [DOI] [PubMed] [Google Scholar]
- Biernat J, Mandelkow EM, Schroter C, Lichtenberg-Kraag B, Steiner B, Berling B, Meyer H, Mercken M, Vandermeeren A, Goedert M, et al. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. The EMBO journal. 1992;11(4):1593–1597. doi: 10.1002/j.1460-2075.1992.tb05204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, Berry RW. Tau, tangles, and Alzheimer’s disease. Biochimica et biophysica acta. 2005;1739(2–3):216–223. doi: 10.1016/j.bbadis.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Boekhoorn K, Joels M, Lucassen PJ. Increased proliferation reflects glial and vascular-associated changes, but not neurogenesis in the presenile Alzheimer hippocampus. Neurobiol Dis. 2006;24(1):1–14. doi: 10.1016/j.nbd.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17(5):1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron. 1993;10(6):1089–1099. doi: 10.1016/0896-6273(93)90057-x. [DOI] [PubMed] [Google Scholar]
- Brinton RD, Wang JM. Therapeutic potential of neurogenesis for prevention and recovery from Alzheimer’s disease: allopregnanolone as a proof of concept neurogenic agent. Curr Alzheimer Res. 2006;3(3):185–190. doi: 10.2174/156720506777632817. [DOI] [PubMed] [Google Scholar]
- Caille I, Allinquant B, Dupont E, Bouillot C, Langer A, Muller U, Prochiantz A. Soluble form of amyloid precursor protein regulates proliferation of progenitors in the adult subventricular zone. Development (Cambridge, England) 2004;131(9):2173–2181. doi: 10.1242/dev.01103. [DOI] [PubMed] [Google Scholar]
- Chen Q, Nakajima A, Choi SH, Xiong X, Sisodia SS, Tang YP. Adult neurogenesis is functionally associated with AD-like neurodegeneration. Neurobiology of disease. 2008;29(2):316–326. doi: 10.1016/j.nbd.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevallier NL, Soriano S, Kang DE, Masliah E, Hu G, Koo EH. Perturbed neurogenesis in the adult hippocampus associated with presenilin-1 A246E mutation. Am J Pathol. 2005;167(1):151–159. doi: 10.1016/S0002-9440(10)62962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398(6727):518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- Donovan MH, Yazdani U, Norris RD, Games D, German DC, Eisch AJ. Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer’s disease. The Journal of comparative neurology. 2006;495(1):70–83. doi: 10.1002/cne.20840. [DOI] [PubMed] [Google Scholar]
- Ermini FV, Grathwohl S, Radde R, Yamaguchi M, Staufenbiel M, Palmer TD, Jucker M. Neurogenesis and alterations of neural stem cells in mouse models of cerebral amyloidosis. The American journal of pathology. 2008;172(6):1520–1528. doi: 10.2353/ajpath.2008.060520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure A, Verret L, Bozon B, El Tannir, El Tayara N, Ly M, Kober F, Dhenain M, Rampon C, Delatour B. Impaired neurogenesis, neuronal loss, and brain functional deficits in the APPxPS1-Ki mouse model of Alzheimer’s disease. Neurobiology of aging. 2009 doi: 10.1016/j.neurobiolaging.2009.03.009. [DOI] [PubMed] [Google Scholar]
- Feng R, Rampon C, Tang YP, Shrom D, Jin J, Kyin M, Sopher B, Miller MW, Ware CB, Martin GM, Kim SH, Langdon RB, Sisodia SS, Tsien JZ. Deficient neurogenesis in forebrain-specific presenilin-1 knockout mice is associated with reduced clearance of hippocampal memory traces. Neuron. 2001;32(5):911–926. doi: 10.1016/s0896-6273(01)00523-2. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. The Biochemical journal. 2001;359(Pt 1):1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L, Qiao S, Lan X, Chi L, Luo C, Lien L, Yan Liu Q, Liu R. Neurogenic responses to amyloid-beta plaques in the brain of Alzheimer’s disease-like transgenic (pPDGF-APPSw, Ind) mice. Neurobiology of disease. 2008;29(1):71–80. doi: 10.1016/j.nbd.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoshal N, Garcia-Sierra F, Wuu J, Leurgans S, Bennett DA, Berry RW, Binder LI. Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer’s disease. Experimental neurology. 2002;177(2):475–493. doi: 10.1006/exnr.2002.8014. [DOI] [PubMed] [Google Scholar]
- Godemann R, Biernat J, Mandelkow E, Mandelkow EM. Phosphorylation of tau protein by recombinant GSK-3beta: pronounced phosphorylation at select Ser/Thr-Pro motifs but no phosphorylation at Ser262 in the repeat domain. FEBS letters. 1999;454(1–2):157–164. doi: 10.1016/s0014-5793(99)00741-3. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Crowther RA, Six J, Lubke U, Vandermeeren M, Cras P, Trojanowski JQ, Lee VM. The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(11):5066–5070. doi: 10.1073/pnas.90.11.5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neuroscience letters. 1995;189(3):167–169. doi: 10.1016/0304-3940(95)11484-e. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Liu D, Nath A, Borchard AC, Mattson MP. Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid beta-peptide: implications for the pathogenesis of Alzheimer’s disease. Neuromolecular Med. 2002a;1(2):125–135. doi: 10.1385/NMM:1:2:125. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Chan SL, Borchard AC, Rao MS, Mattson MP. Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer’s disease. J Neurochem. 2002b;83(6):1509–1524. doi: 10.1046/j.1471-4159.2002.01267.x. [DOI] [PubMed] [Google Scholar]
- Hernandez F, Lucas JJ, Cuadros R, Avila J. GSK-3 dependent phosphoepitopes recognized by PHF-1 and AT-8 antibodies are present in different tau isoforms. Neurobiology of aging. 2003;24(8):1087–1094. doi: 10.1016/j.neurobiolaging.2003.04.002. [DOI] [PubMed] [Google Scholar]
- Imayoshi I, Sakamoto M, Ohtsuka T, Takao K, Miyakawa T, Yamaguchi M, Mori K, Ikeda T, Itohara S, Kageyama R. Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nature neuroscience. 2008;11(10):1153–1161. doi: 10.1038/nn.2185. [DOI] [PubMed] [Google Scholar]
- Jagasia R, Song H, Gage FH, Lie DC. New regulators in adult neurogenesis and their potential role for repair. Trends in molecular medicine. 2006;12(9):400–405. doi: 10.1016/j.molmed.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Melnikova T, Fadale DJ, Xu GM, Slunt HH, Gonzales V, Younkin LH, Younkin SG, Borchelt DR, Savonenko AV. Environmental enrichment mitigates cognitive deficits in a mouse model of Alzheimer’s disease. J Neurosci. 2005;25(21):5217–5224. doi: 10.1523/JNEUROSCI.5080-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomolecular engineering. 2001;17(6):157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Jin K, Galvan V, Xie L, Mao XO, Gorostiza OF, Bredesen DE, Greenberg DA. Enhanced neurogenesis in Alzheimer’s disease transgenic (PDGF-APPSw, Ind) mice. Proc Natl Acad Sci U S A. 2004a;101(36):13363–13367. doi: 10.1073/pnas.0403678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA. Increased hippocampal neurogenesis in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004b;101(1):343–347. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GV, Stoothoff WH. Tau phosphorylation in neuronal cell function and dysfunction. J Cell Sci. 2004;117(Pt 24):5721–5729. doi: 10.1242/jcs.01558. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Brandon EP, Gage FH. Environmental stimulation of 129/SvJ mice causes increased cell proliferation and neurogenesis in the adult dentate gyrus. Curr Biol. 1998;8(16):939–942. doi: 10.1016/s0960-9822(07)00377-6. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Gast D, Gage FH. Neuroplasticity in old age: sustained fivefold induction of hippocampal neurogenesis by long-term environmental enrichment. Ann Neurol. 2002;52(2):135–143. doi: 10.1002/ana.10262. [DOI] [PubMed] [Google Scholar]
- Kesslak JP, Cotman CW, Chui HC, Van den Noort S, Fang H, Pfeffer R, Lynch G. Olfactory tests as possible probes for detecting and monitoring Alzheimer’s disease. Neurobiol Aging. 1988;9(4):399–403. doi: 10.1016/s0197-4580(88)80087-3. [DOI] [PubMed] [Google Scholar]
- Kolecki R, Lafauci G, Rubenstein R, Mazur-Kolecka B, Kaczmarski W, Frackowiak J. The effect of amyloidosis-beta and ageing on proliferation of neuronal progenitor cells in APP-transgenic mouse hippocampus and in culture. Acta neuropathologica. 2008;116(4):419–424. doi: 10.1007/s00401-008-0380-4. [DOI] [PubMed] [Google Scholar]
- Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. The Journal of neuroscience. 1996;16(6):2027–2033. doi: 10.1523/JNEUROSCI.16-06-02027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurt MA, Davies DC, Kidd M, Duff K, Howlett DR. Hyperphosphorylated tau and paired helical filament-like structures in the brains of mice carrying mutant amyloid precursor protein and mutant presenilin-1 transgenes. Neurobiology of disease. 2003;14(1):89–97. doi: 10.1016/s0969-9961(03)00084-6. [DOI] [PubMed] [Google Scholar]
- Lalonde R, Kim HD, Maxwell JA, Fukuchi K. Exploratory activity and spatial learning in 12-month-old APP(695)SWE/co+PS1/DeltaE9 mice with amyloid plaques. Neuroscience letters. 2005;390(2):87–92. doi: 10.1016/j.neulet.2005.08.028. [DOI] [PubMed] [Google Scholar]
- Lazarov O, Marr R. Neurogenesis and Alzheimer’s disease: At the Crossroads. Experimental Neurology. 2009a doi: 10.1016/j.expneurol.2009.08.009. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O, Morfini GA, Lee EB, Farah MH, Szodorai A, DeBoer SR, Koliatsos VE, Kins S, Lee VM, Wong PC, Price DL, Brady ST, Sisodia SS. Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: revisited. The Journal of neuroscience. 2005;25(9):2386–2395. doi: 10.1523/JNEUROSCI.3089-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O, Marr R. Neurogenesis and Alzheimer’s disease: At the crossroads. Experimental Neurology. 2009 doi: 10.1016/j.expneurol.2009.08.009. (In Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CW, Lau KF, Miller CC, Shaw PC. Glycogen synthase kinase-3 beta-mediated tau phosphorylation in cultured cell lines. Neuroreport. 2003;14(2):257–260. doi: 10.1097/00001756-200302100-00020. [DOI] [PubMed] [Google Scholar]
- Lee MK, Borchelt DR, Kim G, Thinakaran G, Slunt HH, Ratovitski T, Martin LJ, Kittur A, Gandy S, Levey AI, Jenkins N, Copeland N, Price DL, Sisodia SS. Hyperaccumulation of FAD-linked presenilin 1 variants in vivo. Nat Med. 1997;3(7):756–760. doi: 10.1038/nm0797-756. [DOI] [PubMed] [Google Scholar]
- Li D, Tang J, Xu H, Fan X, Bai Y, Yang L. Decreased hippocampal cell proliferation correlates with increased expression of BMP4 in the APPswe/PS1DeltaE9 mouse model of Alzheimer’s disease. Hippocampus. 2008;18(7):692–698. doi: 10.1002/hipo.20428. [DOI] [PubMed] [Google Scholar]
- Lie DC, Colamarino SA, Song HJ, Desire L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR, Gage FH. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437(7063):1370–1375. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- Lindwall G, Cole RD. Phosphorylation affects the ability of tau protein to promote microtubule assembly. The Journal of biological chemistry. 1984;259(8):5301–5305. [PubMed] [Google Scholar]
- Lois C, Alvarez-Buylla A. Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc Natl Acad Sci U S A. 1993;90(5):2074–2077. doi: 10.1073/pnas.90.5.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Toledano MA, Shelanski ML. Increased neurogenesis in young transgenic mice overexpressing human APP(Sw, Ind) J Alzheimers Dis. 2007;12(3):229–240. doi: 10.3233/jad-2007-12304. [DOI] [PubMed] [Google Scholar]
- Mercken M, Vandermeeren M, Lubke U, Six J, Boons J, Van de Voorde A, Martin JJ, Gheuens J. Monoclonal antibodies with selective specificity for Alzheimer Tau are directed against phosphatase-sensitive epitopes. Acta neuropathologica. 1992;84(3):265–272. doi: 10.1007/BF00227819. [DOI] [PubMed] [Google Scholar]
- Mirochnic S, Wolf S, Staufenbiel M, Kempermann G. Age effects on the regulation of adult hippocampal neurogenesis by physical activity and environmental enrichment in the APP23 mouse model of Alzheimer disease. Hippocampus. 2009 doi: 10.1002/hipo.20560. [DOI] [PubMed] [Google Scholar]
- Mitchell TW, Mufson EJ, Schneider JA, Cochran EJ, Nissanov J, Han LY, Bienias JL, Lee VM, Trojanowski JQ, Bennett DA, Arnold SE. Parahippocampal tau pathology in healthy aging, mild cognitive impairment, and early Alzheimer’s disease. Annals of neurology. 2002;51(2):182–189. doi: 10.1002/ana.10086. [DOI] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Beffert U, Busciglio J, Brady ST. Fast axonal transport misregulation and Alzheimer’s disease. Neuromolecular medicine. 2002a;2(2):89–99. doi: 10.1385/NMM:2:2:089. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. The EMBO journal. 2002b;21(3):281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niidome T, Taniuchi N, Akaike A, Kihara T, Sugimoto H. Differential regulation of neurogenesis in two neurogenic regions of APPswe/PS1dE9 transgenic mice. Neuroreport. 2008;19(14):1361–1364. doi: 10.1097/WNR.0b013e32830e6dd6. [DOI] [PubMed] [Google Scholar]
- Ohsawa I, Takamura C, Morimoto T, Ishiguro M, Kohsaka S. Amino-terminal region of secreted form of amyloid precursor protein stimulates proliferation of neural stem cells. The European journal of neuroscience. 1999;11(6):1907–1913. doi: 10.1046/j.1460-9568.1999.00601.x. [DOI] [PubMed] [Google Scholar]
- Otvos L, Jr, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. Journal of neuroscience research. 1994;39(6):669–673. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- Paton JA, Nottebohm FN. Neurons generated in the adult brain are recruited into functional circuits. Science (New York, NY. 1984;225(4666):1046–1048. doi: 10.1126/science.6474166. [DOI] [PubMed] [Google Scholar]
- Peterson DA. Quantitative histology using confocal microscopy: implementation of unbiased stereology procedures. Methods. 1999;18(4):493–507. doi: 10.1006/meth.1999.0818. [DOI] [PubMed] [Google Scholar]
- Peterson DA. Stem cells in brain plasticity and repair. Current opinion in pharmacology. 2002;2(1):34–42. doi: 10.1016/s1471-4892(01)00118-7. [DOI] [PubMed] [Google Scholar]
- Price DL, Sisodia SS, Borchelt DR. Genetic neurodegenerative diseases: the human illness and transgenic models. Science. 1998;282(5391):1079–1083. doi: 10.1126/science.282.5391.1079. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science (New York, NY. 1992;255(5052):1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- Rossi G, Dalpra L, Crosti F, Lissoni S, Sciacca FL, Catania M, Di Fede G, Mangieri M, Giaccone G, Croci D, Tagliavini F. A new function of microtubule-associated protein tau: involvement in chromosome stability. Cell cycle (Georgetown, Tex. 2008;7(12):1788–1794. doi: 10.4161/cc.7.12.6012. [DOI] [PubMed] [Google Scholar]
- Sardi SP, Murtie J, Koirala S, Patten BA, Corfas G. Presenilin-dependent ErbB4 nuclear signaling regulates the timing of astrogenesis in the developing brain. Cell. 2006;127(1):185–197. doi: 10.1016/j.cell.2006.07.037. [DOI] [PubMed] [Google Scholar]
- Seki T, Arai Y. Age-related production of new granule cells in the adult dentate gyrus. Neuroreport. 1995;6(18):2479–2482. doi: 10.1097/00001756-199512150-00010. [DOI] [PubMed] [Google Scholar]
- Serby M. Olfactory deficits in Alzheimer’s disease. J Neural Transm Suppl. 1987;24:69–77. [PubMed] [Google Scholar]
- Seubert P, Mawal-Dewan M, Barbour R, Jakes R, Goedert M, Johnson GV, Litersky JM, Schenk D, Lieberburg I, Trojanowski JQ, et al. Detection of phosphorylated Ser262 in fetal tau, adult tau, and paired helical filament tau. The Journal of biological chemistry. 1995;270(32):18917–18922. doi: 10.1074/jbc.270.32.18917. [DOI] [PubMed] [Google Scholar]
- Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89(4):629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- Takashima A, Murayama M, Murayama O, Kohno T, Honda T, Yasutake K, Nihonmatsu N, Mercken M, Yamaguchi H, Sugihara S, Wolozin B. Presenilin 1 associates with glycogen synthase kinase-3beta and its substrate tau. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(16):9637–9641. doi: 10.1073/pnas.95.16.9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniuchi N, Niidome T, Goto Y, Akaike A, Kihara T, Sugimoto H. Decreased proliferation of hippocampal progenitor cells in APPswe/PS1dE9 transgenic mice. Neuroreport. 2007;18(17):1801–1805. doi: 10.1097/WNR.0b013e3282f1c9e9. [DOI] [PubMed] [Google Scholar]
- Tesco G, Kim TW, Diehlmann A, Beyreuther K, Tanzi RE. Abrogation of the presenilin 1/beta-catenin interaction and preservation of the heterodimeric presenilin 1 complex following caspase activation. The Journal of biological chemistry. 1998;273(51):33909–33914. doi: 10.1074/jbc.273.51.33909. [DOI] [PubMed] [Google Scholar]
- Tesco G, Tanzi RE. GSK3 beta forms a tetrameric complex with endogenous PS1-CTF/NTF and beta-catenin. Effects of the D257/D385A and FAD-linked mutations. Annals of the New York Academy of Sciences. 2000;920:227–232. doi: 10.1111/j.1749-6632.2000.tb06927.x. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Teplow DB, Siman R, Greenberg B, Sisodia SS. Metabolism of the “Swedish” amyloid precursor protein variant in neuro2a (N2a) cells. Evidence that cleavage at the “beta-secretase” site occurs in the golgi apparatus. The Journal of biological chemistry. 1996;271(16):9390–9397. doi: 10.1074/jbc.271.16.9390. [DOI] [PubMed] [Google Scholar]
- Thuret S, Toni N, Aigner S, Yeo GW, Gage FH. Hippocampus-dependent learning is associated with adult neurogenesis in MRL/MpJ mice. Hippocampus. 2009 doi: 10.1002/hipo.20550. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Lee VM. Paired helical filament tau in Alzheimer’s disease. The kinase connection. The American journal of pathology. 1994;144(3):449–453. [PMC free article] [PubMed] [Google Scholar]
- Trojanowski JQ, Lee VM. Phosphorylation of paired helical filament tau in Alzheimer’s disease neurofibrillary lesions: focusing on phosphatases. The FASEB journal. 1995;9(15):1570–1576. doi: 10.1096/fasebj.9.15.8529836. [DOI] [PubMed] [Google Scholar]
- Tropepe V, Craig CG, Morshead CM, van der Kooy D. Transforming growth factor-alpha null and senescent mice show decreased neural progenitor cell proliferation in the forebrain subependyma. J Neurosci. 1997;17(20):7850–7859. doi: 10.1523/JNEUROSCI.17-20-07850.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH. Functional neurogenesis in the adult hippocampus. Nature. 2002;415(6875):1030–1034. doi: 10.1038/4151030a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret L, Jankowsky JL, Xu GM, Borchelt DR, Rampon C. Alzheimer’s-type amyloidosis in transgenic mice impairs survival of newborn neurons derived from adult hippocampal neurogenesis. The Journal of neuroscience. 2007;27(25):6771–6780. doi: 10.1523/JNEUROSCI.5564-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vescovi AL, Reynolds BA, Fraser DD, Weiss S. bFGF regulates the proliferative fate of unipotent (neuronal) and bipotent (neuronal/astroglial) EGF-generated CNS progenitor cells. Neuron. 1993;11(5):951–966. doi: 10.1016/0896-6273(93)90124-a. [DOI] [PubMed] [Google Scholar]
- Wang JZ, Wu Q, Smith A, Grundke-Iqbal I, Iqbal K. Tau is phosphorylated by GSK-3 at several sites found in Alzheimer disease and its biological activity markedly inhibited only after it is prephosphorylated by A-kinase. FEBS letters. 1998;436(1):28–34. doi: 10.1016/s0014-5793(98)01090-4. [DOI] [PubMed] [Google Scholar]
- Wang R, Dineley KT, Sweatt JD, Zheng H. Presenilin 1 familial Alzheimer’s disease mutation leads to defective associative learning and impaired adult neurogenesis. Neuroscience. 2004;126(2):305–312. doi: 10.1016/j.neuroscience.2004.03.048. [DOI] [PubMed] [Google Scholar]
- Warner MD, Peabody CA, Flattery JJ, Tinklenberg JR. Olfactory deficits and Alzheimer’s disease. Biol Psychiatry. 1986;21(1):116–118. doi: 10.1016/0006-3223(86)90013-2. [DOI] [PubMed] [Google Scholar]
- Weihl CC, Ghadge GD, Kennedy SG, Hay N, Miller RJ, Roos RP. Mutant presenilin-1 induces apoptosis and downregulates Akt/PKB. The Journal of neuroscience. 1999a;19(13):5360–5369. doi: 10.1523/JNEUROSCI.19-13-05360.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihl CC, Ghadge GD, Miller RJ, Roos RP. Processing of wild-type and mutant familial Alzheimer’s disease-associated presenilin-1 in cultured neurons. Journal of neurochemistry. 1999b;73(1):31–40. doi: 10.1046/j.1471-4159.1999.0730031.x. [DOI] [PubMed] [Google Scholar]
- Wen PH, Hof PR, Chen X, Gluck K, Austin G, Younkin SG, Younkin LH, DeGasperi R, Gama Sosa MA, Robakis NK, Haroutunian V, Elder GA. The presenilin-1 familial Alzheimer disease mutant P117L impairs neurogenesis in the hippocampus of adult mice. Experimental neurology. 2004;188(2):224–237. doi: 10.1016/j.expneurol.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Zhang C, McNeil E, Dressler L, Siman R. Long-lasting impairment in hippocampal neurogenesis associated with amyloid deposition in a knock-in mouse model of familial Alzheimer’s disease. Experimental neurology. 2007;204(1):77–87. doi: 10.1016/j.expneurol.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132(4):645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]