

Abstract
Endosomal sorting of the Alzheimer amyloid precursor protein (APP) plays a key role in the biogenesis of the amyloid-β (Aβ) peptide. Genetic lesions underlying Alzheimer's disease (AD) can act by interfering with this physiological process. Specifically, proteins involved in trafficking between endosomal compartments and the trans-Golgi network (TGN) [including the retromer complex (Vps35, Vps26) and its putative receptors (sortilin, SorL1, SorCS1)] have been implicated in the molecular pathology of late-onset AD. Previously, we demonstrated a role for SorCS1 in APP metabolism and Aβ production and, while we implicated a role for the retromer in this regulation, the underlying mechanism remained poorly understood. Here, we provide evidence for a motif within the SorCS1c cytoplasmic tail that, when manipulated, results in perturbed sorting of APP and/or its fragments to endosomal compartments, decreased retrograde TGN trafficking, and increased Aβ production in H4 neuroglioma cells. These perturbations apparently do not involve turnover of the cell surface APP pool, but rather they involve intracellular APP and/or its fragments, downstream of APP endocytosis.
Introduction
Genetic studies of late-onset Alzheimer's disease (LOAD) point to a number of risk factor genes that encode proteins with known functions in trafficking to, and within, endocytic compartments including: SORL1, SORCS1, SORCS2, BIN1, PICALM-1, and CD2AP (Rogaeva et al., 2007; Harold et al., 2009; Liang et al., 2009; Naj et al., 2011). Dysfunction within endocytic compartments is evident in the Alzheimer's disease (AD) brain early in disease progression and is proposed to contribute to the deposition of amyloid-β (Aβ) (Cataldo et al., 2000; Cataldo et al., 2004). Several of these consistently linked genetic risk factors for LOAD are specifically implicated in regulating the intracellular trafficking of amyloid precursor protein (APP) and/or its cognate secretases within the endosomal pathway (for review, see Small and Gandy, 2006).
SORL1, SORCS1, and SORCS2, together with sortilin and SORCS3, belong to the vacuolar protein sorting-10 (Vps10) family of receptors. This receptor family was first identified in yeast as trafficking receptors between endosome and trans-Golgi network (TGN) compartments (for review, see Lane et al., 2012). In mammalian systems, while the Vps10 proteins are highly pleiotropic, SorL1 and SorCS1 are suggested to function in retrograde trafficking of APP (Small et al., 2005; Lane et al., 2010; Vieira et al., 2010; Fjorback et al., 2012) via their interactions with the retromer complex (Small et al., 2005; Lane et al., 2010; Fjorback et al., 2012).
The first member of the Vps10 family of receptors to be implicated as a retromer receptor for APP retrograde trafficking was SorL1 (Small et al., 2005; Fjorback et al., 2012). In vitro and in vivo studies have demonstrated that SorL1 exists in a multimeric complex that includes APP and the retromer, and assembly of this complex is one mechanism through which SorL1 regulates Aβ generation (Fjorback et al., 2012). Reduced expression of Vps35 and/or SorL1 results in increased APP localization to early endosomes leading to increased Aβ42 generation (Offe et al., 2006; Muhammad et al., 2008; Bhalla et al., 2012).
A second member of the Vps10 family of receptors, SorCS1, has also been reported to regulate Aβ generation from cultured cells (Lane et al., 2010; Reitz et al., 2011b). Furthermore, we have implicated SorCS1 as a potential retromer receptor, demonstrating in the brains of wild-type (wt) mice that SorCS1 exists in a complex with APP, SorL1, and Vps35, the core component of the retromer complex (Lane et al., 2010). In the brains of female Sorcs1-deficient mice, we demonstrated increased endogenous levels of Aβ together with decreased levels of Vps35 and SorL1, providing further evidence that Vps35 (and possibly the retromer) is involved in SorCS1-dependent regulation of APP processing and Aβ production (Lane et al., 2010).
While evidence suggests a role for SorCS1 in the modulation of APP/Aβ homeostasis, the molecular basis for this regulation is incompletely understood. We therefore sought to define the roles for sorting motifs in the cytoplasmic tail of the SorCS1c isoform in APP/Aβ homeostasis by investigating potential SorCS1c regulation of APP trafficking into and between endosomal compartments. Through site-directed mutagenesis of canonical sorting motifs in the SorCS1c cytoplasmic tail, we demonstrated that SorCS1c does not directly regulate internalization of APP into endocytic compartments, but appears to regulate the exit of APP and/or APP C-terminal fragments (CTFs) out of endosomal compartments, resulting in increased APP localization to the TGN and decreased secreted Aβ.
Materials and Methods
Antibodies.
Anti-Myc (Cell Signaling Technology), anti-EEA1 (Cell Signaling Technology), anti-TGN38 (BD Biosciences), anti-Rab7 (Cell Signaling Technology), anti-calnexin (Cell Signaling Technology), anti-syntaxin 6 (Cell Signaling Technology), and anti-mouse, anti-rabbit, and anti-goat horseradish peroxidase (HRP) conjugates (Vector Laboratories) were purchased from commercial vendors as indicated. Polyclonal Ab369 (pan-species anti-C-terminal APP antibody) was used to detect human holoAPP and C-terminal fragments (Buxbaum et al., 1990) and 6E10 (Covance) was used to detect human Aβ.
Cell culture studies.
H4 human neuroglioma cell lines stably expressing human wild-type APP (H4 wt APP; a generous gift from Dr. Rudolph Tanzi, Harvard-Mass General Hospital, Boston, MA) were cultured at 37°C/5% CO2 in complete growth medium (DMEM, 10% FBS, 1% penicillin/streptomycin, 1% l-glutamine, 5 mg/ml geneticin). H4 wt APP cells were transiently transfected with cDNA constructs as indicated using LipoD293 (SignaGen Laboratories) at a ratio of 1:4 (cDNA/LipoD293), according to the manufacturer's instructions. The backbone of the SORCS1c-myc cDNA, pcDNA4, was used for all empty vector controls. At 48 h after transfection, lysates were prepared in RIPA buffer (50 mm Tris HCl, pH 7.5, 10 mm NaCl, 1 mm EDTA, 1% NP40, 0.2 mm PMSF, 0.2 mm Na3VO4, 50 mm NaF, 10 mm Na4P207, complete protease inhibitor cocktail; Roche) as previously described (Lane et al., 2010).
Protein concentrations from cell lysates and media were determined using the Bio-Rad Protein Determination Kit. Absorbance was read at 595 nm using a Bio-Rad Microplate Reader (680XR) and analyzed using Microplate Manager version 5.2.1. Samples were subsequently prepared in 5× Laemmli buffer and boiled at 95°C for 5 min. Equal amounts of total protein were loaded onto 10% Bis Tris SDS PAGE gels for electrophoresis with MES-SDS running buffer, and electrophoretically transferred to PVDF membranes. To detect putative phosphorylated immunopositive SorCS1c-myc species, 60 μg of protein lysate was first treated with vehicle or calf intestine alkaline phosphatase (CIP; New England BioLabs) according to manufacturer's instructions and subsequently prepared in 1× Laemmli buffer and analyzed by SDS PAGE and Western blotting. Membranes were analyzed by Western blot using pAb 369 (APP C-terminal) to detect APP holo protein (holoAPP) and presumptive α and β CTFs, and 6E10 to detect Aβ. Identities of APP CTFs were assigned according to molecular weight (Gandy et al., 1992). SorCS1c-myc, Rab7, syntaxin 6, and calnexin were detected, first with the relevant primary antibodies, and visualized using HRP-conjugated, species-specific secondary antibodies. Signals were detected using enhanced chemiluminescence (Pierce). Digital images were captured using LAS3000 (FujiFilm) and were subsequently analyzed using Multi Gauge version 3.1 software.
Immunoprecipitation.
H4 wt APP cells were transiently transfected with cDNAs as indicated, and lysates were prepared as described above. A 300 μg aliquot of cell lysate was used for immunoprecipitation (IP) using A/G plus agarose beads (Santa Cruz Biotechnology) with 2 μg of anti-myc antibody (for immunoprecipitation of SorCS1-myc) according to the manufacturer's instructions. Empty vector (pCDNA4) transfections were included as negative controls.
APP and SorCS1 turnover rates.
For a time course of APP metabolism, H4 wt APPs were transiently transfected with cDNA constructs as indicated. At 8 h after transfection, cells were divided into six-well plates coated with 100 μg/ml polyornithine and allowed to recover for 5 h. Cells were subsequently treated with 50 μg/ml cycloheximide (CHX) in serum-free and antibiotic-free DMEM (Vieira et al., 2010). At the indicated time points, cells were washed in ice-cold 1× PBS and protein harvested in 1× RIPA buffer as described previously (Lane et al., 2010). Lysates were subsequently analyzed by SDS PAGE and Western blotting with immunodetection for holoAPP, APP CTFs, and SorCS1c.
Cell surface biotinylation experiments.
H4 wt APP cells were transfected with the indicated cDNAs. At 48 h after transfection, cells were washed three times in ice-cold 1× PBS (+2.5 mm CaCl + 1 mm MgCl, pH 7.4). Cells were then incubated in either ice-cold 1× PBS (negative control) or ice-cold 1× PBS plus 2 mm Sulfo-NHS-SS-biotin and incubated at 4°C for 30 min. Cells were washed three times in ice-cold 1× PBS (+100 mm glycine, pH 7.4) to quench the reaction before being lysed in 1× RIPA buffer as described previously (Lane et al., 2010). A total of 300 μg of protein lysate from each condition was incubated with 100 μl streptavidin beads (Invitrogen) rotating overnight at 4°C. To isolate the streptavidin beads, samples were centrifuged at 5000 rpm at 4°C for 1 min, the supernatant was retained for analysis of the intracellular fraction (unbound fraction), and the beads were washed five times in ice-cold 1× RIPA buffer and resuspended in 5× Laemmli buffer. Finally, samples were boiled at 95°C for 5 min before analysis by SDS PAGE and immunodetection with the indicated antibodies.
Subcellular fractionation.
Subcellular fractionation of intracellular organelles was performed as previously described (Yu et al., 2000; Yang et al., 2002). Briefly, 3× 15 cm2 plates of H4 wt APP cells were transfected with cDNAs as indicated. At 48 h after transfection, cells were washed and lysed in ice-cold homogenization buffer (130 mm KCl, 25 mm NaCl, 1 mm EGTA, 25 mm Tris, pH 7.4, 1.5 mm MgCl, 0.1%NP40 + complete protease and phosphatase inhibitor cocktail; Roche). Postnuclear supernatants (PNSs) were generated by sequential centrifugation of lysates for 10 min at 1000 × g and 10 min at 3000 × g at 4°C. PNSs were subsequently layered on the top of a 2.5–30% iodixanol gradient (OptiPrep, Axis-Shield) (1 ml of each; 30%, 25%, 15%, 12.5%, 10%, 7.5%, 5%, 2.5%) and centrifuged at 126,000 × g for 30 min. Aliquots of 1 ml each were aspirated from the top of the gradient and analyzed by SDS PAGE and Western blotting as indicated.
Site-directed mutagenesis.
SORCS1cY1132A and SORCS1cM1135A point mutations were generated using the SORCS1c-myc cDNA as a backbone according to Stratagene manufacturer's instructions using the XL quick-change method. The nucleotide sequences of the SORCS1c cDNA point mutants and the open reading frames were confirmed by DNA sequencing.
Immunohistochemistry and microscopy.
H4 wt APP cells were cultured in eight-well chamber slides for 24 h before transfection with SORCS1c-myc, SORCS1cY1132A, or SORCS1cM1135A cDNA as indicated (Lane et al., 2010). At 48 h after transfection, cells were washed three times with ice-cold 1× PBS and fixed in ice-cold 4% PFA at room temperature for 30 min. Cells were subsequently washed three times with ice-cold 1× PBS, and, where indicated, permeabilized in 1× PBS with 0.1% Triton X-100 (PBST) for 10 min at room temperature. Cells were washed three times in 1× PBS and blocked for 1 h at room temperature in PBST (PBS for nonpermeabilized cells) plus 3% goat serum before incubation overnight with the relevant primary antibody (369 at 1:1000 dilution; myc at 1:1000 dilution; rab7, EEA1, and TGN38 at 1:500 dilution) in blocking buffer. After three washes in ice-cold 1× PBS, cells were incubated with the relevant secondary antibody (1:500), Alexa Fluor 488 (APP), or Texas red (SorCS1-myc, rab7, EEA1, TGN38) at room temperature for 1 h. Cells were subsequently washed three times in 1× PBS, the chamber removed, and the slide dried completely before being mounted in ProLong Gold antifade (Invitrogen) reagent with DAPI. Cellular localization of proteins was detected by sequential scanning confocal microscopy using the Leica TCS SP5 DMI and 100× oil objective lens. Images shown are representative of a field of three independent replicates. Images were quantified using ImageJ. For total cellular fluorescence, six random regions of interest (ROIs) were assigned for each iteration, and the corrected total cell fluorescence (CTCF) was calculated using the following equation: CTCF = integrated density − (area of selected ROI × fluorescence of background reading). Data are presented as mean ± SEM.
To calculate the percentage of colocalization, Manders overlap coefficient (thresholded) was calculated using the JACoP plugin (Bolte and Cordelières, 2006) to provide a value of the percentage of pixel overlap of APP with each of the organelle markers (Texas Red). Data are presented as the mean ± SEM.
Statistical analysis.
For analysis of protein expression, densitometric analysis of Western blot bands (D = [B − AU]/mm2) was performed using Multigauge version 3.1 software (Fujifilm). Levels of α/β CTF levels were normalized to holoAPP levels and SorCS1c expression levels, and were expressed as a percentage of the control. Total Aβ levels were analyzed by Western blot, and bands were normalized to SorCS1c expression and expressed as the percentage of Aβ following wild-type SorCS1c expression. In all instances, Levene's test was used to evaluate homogeneity of variance for inclusion in parametric tests (p > 0.05 for Levene's test). Independent sample t tests (parametric design) or Mann–Whitney U tests (nonparametric design) were used to determine significant mean differences between groups. Where two or more variables were compared, a one-way ANOVA (based on parametric inclusion criteria) was used to test group differences, and Bonferroni's correction was used to correct for multiple comparisons. For all analyses, independent sample t tests were used to determine significant mean differences between either the two transfection conditions or versus control. Significance for all results is reported as p < 0.05 using two-tailed tests with an α-level of 0.05 using GraphPad Prism 5.0.
Results
Disruption of the YAQM motif in the SorCS1c intracellular domain decreases internalization of SorCS1c but not of APP
APP is proteolyzed within multiple pathways that are spatially separated across different cellular compartments, with endosomes implicated as the primary compartment for Aβ production. Aβ is generated through sequential cleavage of APP by BACE-1, producing the soluble APPβ fragment and the βCTF. Last, γ-secretase cleavage releases the Aβ peptide, of varying lengths, and the APP intracellular domain (for review, see Small and Gandy, 2006).
One model of SorCS1 action suggests that this protein regulates the rate of internalization of APP into endosomal compartments (Hermey, 2011), thereby regulating APP metabolism and Aβ generation. Therefore, we sought to dissect the potential involvement of the SorCS1c isoform (referred to in figures as “SorCS1”) in regulating APP internalization through manipulation of the tyrosine-based internalization motif 1132YAQM1135 in the SorCS1c cytoplasmic tail. In cell surface biotinylation experiments, alanine substitution for tyrosine (SORCS1cY1132A), as previously reported (Nielsen et al., 2008), resulted in a 19.5 ± 2.7% increase (p = < 0.01) in fractional distribution of SorCS1c to the plasma membrane (PM) in transiently transfected H4 wt APP cells (SorCS1c PM fraction: 30.5 ± 3.4%; SorCS1cY1132A PM fraction: 50 ± 2.7%; p = < 0.01) (Fig. 1Bii,C). Interestingly, the tyrosine residue appeared to be the essential residue for internalization, as mutation of the methionine (SORCS1cM1135A) residue alone did not significantly increase steady-state levels of SorCS1cM1135A in the PM fraction (SorCS1c PM fraction: 30.5 ± 3.4%; SorCS1cM1135A PM fraction: 33.2 ± 3.1% n = 3) (Fig. 1Bii,C).
Figure 1.
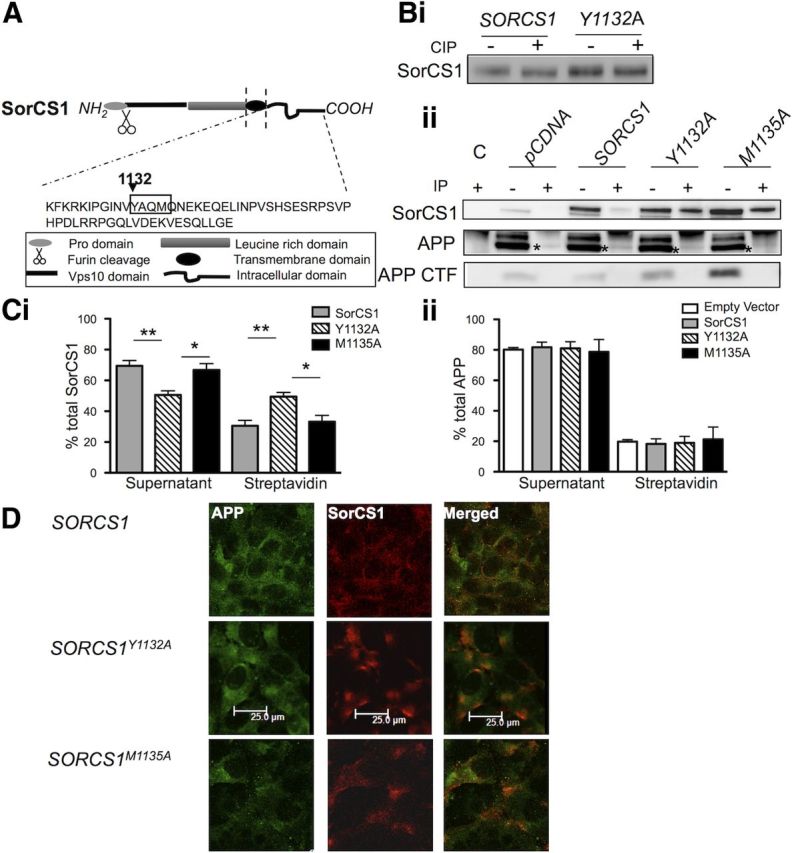
Tyrosine-based internalization motif YAQM within the SorCS1c cytoplasmic domain regulates internalization of SorCS1c but not APP. A, Schematic representation of SorCS1c, highlighting the sequence of the SorCS1c cytoplasmic tail. The tyrosine-based internalization motif YAQM is highlighted at positions 1132–1135. Bi, Immunoblot analysis of SorCS1c-myc following incubation of equal concentrations of protein lysate from H4 APP cells expressing SorCS1c-myc or SorCS1cY1132A with (+) or without (−) CIP. Separation of samples on 8% Bis-Tris SDS PAGE resulted in identification of altered migration of SorCS1c-myc but not SorCS1cY1132A myc following dephosphorylation. Bii, Immunoblot analysis of SorCS1c-myc (top), holoAPP (middle), and APP CTF (bottom) following IP with streptavidin beads (+, plasma membrane pool) and the unbound supernatant (−, intracellular pool). Cell lysates were collected from H4 wt APP cells subjected to cell surface biotinylation at 4°C for 30 min following transfection with either pCDNA, wild-type SORCS1C, SORCS1CY1132A (Y1132A), or SORCS1CM1135A (M1135A). A control immunoprecipitation was included from nonbiotinylated H4 wt APP cells transfected with wt SORCS1C (lane 1). C, Densitometric analysis of SorCS1c (Ci) and holoAPP (Cii). Values are presented as the percentage of total protein (intracellular plus plasma membrane associated). The data represents three independent experiments (n = 3) and are presented as the mean ± SEM. *p < 0.05; **p < 0.01. D, Indirect immunofluorescence of APP (first panel), SorCS1c (second panel), and merged (third panel) in nonpermeabilized fixed H4 wt APP cells expressing either wild-type SORCS1C (first row), SORCS1CY1132A (second row), or SORCS1CM1135A (third row). Cells are representative of three independent experiments. Scale bar, 25 μm.
CIP treatment of protein lysates from wt H4 APP cells overexpressing SorCS1cY1132A consistently abolished the electrophoretic shift that was observed in lysates from cells overexpressing wild-type SorCS1c (Fig. 1Bi). These data suggest that some steps in the life cycles of APP and SorCS1c are likely to be modulated by SorCS1c phosphorylation. Preventing SorCS1c internalization did not, however, appear to alter the steady-state level of biotinylated APP in the PM fraction (SorCS1c expression, APP PM fraction: 18.25 ± 3.3%; SorCS1cY1132A expression, APP PM fraction: 18.9%±4.3 n = 3; Fig. 1Bii,C). In line with this conclusion, overexpression of wild-type SorCS1c also did not significantly increase the level of biotinylated APP in the PM fraction compared with that detected in the presence of the empty vector control (pCDNA expression, APP PM fraction: 19.7 ± 1.29%; SorCS1c expression, APP PM fraction: 18.25 ± 3.35%, n = 5).
Using cell surface localization experiments as an independent method of analysis, we observed a selective increase in SorCS1cY1132Amyc signal at the plasma membrane in nonpermeabilized cells (wt SorCS1c signal: 0.62 ± 0.1; SorCS1cY1132A signal: 1.33 ± 0.14) in the absence of any apparent increase in overall APP signal (SORCS1: APP = 2.07 ± 0.47; SORCS1Y1132A: APP = 1.73 ± 0.19) (Fig. 1D; Table 1). In agreement with these data, expression of SorCS1cY1132A or SorCS1cM1135A did not increase the recovery of biotinylated APP αCTF recovery (streptavidin IP), which might have been predicted due to α-secretase activity at this location (Fig. 1 Bii, panel 3). Increased α/β CTF levels were observed, however, within intracellular fractions (unbound fraction) upon expression of SorCS1cY1132A and SorCS1cM1135A (Fig. 1Bii, panel 3). We observed a fourfold increase of α/β CTFs (407 ± 116%, p = 0.052, n = 3) upon expression of SorCS1cY1132A and a 3.7-fold increase of α/β CTFs (371 ± 71.8%, p = 0.016, n = 3) upon expression of SorCS1cM1135A. These data indicate that the tyrosine and methionine residues within the SorCS1c cytoplasmic tail are critical for SorCS1c regulation of APP metabolism and that this regulation is independent of APP endocytosis. Furthermore, this regulation appears to primarily involve differential distribution among intracellular compartments without directly affecting levels of APP or APP CTFs at the cell surface (Fig. 1B,C,D).
Table 1.
Tyrosine-based internalization motif YAQM within the SorCS1c cytoplasmic domain regulates internalization of SorCS1c but not APP
| pCDNA APP |
SORCS1 |
SORCS1Y1132A |
SORCS1M1135A |
|||
|---|---|---|---|---|---|---|
| APP | SorCS1 | APP | SorCS1 | APP | SorCS1 | |
| 2.43 ± 0.27 | 2.07 ± 0.48 | 0.62 ± 0.1 | 1.73 ± 0.19 | 1.33 ± 0.14 | 1.58 ± 0.05 | 0.86 ± 0.06 |
Cell surface fluorescence for APP and SorCS1-myc shown in Figure 1D was quantified using ImageJ software and is expressed as the CTCF ± SEM.
Disruption of the SorCS1c YAQM motif impairs the ability of SorCS1c to regulate Aβ generation
As described above, expression of SorCS1cY1132A and SorCS1cM1135A resulted in increased levels of intracellular α/β CTFs (Fig. 1Bii, panel 3). Therefore, we next sought to determine whether this increase in intracellular APP α/β CTFs resulted in altered levels of secreted Aβ. Indeed, when compared with the expression of wild-type SorCS1c, the expression of SorCS1cY1132A and SorCS1cM1135A resulted in a 1.45-fold increase (145.5 ± 12.12%, p = 0.01, n = 7) and 1.3-fold increase (132.2 ± 11.02%, p = 0.03, n = 6) in Aβ generation, respectively (Fig. 2). These data indicate that manipulating the SorCS1c intracellular sorting motif modifies the ability of SorCS1c to regulate Aβ production (Fig. 2).
Figure 2.

SorCS1c YAQM motif is necessary for SorCS1c reduction of Aβ production. Immunoblot and densitometric analysis of total soluble secreted Aβ from H4 wt APP cells expressing pDNA4, SORCS1C, SORCS1CY1132A, or SORCS1CM1135A. Values are presented as the percentage of Aβ from the SORCS1C dataset. The results represent six independent experiments. The data are presented as the mean ± SEM. **p < 0.05.
Mutation of the YAQM motif does not disrupt complex formation among SorCS1, APP, or Vps35
To determine whether mutation of the YAQM motif results in altered Aβ levels through disruption of the SorCS1/APP/Vps35 complex previously reported (Lane et al., 2010), we tested the ability of SorCS1cY1132A to coimmunoprecipitate with APP and Vps35. In immunoprecipitation/immunoblotting experiments using transfected H4 wt APP cell lines, we observed recovery of holoAPP, Vps35, and SorL1 in SorCS1c-myc and SorCS1cY1132Amyc precipitates (Fig. 3A). These data suggest that mutation of the tyrosine residue of the YAQM motif does not regulate Aβ levels through disruption of SorCS1c association with APP, SorL1, or Vps35.
Figure 3.

Disruption of the SorCS1c YAQM does not affect SorCS1–APP–Vps35 complex formation. A, B, H4 wt APP cells were transfected with pCDNA4 (control transfection), SORCS1c-myc, or SORCS1cY1132A, as indicated. Lysates were prepared as described, and a 300 μg aliquot of protein lysate was used for immunoprecipitation using A/G plus agarose beads (Santa Cruz Biotechnology) with 2 μg of anti-myc antibody according to the manufacturers instructions. B, Control IgG immunoprecipitation.
Disruption of the SorCS1c YAQM motif alters APP subcellular distribution
Next, we used independent methods to determine how disruption of the tyrosine and methionine residues of the SorCS1c YAQM motif impacts SorCS1c, APP, and APP α/β CTF localization within intracellular compartments by examining the distribution of these analytes on a 2.5–30% iodixanol OptiPrep gradient (Fig. 4) and through fluorescence microscopy (Fig. 5; Table 2). Following overexpression of SorCS1c, holoAPP was seen to concentrate in the heavier TGN-rich fractions (fractions 6–8) (Fig. 4Aii, panel 2). Increased levels of certain mature APP species were also evident in fractions 7 and 8, reflecting post-translationally modified holoAPP, consistent with an increase in APP localization to the TGN (Fig. 4Aii, panel 2). Two populations of SorCS1c were identified, one in the lightest membrane fractions (fraction 1 and 2) and another codistributing with APP in the TGN-rich fractions (fractions 6–8) (Fig. 4Aii, panel 1). These data are consistent with previous observations of a prominent colocalization between SorCS1c and APP in perinuclear TGN regions (Lane et al., 2010).
Figure 4.
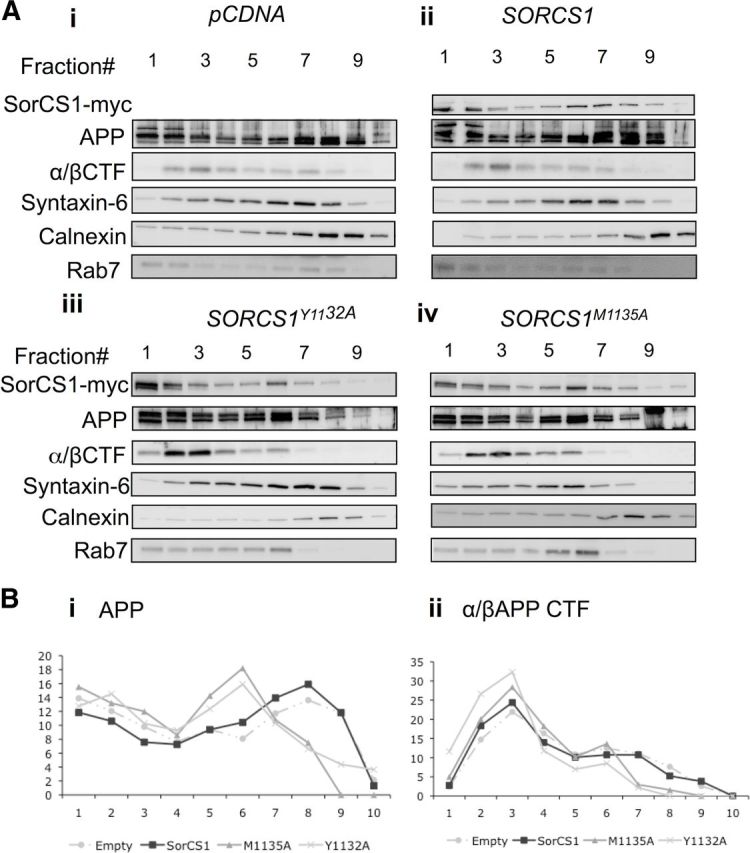
Disruption of SorCS1c YAQM results in a shift of APP and APP CTFs to lighter compartments. A, Immunoblot analysis of SorCS1c (top), holoAPP (second panel), and APP α/β CTF (C99, third panel) from H4 wt APP cells expressing pCDNA4 (Control) (Ai), SORCS1C (Aii), SORCS1CY1132A (Aiii), or SORCS1CM1135A (Aiv). Cell lysates were separated on a 2.5–30% iodixanol gradient before analysis by Western blotting. Fractions were collected from the top of the gradient (1 = lightest; 10 = heaviest). Membranes were additionally blotted for calnexin (ER marker), syntaxin 6 (TGN marker), and Rab7 (late endosome marker) to demonstrate separation of cellular fractions. The Western blots shown are representative of two independent experiments. B, Densitometric analysis of holoAPP (Bi), and APP CTF (Bii), within each fraction (1–10) of a 10-step gradient for each treatment condition. Fractions were collected from the top of the gradient (1 = lightest; 10 = heaviest). Values are presented as the percentage of total analyte in all fractions.
Figure 5.

Disruption of the SorCS1c YAQM motif decreases APP localization to the TGN. A–C, Indirect immunofluorescence of TGN38 (A, first panel), APP (A, second panel), and merged (A, third panel); EEA1 (B, first panel), APP (B, second panel), and merged (B, third panel); Rab7 (C, first panel), APP (C, second panel), and merged (C, third panel) in permeabilized fixed H4 wt APP cells expressing pCDNA4 (C, first panel), wt SORCS1C (C, second panel), SORCS1CY1132A (C, third panel), or SORCS1CM1135A (fourth panel). Cells are representative of a field of three independent experiments. Scale bar, 25 μm. See Table 2 for quantification.
Table 2.
Disruption of the SorCS1c YAQM motif decreases APP localization to the TGN
| pCDNA | SORCS1 | SORCS1Y1132A | SORCS1M1135A | |
|---|---|---|---|---|
| TGN38 | 33 ± 5% | 52 ± 4% | 40 ± 3% | 40 ± 3% |
| EEA1 | 39 ± 4% | 36 ± 5% | 37 ± 2% | 42 ± 7% |
| RAB7 | 68 ± 5% | 40 ± 4% | 48 ± 5% | 29 ± 3% |
Colocalization was quantified using ImageJ and the JACoP plugin (Bolte et al., 2006). The percentage of colocalization is expressed as the percentage of APP pixel overlap with each of the organelle markers ± SEM.
When SorCS1cY1132A was expressed, in agreement with the cell surface localization experiments, the majority of SorCS1cY1132A shifted toward the lightest PM fractions of the gradient (fraction 1–2) (Fig. 4Aii, panel 1), while holoAPP left shifted toward fractions 4–7 with decreased localization to TGN-rich fractions (Fig. 4Aiii, panel 2, Bi). The level of APP α/β CTFs also concentrated in these lighter compartments (fractions 2–6) (Fig. 4Aiii, panel 3, Bii).
The SorCS1cM1135A mutant appeared in virtually all fractions of the gradient (Fig. 4Aiv, panel 1). This was different from the prominent concentration of the SorCS1cY1132A in the lightest PM fractions (Fig. 4Aiii, panel 1). These subcellular fractionation data are consistent with our cell surface biotinylation data (Fig. 2; Table 1), where SorCS1cM1135A did not significantly interfere with SorCS1c endocytosis. Interestingly, SorCS1M1135A expression also resulted in a decrease in holoAPP localization to TGN-rich fractions, with APP now also localizing to fractions 4–7 (Fig. 4Aiv, panel 2, Bi). Again, APP α/β CTFs were concentrated in these lighter fractions (fractions 2–6) compared with conditions of wt SorCS1 expression (Fig. 4Aiv, panel 3, Bii).
We next sought to confirm these localization data using confocal microscopy. Upon overexpression of wild-type SorCS1c in wt H4 APP cells, a 19% increase in colocalization of APP to TGN38-positive regions was observed (Fig. 5A; Table 2) (percentage of APP pixels overlapping TGN38: pcDNA, 33 ± 5%; SORCS1, 52 ± 4%). This is consistent with previous colocalization between APP and SorCS1c in HEK293t cells (Lane et al., 2010) and with the subcellular fractionation data described above (Fig. 4). However, upon expression of SorCS1cY1132A, this obvious colocalization of APP to TGN38-immunoreactive compartments was decreased (Table 2; Fig. 5A) (SORCS1, 52 ± 4%; SORCS1Y1132A, 40 ± 3%). Similarly, expression of SorCS1cM1135A resulted in decreased APP colocalization to TGN38-positive compartments (SORCS1, 52 ± 4%; SORCS1M1135A, 40 ± 3%) (Fig. 5A; Table 2). Interestingly, a decrease in colocalization to rab7-positive vesicles was observed under conditions of SorCS1M1135A expression (SORCS1, 40 ± 4%; SORCS1M1135A, 29 ± 3%), and, under these conditions, APP appeared to colocalize to distinct enlarged vesicles (Fig. 5B). Together, these data demonstrate that expression of wt SorCS1 results in increased sorting of APP to the TGN, and disruption of the SorCS1 YAQM internalization motif results in mis-sorting of APP within endosomes and reduced APP localization to the TGN, downstream of APP endocytosis.
SorCS1cM1135A results in altered dynamics of APP metabolism
To investigate the mechanisms through which the methionine residue might regulate APP localization and metabolism, we used CHX time course experiments to investigate how SorCS1cM1135A regulates the rate of turnover of APP into APP α/β CTFs compared with wild-type SorCS1c expression or empty vector control (Fig. 6). First, in agreement with previous observations, wt SorCS1c expression resulted in decreased APP α/β CTF levels at all time points studied (Fig. 6A, fifth panel down, B). Expression of SorCS1cM1135A, however, resulted in an ∼3.2-fold increase (316 ± 101.6%, p = 0.049, n = 3) in levels of APP α/β CTF at 180 min compared with levels identified in conditions of expression of wt SorCS1c or empty vector control (Fig. 6A, panel 6, B). Together with the fractionation and localization data, these data suggest that wild-type SorCS1c functions as a chaperone to sort APP both away from endosomal compartments and sites of high β-secretase activity to the TGN and as a clearance receptor for APP CTFs (for a model, see Fig. 7).
Figure 6.

SorCS1cM1135A results in the accumulation of APP CTFs. A, Immunoblot analysis of SorCS1c (top), holoAPP (middle), and APP α/β CTF (bottom) turnover rate in the presence of CHX in H4 wt APP cells expressing empty vector (pCDNA 4, control), SorCS1c-myc, or SorCS1cM1135A in H4 cells stably expressing APP. B, Densitometric analysis of APP CTF accumulation in the presence of (50 μg/ml) CHX at 60, 120, and 180 min. The results represent three independent experiments, and the data are presented as the mean ± SEM. *p < 0.05.
Figure 7.

Model for SorCS1c-regulated sorting of APPs though the amyloidogenic pathway. 1, Upon exit from the TGN, APP is sorted into either secretory vesicles to the plasma membrane or into clathrin-coated vesicles into the endosomal pathway. Within the secretory pathway and at the plasma membrane (PM), APP is cleaved by the α-secretases (ADAM10, ADAM17) and by the γ-secretase machinery, precluding the generation of the Aβ peptide. 2, α-Secretase processing of APP at the PM is rate limiting, and unprocessed APP is internalized into the endosomal pathway. Within endosomal compartments and regions of low pH, APP is sequentially cleaved by β-secretase and γ-secretase, resulting in the formation of the Aβ peptide (of varying lengths). 3, APP is recycled from the endosomal compartments (EE, early endosomes; LE, late endosomes; RE, recycling endosomes) to the TGN via the retromer complex and its receptors, the Vps10 family. Expression of wt SorCS1c (black bar) results in increased APP localization to the TGN and decreased Aβ production. Previous studies implicate the retromer in this regulation, consistent with other VPS10 receptors, including SorL1. Expression of SorCS1cY1132A (light gray bar) results in increased localization of SorCS1cY1132A to the plasma membrane and mislocalization of APP to endosomal compartments, decreased recycling to the TGN, and increased Aβ production. While expression of SORCS1cM1135A (dark gray bar) does not significantly alter the rate of SorCS1c or APP internalization into endosomes, it does, however, result in decreased recycling of APP to the TGN, APP localization to enlarged vesicles, and increased APP CTF and Aβ production. We propose here a model whereby SorCS1 regulates trafficking of APP between endosomes and the TGN. Preventing SorCS1 internalization into endosomal compartments (SorCS1cY1132A) or out of endosomes (SorCS1cM1135A) prevents SorCS1-regulated sorting of APP to the TGN and possibly to the lysosome. ER, Endoplasmic reticulum.
Discussion
Multiple proteins involved in regulating trafficking within endosomal compartments have now been linked, through genome-wide association study (GWAS) with risk for LOAD, including SORL1 and SORCS1 (Rogaeva et al., 2007; Liang et al., 2009). Importantly, several of the single-nucleotide polymorphisms identified from GWAS in SORL1 and SORCS1 have now also been demonstrated to influence their expression levels and are correlated with disease endophenotypes, including hippocampal atrophy and memory retention (Reitz et al., 2011a,c). Furthermore, pathogenic mutations in SorL1 have now been identified to be causative of a form of familial AD (Pottier et al., 2012). The retromer itself is also of particular interest in LOAD. Decreased levels of Vps35 and Vps26 were identified in vulnerable regions of the AD brain (Small et al., 2005), and SNX1, SNX3, and RAB7A, genes that encode proteins essential for membrane association of the retromer, are possible AD risk genes (Vardarajan et al., 2012).
In cell biology studies, SorL1 was implicated in APP sorting within endosomal compartments (Offe et al., 2006), and in endosomes to TGN sorting via an interaction with the retromer complex (Small et al., 2005; Fjorback et al., 2012), with the complex itself showing decreased expression in vulnerable regions of the AD brain (Small et al., 2005). However, the role for SorL1 function in APP trafficking has been shown to be pleiotropic, with additional roles described for SorL1 in regulating APP exit from the TGN (Schmidt et al., 2007) and/or in regulation of APP oligomerization (Lao et al., 2012; Schmidt et al., 2012). We and others previously demonstrated that SorCS1 also regulates APP metabolism and Aβ production (Lane et al., 2010; Reitz et al., 2011b) potentially through an interaction with the retromer complex (Lane et al., 2010). In cells and in mice, we demonstrated SorCS1c regulation of Aβ production, isolated complexes containing SorCS1c, APP, and Vps35, and observed decreased Vps35 protein levels in Sorcs1-deficient mice (Lane et al., 2010). Importantly, the increases in endogenous Aβ levels were only identified in the brains of female Sorcs1-deficient mice, mirroring genetic data that reported a stronger correlation for this gene in the female population with AD (Liang et al., 2009) and T2DM (Goodarzi et al., 2007).
Herein we sought to dissect the role of SorCS1 in regulating trafficking pathways to elucidate potential common mechanisms through which SorCS1 contributes to APP metabolism and subsequent Aβ production. Here, we provide evidence for a trafficking role for SorCS1c in the regulation of APP metabolism and Aβ generation. This last event appeared dependent on (1) the presence of an intact SorCS1c YAQM motif and (2) downstream interaction of APP and SorCS1c within the endosomal/lysosomal pathway. A model for SorCS1c regulation of APP sorting is shown in Figure 7.
One model of SorCS1 action suggests that this protein regulates APP metabolism and Aβ generation through regulating the rate of internalization of APP into endosomal compartments (Hermey, 2011). Nielsen et al. (2008) previously reported that the canonical tyrosine YAQM internalization motif within the SorCS1c intracellular domain is required for SorCS1c endocytosis via the AP2 adaptor complex. Therefore, to dissect the mechanism by which SorCS1 regulates Aβ production, we first sought to investigate whether SorCS1c regulates trafficking of APP into the endosomal pathway through manipulation of this internalization motif. In agreement with Nielsen et al. (2008), alanine substitution of the tyrosine residue at position 1132 (SorCS1cY1132A) increased the fractional distribution of SorCS1c to the plasma membrane as demonstrated through cell surface biotinylation experiments, subcellular fractionation, and cell surface indirect immunofluorescence. However, retention of SorCS1c at the plasma membrane did not alter the rate of APP internalization by any method of analyses, nor did it result in increased production of APP αCTF within the plasma membrane fraction. These data suggest that SorCS1c does not regulate Aβ production through control of APP endocytosis.
Subcellular fractionation and localization experiments presented here, demonstrate that overexpression of wt SorCS1 results in increased localization of APP to TGN38-positive compartments. This observation is consistent with previous implications for the retromer in SorCS1 regulation of Aβ production (Lane et al., 2010). Interestingly, while disruption of the YAQM internalization motif did not directly affect APP internalization into endocytic compartments, it did result in decreased APP localization to the TGN as determined by subcellular fractionation and fluorescence microscopy. This mislocalization of APP resulted in increased intracellular CTF levels and increased secreted Aβ, consistent with increased APP localization to endocytic compartments. Disruption of the YAQM motif did not, however, disrupt SorCS1–APP–Vps35 complex formation. These data suggest that while this motif is not directly required for interaction with the retromer, it is required for correct sorting of SorCS1 within endosomal compartments. Most likely, disruption of the tyrosine residue reduces SorCS1 localization to endocytic compartments and thereby prevents recycling of APP from endosomes to the TGN. Disruption of the methionine residue, however, results in the accumulation of APP CTFs, SorCS1M1135A, and production of Aβ, potentially due to impaired lysosomal clearance of both SorCS1 and APP metabolites. In fact, previous studies by Nielsen et al. (2008) have implicated a role for SorCS1 in lysosomal trafficking, and several studies now implicate this family of proteins as clearance receptors for both progranulin (Hu et al., 2010) and Aβ (Carlo et al., 2013).
Importantly, a genetic interaction between SORCS1 and APOE has been reported in risk for LOAD (Wang et al., 2012) and SORCS1 is a risk factor for insulin resistance-T2DM (Goodarzi et al., 2007), another well known risk factor for LOAD. While our data implicate SorCS1c in retrograde trafficking of APP, dependent on and independent of the retromer, regulation of trafficking of other ligands and/or receptors might also explain the contribution of SorCS1 to clinical phenotypes of both AD and T2DM. For example, SorCS1 may modulate trafficking of insulin and/or of the insulin receptor. Sortilin, SorL1, and Vps35 have been implicated in the trafficking of insulin-responsive vesicles (IRVs) (Jedrychowski et al., 2010), while sortilin and SorL1 have now been implicated as clearance receptors for apoE–Aβ complexes (Carlo et al., 2013). These data highlight the involvement of the Vps10 receptors and retromer-regulated trafficking pathways in the regulation of both Aβ generation and clearance and insulin signaling. Further elucidation of these pathways should provide new insights into the pathogenesis of both AD and T2DM.
Further investigation is ultimately required to elucidate the complete regulatory mechanism for SorCS1 in the endosomal sorting of APP and regulation of Aβ generation. The Vps10 receptors clearly play pleiotropic roles in the intracellular sorting of multiple ligands and receptors (Nielsen et al., 2008; Lane et al., 2012), and it is highly likely that SorCS1 functions to regulate APP metabolism through multiple pathways. Here, we have identified a motif within the SorCS1c cytoplasmic tail that, when mutated, results in mis-sorting of APP within endosomal compartments, decreased endosome-to-TGN trafficking of APP, and increased Aβ production, independent of regulating APP endocytosis (Fig. 7). Future studies are required to (1) identify the retromer binding domain within the SorCS1c cytoplasmic domain; (2) confirm a role for SorCS1c-dependent endosome to lysosome trafficking of APP/APP CTFs; and (3) determine whether common SorCS1-regulated pathways exist for both APP sorting and metabolism, and IRV sorting/insulin signaling pathways. Identification of these common pathways may ultimately lead to the identification of molecular mechanisms that contribute to combined risk for developing both AD and T2DM.
Footnotes
This research was supported by National Institutes of Health (NIH) Grant NS075685 (to S.G.), the Cure Alzheimer's Fund (to S.G.), the U.S. Department of Veterans Affairs (to S.G.), National Institute of Diabetes and Digestive and Kidney Diseases Grants DK58037 and DK66369 (to A.D.A.), and an American Diabetes Association Research Award (to M.E.E). J.W.S. was a trainee in the Integrated Pharmacological Sciences Training Program supported by Grant T32GM062754 from the National Institute of General Medical Sciences to Terry Krulwich (Icahn School of Medicine at Mount Sinai). Confocal laser scanning microscopy was performed at the Icahn School of Medicine at Mount Sinai Microscopy Shared Resource Facility, supported with funding from NIH-National Cancer Institute Shared Resources Grant 5R24 CA095823-04, National Science Foundation Major Research Instrumentation Grant DBI-9724504, and NIH Shared Instrumentation Grant 1 S10 RR0 9145-01. We thank Rudolph E. Tanzi for cDNA constructs.
The authors declare no competing financial interests.
References
- Bhalla A, Vetanovetz CP, Morel E, Chamoun Z, Di Paolo G, Small SA. The location and trafficking routes of the neuronal retromer and its role in amyloid precursor protein transport. Neurobiol Dis. 2012;47:126–134. doi: 10.1016/j.nbd.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Gandy SE, Cicchetti P, Ehrlich ME, Czernik AJ, Fracasso RP, Ramabhadran TV, Unterbeck AJ, Greengard P. Processing of Alzheimer beta/A4 amyloid precursor protein: modulation by agents that regulate protein phosphorylation. Proc Natl Acad Sci U S A. 1990;87:6003–6006. doi: 10.1073/pnas.87.15.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlo AS, Gustafsen C, Mastrobuoni G, Nielsen MS, Burgert T, Hartl D, Rohe M, Nykjaer A, Herz J, Heeren J, Kempa S, Petersen CM, Willnow TE. The pro-neurotrophin receptor sortilin is a major neuronal apolipoprotein E receptor for catabolism of amyloid-β peptide in the brain. J Neurosci. 2013;33:358–370. doi: 10.1523/JNEUROSCI.2425-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157:277–286. doi: 10.1016/S0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Petanceska S, Terio NB, Peterhoff CM, Durham R, Mercken M, Mehta PD, Buxbaum J, Haroutunian V, Nixon RA. Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol Aging. 2004;25:1263–1272. doi: 10.1016/j.neurobiolaging.2004.02.027. [DOI] [PubMed] [Google Scholar]
- Fjorback AW, Seaman M, Gustafsen C, Mehmedbasic A, Gokool S, Wu C, Militz D, Schmidt V, Madsen P, Nyengaard JR, Willnow TE, Christensen EI, Mobley WB, Nykjær A, Andersen OM. Retromer binds the FANSHY sorting motif in SorLA to regulate amyloid precursor protein sorting and processing. J Neurosci. 2012;32:1467–1480. doi: 10.1523/JNEUROSCI.2272-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandy SE, Buxbaum JD, Suzuki T, Ramabhadran TV, Caporaso GL, Nairn AC, Greengard P. The nature and metabolism of potentially amyloidogenic carboxyl-terminal fragments of the Alzheimer beta/A4-amyloid precursor protein: some technical notes. Neurobiol Aging. 1992;13:601–603. doi: 10.1016/0197-4580(92)90063-4. [DOI] [PubMed] [Google Scholar]
- Goodarzi MO, Lehman DM, Taylor KD, Guo X, Cui J, Quiñones MJ, Clee SM, Yandell BS, Blangero J, Hsueh WA, Attie AD, Stern MP, Rotter JI. SORCS1: a novel human type 2 diabetes susceptibility gene suggested by the mouse. Diabetes. 2007;56:1922–1929. doi: 10.2337/db06-1677. [DOI] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermey G. Targeting amyloid precursor protein. Ann Neurol. 2011;69:8–10. doi: 10.1002/ana.22342. [DOI] [PubMed] [Google Scholar]
- Hu F, Padukkavidana T, Vægter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, Strittmatter SM. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein,progranulin. Neuron. 2010;68:654–667. doi: 10.1016/j.neuron.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedrychowski MP, Gartner CA, Gygi SP, Zhou L, Herz J, Kandror KV, Pilch PF. Proteomic analysis of GLUT4 storage vesicles reveals LRP1 to be an important vesicle component and target of insulin signaling. J Biol Chem. 2010;285:104–114. doi: 10.1074/jbc.M109.040428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane RF, Raines SM, Steele JW, Ehrlich ME, Lah JA, Small SA, Tanzi RE, Attie AD, Gandy S. Diabetes-associated SorCS1 regulates Alzheimer's amyloid-β metabolism: evidence for involvement of SorL1 and the retromer complex. J Neurosci. 2010;30:13110–13115. doi: 10.1523/JNEUROSCI.3872-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane RF, St George-Hyslop P, Hempstead BL, Small SA, Strittmatter SM, Gandy S. Vps10 family proteins and the retromer complex in aging-related neurodegeneration and diabetes. J Neurosci. 2012;32:14080–14086. doi: 10.1523/JNEUROSCI.3359-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao A, Schmidt V, Schmitz Y, Willnow TE, Wolkenhauer O. Multi-compartmental modeling of SORLA's influence on amyloidogenic processing in Alzheimer's disease. BMC Syst Biol. 2012;6:74. doi: 10.1186/1752-0509-6-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Slifer M, Martin ER, Schnetz-Boutaud N, Bartlett J, Anderson B, Züchner S, Gwirtsman H, Gilbert JR, Pericak-Vance MA, Haines JL. Genomic convergence to identify candidate genes for Alzheimer disease on chromosome 10. Hum Mutat. 2009;30:463–471. doi: 10.1002/humu.20953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhammad A, Flores I, Zhang H, Yu R, Staniszewski A, Planel E, Herman M, Ho L, Kreber R, Honig LS, Ganetzky B, Duff K, Arancio O, Small SA. Retromer deficiency observed in Alzheimer's disease causes hippocampal dysfunction, neurodegeneration, and Abeta accumulation. Proc Natl Acad Sci U S A. 2008;105:7327–7332. doi: 10.1073/pnas.0802545105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen MS, Keat SJ, Hamati JW, Madsen P, Gutzmann JJ, Engelsberg A, Pedersen KM, Gustafsen C, Nykjaer A, Gliemann J, Hermans-Borgmeyer I, Kuhl D, Petersen CM, Hermey G. Different motifs regulate trafficking of SorCS1 isoforms. Traffic. 2008;9:980–994. doi: 10.1111/j.1600-0854.2008.00731.x. [DOI] [PubMed] [Google Scholar]
- Offe K, Dodson SE, Shoemaker JT, Fritz JJ, Gearing M, Levey AI, Lah JJ. The lipoprotein receptor LR11 regulates amyloid beta production and amyloid precursor protein traffic in endosomal compartments. J Neurosci. 2006;26:1596–1603. doi: 10.1523/JNEUROSCI.4946-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottier C, Hannequin D, Coutant S, Rovelet-Lecrux A, Wallon D, Rousseau S, Legallic S, Paquet C, Bombois S, Pariente J, Thomas-Anterion C, Michon A, Croisile B, Etcharry-Bouyx F, Berr C, Dartigues JF, Amouyel P, Dauchel H, Boutoleau-Bretonnière C, Thauvin C, et al. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol Psychiatry. 2012;17:875–879. doi: 10.1038/mp.2012.15. [DOI] [PubMed] [Google Scholar]
- Reitz C, Cheng R, Rogaeva E, Lee JH, Tokuhiro S, Zou F, Bettens K, Sleegers K, Tan EK, Kimura R, Shibata N, Arai H, Kamboh MI, Prince JA, Maier W, Riemenschneider M, Owen M, Harold D, Hollingworth P, Cellini E, et al. Meta-analysis of the association between variants in SORL1 and Alzheimer disease. Arch Neurol. 2011a;68:99–106. doi: 10.1001/archneurol.2010.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C, Tokuhiro S, Clark LN, Conrad C, Vonsattel JP, Hazrati LN, Palotás A, Lantigua R, Medrano M, Jiménez-Velázquez Z, Vardarajan B, Simkin I, Haines JL, Pericak-Vance MA, Farrer LA, Lee JH, Rogaeva E, George-Hyslop PS, Mayeux R. SORCS1 alters amyloid precursor protein processing and variants may increase Alzheimer's disease risk. Ann Neurol. 2011b;69:47–64. doi: 10.1002/ana.22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C, Lee JH, Rogers RS, Mayeux R. Impact of genetic variation in SORCS1 on memory retention. PLoS One. 2011c;6:e24588. doi: 10.1371/journal.pone.0024588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, Chen F, Shibata N, Lunetta KL, Pardossi-Piquard R, Bohm C, Wakutani Y, Cupples LA, Cuenco KT, Green RC, Pinessi L, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt V, Sporbert A, Rohe M, Reimer T, Rehm A, Andersen OM, Willnow TE. SorLA/LR11 regulates processing of amyloid precursor protein via interaction with adaptors GGA and PACS-1. J Biol Chem. 2007;282:32956–32964. doi: 10.1074/jbc.M705073200. [DOI] [PubMed] [Google Scholar]
- Schmidt V, Baum K, Lao A, Rateitschak K, Schmitz Y, Teichmann A, Wiesner B, Petersen CM, Nykjaer A, Wolf J, Wolkenhauer O, Willnow TE. Quantitative modelling of amyloidogenic processing and its influence by SORLA in Alzheimer's disease. EMBO J. 2012;31:187–200. doi: 10.1038/emboj.2011.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Gandy S. Sorting through the cell biology of Alzheimer's disease: intracellular pathways to pathogenesis. Neuron. 2006;52:15–31. doi: 10.1016/j.neuron.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Kent K, Pierce A, Leung C, Kang MS, Okada H, Honig L, Vonsattel JP, Kim TW. Model-guided microarray implicates the retromer complex in Alzheimer's disease. Ann Neurol. 2005;58:909–919. doi: 10.1002/ana.20667. [DOI] [PubMed] [Google Scholar]
- Vardarajan BN, Bruesegem SY, Harbour ME, George-Hyslop PS, Seaman MN, Farrer LA. Identification of Alzheimer disease-associated variants in genes that regulate retromer function. Neurobiol Aging. 2012;33:2231.e15–2231.e30. doi: 10.1016/j.neurobiolaging.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira SI, Rebelo S, Esselmann H, Wiltfang J, Lah J, Lane R, Small SA, Gandy S, da Cruz E Silva EF, da Cruz E Silva OA. Retrieval of the Alzheimer's amyloid precursor protein from the endosome to the TGN is S655 phosphorylation state-dependent and retromer-mediated. Mol Neurodegener. 2010;5:40. doi: 10.1186/1750-1326-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HF, Yu JT, Zhang W, Wang W, Liu QY, Ma XY, Ding HM, Tan L. SORCS1 and APOE polymorphisms interact to confer risk for late-onset Alzheimer's disease in a Northern Han Chinese population. Brain Res. 2012;1448:111–116. doi: 10.1016/j.brainres.2012.01.067. [DOI] [PubMed] [Google Scholar]
- Yang DS, Tandon A, Chen F, Yu G, Yu H, Arawaka S, Hasegawa H, Duthie M, Schmidt SD, Ramabhadran TV, Nixon RA, Mathews PM, Gandy SE, Mount HT, St George-Hyslop P, Fraser PE. Mature glycosylation and trafficking of nicastrin modulate its binding to presenilins. J Biol Chem. 2002;277:28135–28142. doi: 10.1074/jbc.M110871200. [DOI] [PubMed] [Google Scholar]
- Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, Supala A, Levesque L, Yu H, Yang DS, Holmes E, Milman P, Liang Y, Zhang DM, Xu DH, Sato C, et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature. 2000;407:48–54. doi: 10.1038/35024009. [DOI] [PubMed] [Google Scholar]