

Abstract
Background and Purpose
Small molecule glucokinase activators (GKAs) have been associated with potent antidiabetic efficacy and hepatic steatosis in rodents. This study reports the discovery of S 50131 and S 51434, two novel GKAs with an original scaffold and an atypical pharmacological profile.
Experimental Approach
Activity of the compounds was assessed in vitro by measuring activation of recombinant glucokinase, stimulation of glycogen synthesis in rat hepatocytes and increased insulin secretion from rat pancreatic islets of Langerhans. Efficacy and safety in vivo were evaluated after oral administration in db/db mice by measuring glycaemia, HbA1c and dyslipidaemia-associated events.
Key Results
S 50131 and S 51434 activated GK and stimulated glycogen synthesis in hepatocytes and insulin secretion from pancreatic islets. Unexpectedly, while both compounds effectively lowered glycaemia after acute oral administration, they did not decrease HbA1c after a 4-week treatment in db/db mice. This lack of antidiabetic efficacy was associated with increased plasma free fatty acids (FFAs), contrasting with the effect of GKA50 and N00236460, two GKAs with sustained HbA1c lowering activity but neutral regarding plasma FFAs. S 50131, but not S 51434, also induced hepatic steatosis, as did GKA50 and N00236460. However, a shorter, 4-day treatment resulted in increased hepatic triglycerides without changing the plasma FFA levels, demonstrating dynamic alterations in the lipid profile over time.
Conclusions and Implications
In addition to confirming the occurrence of dyslipidaemia with GKAs, these findings provide new insights into understanding how such compounds may sustain or lose efficacy over time.
Keywords: glucokinase, free fatty acids, triglycerides, type 2 diabetes, glucose homeostasis, safety
Introduction
Type 2 diabetes is a consequence of impaired beta cell function and increased insulin resistance (Kahn, 2003) and is associated with increased risk factors such as hypertension, cardiovascular disease and dyslipidaemia (Fuller and Stevens, 1991; Stamler et al., 1993; Chahil and Ginsberg, 2006). Currently available antidiabetic treatments are frequently associated with loss of efficacy over time and side effects such as hypoglycaemia, weight gain and gastrointestinal disorders (Tahrani et al., 2011), justifying the need for new and safer treatments (Verspohl, 2012). Among the numerous approaches proposed to lower glycaemia in type 2 diabetic patients, glucokinase (GK), also called hexokinase IV, emerged as one of the most attractive targets because of its dual role in stimulating glucose uptake by hepatocytes and insulin secretion from pancreatic beta cells (Bontemps et al., 1978; Meglasson and Matschinsky, 1986). GK is somewhat different to the other known hexokinases as it has a low affinity for glucose (S0.5, 7–9 mM) and is not inhibited by physiological concentrations of glucose-6-phosphate. These characteristics, plus the fact that glucose entry into hepatocytes and beta cells is not limited by glucose transporters, have led to the proposal that GK acts as a glucose sensor for these two cell types (Matschinsky, 1990; Schuit et al., 2001).
Co-crystallization studies of GK with glucose and a synthetic activator led to the discovery of an allosteric site at a region distant from the substrate binding site (Kamata et al., 2004). This finding suggested exciting possibilities to manipulate GK, and several academic laboratories and pharmaceutical companies launched medicinal chemistry programmes to find small glucokinase activators (GKAs) (Grimsby et al., 2003; Brocklehurst et al., 2004; Efanov et al., 2005). Generally, these molecules lowered plasma glucose levels with good efficacy in preclinical rodent models, similar to that seen with metformin (Grimsby et al., 2003; Coope et al., 2006; Fyfe et al., 2007; Ohyama et al., 2010; Winzell et al., 2011; De Ceuninck et al., 2013). However, most GKAs stimulated insulin secretion from pancreatic beta cells at inappropriately low glucose levels, raising concerns about the possibility of hypoglycaemic episodes. For this reason, a number of groups focused their efforts on compounds primarily directed at the liver (Bebernitz et al., 2009; Pfefferkorn et al., 2012). However, the risks associated with this strategy were a matter of debate (Agius, 2009; Rees and Gloyn, 2013). For example, it was reported that liver-specific overexpression of Gk in rodents, while lowering blood glucose, was associated with dyslipidaemia, increased hepatic lipogenesis and insulin resistance, with subtle differences observed depending on the model used (O'Doherty et al., 1999; Jackerott et al., 2002; Ferre et al., 2003). In humans, single nucleotide polymorphisms in the gene encoding glucokinase regulatory protein (GCKR), a liver-specific GK inhibitory protein, increased hepatic GK activity and decreased plasma glucose levels, but were associated with dyslipidaemia and fatty liver (Saxena et al., 2007; Santoro et al., 2012). Furthermore, the mRNA expression of hepatic GK was tightly linked with de novo lipogenesis and liver fat content (Peter et al., 2011). Finally, we recently reported that GKAs belonging to three different chemical classes induced dyslipidaemia and hepatic steatosis after chronic oral administration in normoglycaemic and hyperglycaemic rodent models (De Ceuninck et al., 2013). Furthermore, the observed hepatic steatosis was closely related to the antidiabetic efficacy. In the present study, we report the discovery of a novel chemical class of GKAs with high acute antidiabetic efficacy, but which surprisingly lacked activity after chronic treatment in hyperglycaemic db/db mice. We suggest that the altered profile of circulating free fatty acids (FFAs) is likely to be the cause of this loss of activity.
Methods
GK activation assay
GK activity was measured as previously described (De Ceuninck et al., 2013) in buffer containing 50 mM HEPES, 100 mM KCl, 5 mM MgCl2, 5 mM DTT, pH 7.4, 0.75 μg·mL−1 recombinant GK (BioXtal, Marseille, France), and various compound and glucose concentrations. The reaction was initiated by the addition of 10 mM ATP, 1 U·mL−1 glucose-6-phosphate dehydrogenase and 1 mM NADP. The production of NADPH was measured by the absorbance at 340 nm after 45 min. Results were calculated after subtraction of the absorbance at time zero. In order to determine EC50 (half-maximal potency) and Emax (corresponding to the plateau, where maximum potency is reached), each compound (0–100 μM) was incubated in reaction buffer containing 8 mM glucose. To measure the effect of compounds (6 nM to 60 μM) on the affinity of GK for glucose (S0.5), glucose was used at concentrations ranging from 1.1 to 55 mM. Experiments were performed twice for each compound, producing similar results.
Animals
All animal care and experimental procedures were reviewed and approved by our internal ethics committee. All studies involving animals are reported in accordance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010). A total of 422 animals were used in the experiments described here.
Rat hepatocyte preparation and primary culture
Sprague-Dawley rats (Charles River Laboratories, Saint-Germain-sur-l'Arbresle, France) were anaesthetized by i.p. injection of 54.7 mg kg-1 of sodium pentobarbital (CEVA santé animale, Libourne, France) Hepatocytes were isolated by the two-step in situ perfusion technique (Seglen, 1976). Briefly, the liver was perfused via the portal vein for 2 min with Krebs buffer containing 2 mM EDTA and 10 IU·mL−1 heparin at a flow rate of 40 mL·min−1, then for 2 min with Krebs buffer at 40 mL·min−1 and for 5 min at 35 mL·min−1 with a Krebs solution containing 166 mU·mL−1 collagenase A. After perfusion, the liver was excised and cells were dissociated with Krebs buffer supplemented with 1.8 mM CaCl2, recovered by centrifugation at 30× g for 2 min, and separated by Percoll density-gradient centrifugation at 100× g for 20 min at room temperature. Cells were accepted for studies when viability was >85% as assessed by Trypan blue dye exclusion. Hepatocytes were cultured in 24-well plates at a density of 75 × 103 cells·cm−2 and incubated for 24 h at 37°C under 5% CO2 in DMEM containing 4.5 g·L−1 glucose, 10% (v/v) fetal calf serum, 100 nM dexamethasone, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin (complete medium).
Glycogen synthesis in hepatocytes maintained in primary culture
Hepatocytes grown for 24 h in complete medium were washed and incubated for 1 h in serum-free, dexamethasone-free DMEM containing 1 g·L−1 glucose and 0.1% (w/v) BSA. The medium was renewed and 74 kBq·mL−1 D-[U-14C] glucose plus vehicle or compound (0.625 to 10 μM) were added to each well. Hepatocytes were incubated further for 3 h at 37°C, then washed three times in ice-cold PBS, and solubilized with 1 N NaOH under gentle agitation at room temperature. A sample was taken to measure the protein content by the method of Bradford. For glycogen isolation (Ursø et al., 1999), the remaining homogenates were transferred to polypropylene tubes containing 1.7 mg·mL−1 rabbit type III glycogen as a carrier. The mixtures were sequentially boiled for 30 min, cooled on ice, precipitated for 16 h at −20°C with 70% ice-cold pure ethanol, and centrifuged at 2000× g for 10 min at 4°C. The pellets were washed with 70% pure ethanol, then resuspended in 250 μL water, at 70°C. The mixtures were transferred to vials containing 4.75 mL of scintillation liquid and counted using a Packard liquid scintillation counter. Each compound was tested in quadruplicate wells in four independent experiments.
Culture of rat pancreatic islets and insulin secretion assay
For the isolation of islets of Langerhans, Wistar rats (Charles River Laboratories) were anaesthetized by i.p. injection of 54.7 mg·kg−1 of sodium pentobarbital. The pancreatic duct was ligated distally and injected with an ice-cooled solution of Hank's balanced salt solution (HBSS) containing 0.5 mg·mL−1 type V collagenase (Sigma-Aldrich, Lyon, France). The pancreas was removed, incubated in this solution for 9 min at 37°C, then the digestion stopped by dilution in ice-cold HBSS. The resulting solution was further homogenized by several passes through a 12-gauge needle. The homogenate was centrifuged, resuspended in ice-cold HBSS, and passed through a 500-μm filter. The filtrate was pelleted and resuspended in Histopaque 1077 (Sigma-Aldrich), centrifuged, and the islets recovered from the interface and washed in HBSS. Islets were hand-picked and distributed in 96-well plates at 4 islets·in 200 μL−1·per well in RPMI 1640 medium containing 1% penicillin/streptomycin at glucose concentrations ranging from 2.8 to 16.7 mM, in the presence of vehicle or 10 μM test compound. Insulin secreted into the medium was measured after a 90-min incubation using a rat high-range commercial ELISA (Mercodia, Uppsala, Sweden). Each condition was tested in eight different wells in three independent experiments.
Prediction of intestinal absorption and metabolic bioavailability
The intestinal absorption was predicted using Caco-2 cells (Yee, 1997; American Type Culture Collection, Manassas, VA) with adaptation of the experimental protocol as previously described (De Ceuninck et al., 2013). The prediction of metabolic bioavailability was calculated from in vitro metabolic stability measurements using rat or mouse hepatic microsomes as detailed previously (De Ceuninck et al., 2013), based on a model initially described by Bertrand et al. (2000).
Studies in vivo
Male diabetic db/db mice (C57BLKS/J background) from Jackson Laboratories (Bar Harbor, ME, USA) were housed in internal facilities 1 week before starting the treatment. Mice were fed ad libitum with five K52 pellets purchased from Scientific Animal Food & Engineering (SAFE, Augy, France) with free access to water. Five K52 pellets are similar in formulation to 5K0Q, the primary diet used at the Jackson Laboratories, with calories being provided by protein (mean 22%), fat (mean 16%) and carbohydrates (mean 62%). Experiments were carried out using 10-week and 7-week-old mice for acute and chronic studies, respectively. Groups were randomized on basal glycaemia levels just before treatment. For acute studies, mice were fasted for 2 h and compounds given orally in 1% (w/v) hydroxy ethyl cellulose (HEC). Glycaemia was measured using the Accu-check glucometer (Roche Diagnostics, Meylan, France), by sampling blood from the tail vein 1, 2, 4 and 6 h after compound administration. For chronic studies, mice were treated orally (1% HEC) for 4 days or 4 weeks. The last dose of the compound was given 1 day before killing the mice. On the final day, mice were fasted at 9 a.m. for 4 h, then anaesthetized with 5% isofluorane (Baxter, Maurepas, France). Blood was taken via intracardiac puncture and the livers removed and snap-frozen in liquid nitrogen, then stored at −80°C. Plasma HbA1c, triglyceride (TGs), alanine aminotransferase (ALAT) and aspartyl aminotransferase (ASAT) levels were measured using Kits from Horiba-ABX (Montpellier, France) and plasma FFAs were measured using the NEFA-HR kit from Wako Chemicals (Neuss, Germany).
Measurement of hepatic TG and glycogen content
Hepatic TGs were measured as previously described (De Ceuninck et al., 2013). Briefly, 100 mg of liver was extracted in 350 μL of ethanol containing 15% potassium hydroxide (w/v) for 16 h at 55°C. The homogenate was completed to 1 mL with 50% ethanol and centrifuged at 150× g for 10 min at 4°C. The supernatant was removed and completed to 1.2 mL with 50% ethanol and centrifuged at 150× g for 10 min at 4°C. A 200-μL sample of the supernatant was supplemented with 0.5 M MgCl2. The mixtures were kept on ice then centrifuged at 1000× g for 10 min at 4°C. The TG concentration in the supernatant was measured using the TG kit from Horiba-ABX. The hepatic glycogen content was assessed by the method of Keppler and Decker (1974). Briefly, 200 mg of liver was incubated in 1 mL of 1 N KOH for 30 min at 85°C. After cooling, the pH was lowered to pH 4.75 by adding one volume of 1.5 N acetic acid. Denatured proteins were removed by centrifugation at 2000× g for 5 min at 4°C. Glycogen in the supernatant was digested by adding 8.3 units·mL−1 of amyloglucosidase (Sigma-Aldrich) in 0.1 M acetate buffer, pH 4.5 for 30 min at 37°C. The resulting glucose was measured using the oxidase method on a Cobas automated system. Samples were quantified using glycogen from rabbit liver (Sigma-Aldrich) subjected to the same digestion protocol.
Data analysis
All data were analysed using the GraphPad Prism 5.04 software (La Jolla, CA, USA). EC50 values were calculated using a four parameter logistic analysis (dose-response, stimulation, variable slope, four parameters) from at least four independent experiments. Values of S0.5 are reported as the means and range of values from two experiments, and shown for one representative experiment. All other data are reported as the means and SEM; significant differences between means were assessed by Student's t-test or one-way ANOVA as appropriate, with Dunnett's post hoc test.”
Materials
S 49164 and S 50612 were synthesized by the Department of Medicinal Chemistry at Servier and isolation of their enantiomers S 50131, S 50133, S 51434 and S 51435 was achieved by chiral HPLC. N00236460 (De Ceuninck et al., 2013) and GKA50 (McKerrecher et al., 2006) were also synthesized in-house. Spectroscopic and physical characterization data (1H, 13C NMR spectra, IR, CHN, MS) were in agreement with the reported structure of all compounds.
Results
Chemical structure and enzymatic properties of tetrahydrocyclopropa[a]indene compounds
The chemical structures of the racemic compounds S 49164 (6-({[(1R,1aR,6aR),(1S,1aS,6aS)-1a-methyl-1,1a,6,6a-tetrahydrocyclopropa[a]inden-1-yl]carbonyl}amino)pyridine-3-carboxylic acid; general formula C18H16N2O3) and S 50612 (6-({[(1R,1aR,6aR),(1S,1aS,6aS)-1a-propyl-1,1a,6,6a-tetrahydrocyclopropa[a]inden-1-yl]carbonyl}amino)pyridine-3-carboxylic acid; general formula C20H20N2O3) and their enantiomers are shown in Figure 1. Their molecular weights (free bases) were 308.34 and 336.39 g·mol−1, respectively.
Figure 1.
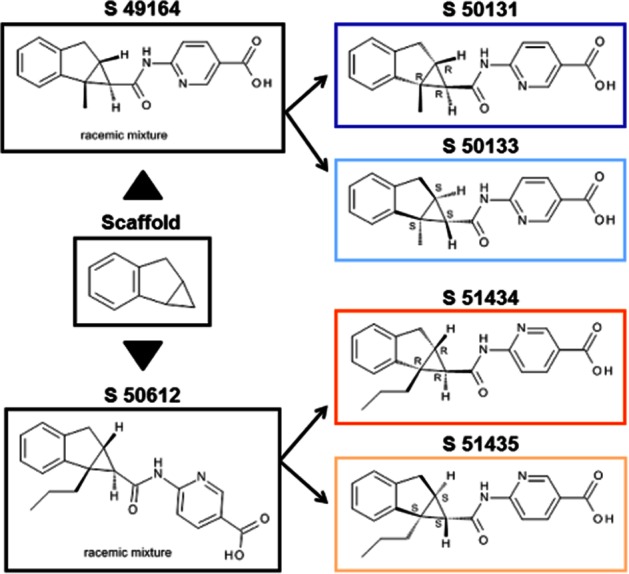
Chemical structures of the novel glucokinase activators S 49164, S 50612 and their enantiomers.
The pharmacological potency of the racemic compounds and their enantiomers was tested on recombinant GK at glucose concentrations ranging from 1.1 mM to 55 mM (Figure 2). Both S 49164 and S 50612 increased the enzymic activity of GK in a dose-dependent manner. The potency was improved with the enantiomers S 50131 and S 51434, whereas the S 50133 and S 51435 enantiomers were only weakly active. As measured in two independent experiments, 6 μM S 50131 highly increased the affinity of GK for glucose with an S0.5 falling from a mean of 10.01 mM (range 9.76–10.26 mM) to a mean of 2.19 mM (range 1.73–2.66 mM). Similarly, 6 μM S 51434 decreased the S0.5 of GK for glucose from 8.42 mM (range 7.83–9.02 mM) to 0.60 mM (range 0.39–0.82 mM). No clear modification of the Vmax was observed with any compound. The half-maximal efficacy (EC50) and maximal potency (Emax) of S 50131 and S 51434 were measured in separate experiments, at a fixed glucose concentration of 8 mM (not shown). S 50131 had an EC50 of 726 ± 177 nM and activated GK with an Emax of 150 ± 17% at 100 μM (means ± SEM, n = 5). The EC50 of S 51434 was 2.24 ± 1.04 μM and the Emax was 120 ± 14% at 100 μM (means ± SEM, n = 4).
Figure 2.
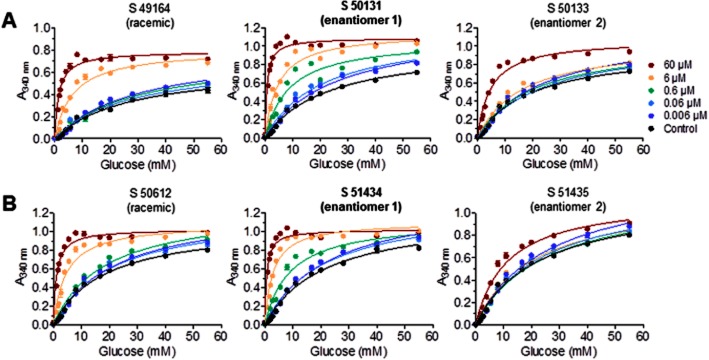
Glucose-dependent and dose-dependent effect of S 49164 and its enantiomers (A), and S 50612 and its enantiomers (B) on human recombinant glucokinase. Data are means ± SEM of one representative experiment (out of two) performed in four replicates for each point.
S 49164, S 50612, and their active enantiomers S 50131 and S 51434, stimulate glycogen synthesis in cultured hepatocytes and insulin secretion from pancreatic islets of Langerhans
Both racemic compounds and their active enantiomers increased glycogen synthesis in cultured rat hepatocytes in a dose-dependent manner. At the concentration of 10 μM, S 49164 and S 50612 stimulated glycogen synthesis by 183 ± 9% (mean ± SEM, P < 0.001; n = 4) and 178 ± 18% (P < 0.001; n = 4), respectively, compared to control vehicle-treated cells (Figure 3A,C). The potency was further increased with enantiomers S 50131 and S 51434, reaching up to 234 ± 19% (P < 0.001; n = 4) and 345 ± 39% (P < 0.001; n = 4) over controls, respectively. When tested at 10 μM, all four compounds stimulated insulin secretion from rat pancreatic islets (Figure 3B,D). Maximal stimulation was observed at 5.5 mM glucose, where S 50131 and S 51434 increased insulin secretion by 6.5- and 21.7-fold compared to controls (P < 0.001, n = 3). At 8.0 mM glucose, insulin secretion was generally increased by about twofold for S 49164, S 50612, S 50131 and S 51434 (P < 0.001, n = 3) compared to controls. Insulin secretion was unaffected by any compound at glucose concentrations below 5.5 mM and only weakly increased at 11 mM glucose. As expected, S 50133 and S 51435 were inactive in the hepatocyte and pancreatic islet assays. Based on assays using Caco-2 cells, S 49164, S 50131, S 50612 and S 51434 were predicted to have excellent intestinal permeabilities (expressed as predicted absorbed fraction, Fabs) as well as very good metabolic stabilities (expressed as metabolic bioavailabilities, MF) when incubated with mouse or rat microsomes (Table 1), making them good candidates for in vivo evaluation.
Figure 3.

In vitro pharmacological profiles of S 49164, S 50612 and their enantiomers (A,C). Glycogen synthesis in primary rat hepatocytes after 3 h of treatment with S 49164 and its enantiomers (A) or S 50612 and its enantiomers (C). Data are means ± SEM of four independent experiments performed in quadruplicate wells. (B, D). Insulin secretion from rat islets of Langherans cells after 90 min of treatment with S 49164 and its enantiomers (B) or S 50612 and its enantiomers (D). Data are means ± SEM of three independent experiments performed in eight replicate wells (containing four islets per well). *P < 0.05, **P < 0.01, ***P < 0.001, significantly different from controls; in A and C, anova and Dunnett's t-test; in B and D, Student's t-test.
Table 1.
Prediction of the intestinal absorption and metabolic availability of tetrahydrocyclopropa[a]indene compounds
| Fabsa (%) | MFb mice (%) | MFb rat (%) | |
|---|---|---|---|
| S 50612 | 100 | 100 | 100 |
| S 51434 | 100 | 95 | 89 |
| S 49164 | 100 | 87 | 92 |
| S 50131 | 100 | 83 | 92 |
Fabs, prediction of the intestinal absorption based on permeability rates from the apical to the basal compartment of cultured Caco-2 cells. Percentages were calculated from a calibration curve obtained with reference compounds for which oral absorption in man was known (Yee, 1997). Values are means of duplicate wells from a single experiment.
MF, prediction of metabolic availability based on in vitro metabolic stability measurements with hepatic microsomes as a function of time. MF values were calculated from a single experiment as detailed previously (De Ceuninck et al., 2013).
S 49164, S 50612, and their active enantiomers S 50131 and S 51434, effectively lower glycaemia after acute administration in db/db mice
In contrast to the inactive enantiomer S 50133, S 50131 lowered glycaemia in db/db mice in a dose-dependent manner, with significant activity reached at the concentration of 45 mg·kg−1 (P < 0.01 after 6 h, Figure 4A). A similar profile was observed for the racemic molecule S 49164 although this compound had a shorter duration of action (P > 0.05 after 6 h). S 51434 and its parent S 50612 lowered glycaemia with higher potency than S 50131 and S 49164, with significant activity still remaining 6 h after oral administration at 15 mg·kg−1 (P < 0.05 and P < 0.01, respectively; Figure 4B). Only residual activity could be measured for the less active S 51435 enantiomer. This compound, as well as S 50133, were discarded for the chronic studies.
Figure 4.
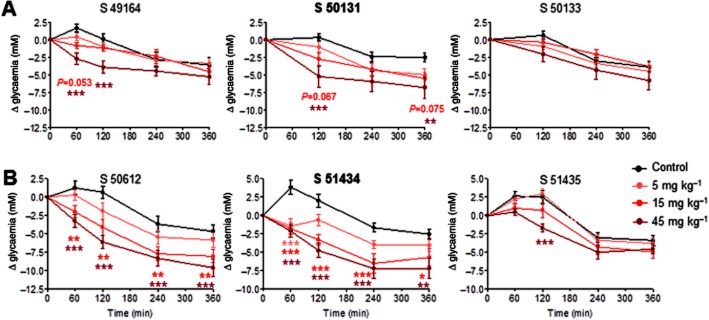
Antihyperglycaemic action of tetrahydrocyclopropa[a]indene glucokinase activators after acute oral administration in db/db mice. Data are means ± SEM of 10 animals. *P < 0.05, **P < 0.01, ***P < 0.001 significantly different from vehicle; Dunnett's t-test. FFA, free fatty acid; TG, triglyceride.
Evaluation of the efficacy/safety profile of S 50131 and S 51434 after 4 weeks or 4 days of oral treatment in db/db mice
The two most active enantiomers, S 50131 and S 51434, were taken forward into chronic studies because their potencies after acute in vivo administration were fully compatible with an expected long-term antidiabetic efficacy. This was evaluated by measuring HbA1c levels after 4 weeks of daily oral administration in db/db mice (Figure 5A,G). Doses were adjusted in order to take into account the 1.5-fold higher relative hypoglycaemic potency of S 51434 observed after 4 h in acute conditions (Figure 4). Thus, S 50131 and S 51434 were given at 60 and 180 μmol·kg−1 (equivalent to 18.5 and 55.5 mg·kg−1) and 40 and 120 μmol·kg−1 (equivalent to 13.5 and 40.4 mg·kg−1), respectively. Safety was evaluated by measuring plasma FFA and TG levels, as well as the hepatic TG content at the end of the study. Both enantiomers were tested in parallel with 120 μmol·kg−1 N00236460 or GKA50 (equivalent to 46.2 mg·kg−1 or 55.8 mg·kg−1, respectively), two reference GKAs previously shown to have excellent in vivo efficacy in rodents (McKerrecher et al., 2006; De Ceuninck et al., 2013). These two GKAs were previously shown to decrease HbA1c levels while increasing hepatic TGs when evaluated in db/db mice (De Ceuninck et al., 2013). In the present study, none of the GKAs tested altered glycaemia measured on days 14 and 28 (Supporting Information Figure S1), most likely because their acute efficacy was no longer present at the time of sampling (21 h after administration). As expected, N00236460 and GKA50 significantly increased the hepatic TG content (P < 0.05 and P < 0.001, respectively; Figure 5F,L) without affecting the plasma lipid levels (Figure 5D,E,J,K), and decreased HbA1c levels, significantly for N00236460 (P < 0.01, Figure 5A) and with a marked downward trend for GKA50 (P = 0.06, Figure 5G). Unexpectedly, S 50131 and S 51434 did not lower HbA1c, despite their efficacy under acute conditions. Furthermore, both compounds increased the plasma FFA levels by a mean of 56 (P < 0.01) and 68% (P < 0.01), respectively, at the highest dose (Figure 5D,J) but did not affect the plasma TG levels (Figure 5E,K). As also observed with N00236460 and GKA50, both tetrahydrocyclopropa[a]indene compounds significantly decreased the hepatic glycogen content (Figure 5C,I). However, only S 50131 increased hepatic TGs, by 48% (P < 0.05), in a similar manner to that seen with N00236460 (Figure 5F). The plasma levels of ALAT and ASAT, HDL-cholesterol and total cholesterol were not affected by treatment with any GKA, except for a slight decrease of total cholesterol observed with GKA50 (Table 2).
Figure 5.

Evaluation of the efficacy and safety of S 50131 and S 51434 compared to GKA50 and N00236460, after 4 weeks of daily oral administration in db/db mice. The doses expressed as mg·kg−1 correspond to 120 μmol·kg−1 for GKA50 and N00236460, to 60 and 180 μmol·kg−1 for S 50131, and to 1.5-fold lower doses (40 and 120 μmol·kg−1) for S 51434, considering its 1.5-fold greater acute hypoglycaemic potency after 4 h, as shown in Figure 4. (A–F) Efficacy and safety profile of S 50131; (G–L) Efficacy and safety profile of S 51434. (A,G) Differences in plasma HbA1c levels between day 1 and day 28; (B,H) Relative liver weights after 4 weeks; (C,I) Hepatic glycogen content after 4 weeks; (D, J) Plasma free fatty acid (FFA) levels after 4 weeks; (E,K) Plasma triglyceride (TG) levels after 4 weeks; (F,L) Hepatic TG content after 4 weeks. Data are means ± SEM of 12 animals per group. *P < 0.05, **P < 0.01, ***P < 0.001 significantly different from vehicle; anova and Dunnett's multiple comparison test for S 50131 and S 51434; Student's t-test for N00236460 or GKA50.
Table 2.
Plasma levels of alanine aminotransferase (ALAT), aspartyl aminotransferase (ASAT), total cholesterol and HDL-cholesterol in the 4-week chronic studies in db/db mice
| ALAT (U·L−1) | ASAT (U·L−1) | Total cholesterol (g·L−1) | HDL-cholesterol (g·L−1) | |
|---|---|---|---|---|
| Vehicle | 151.3 ± 30.7 | 197.5 ± 34.1 | 1.252 ± 0.066 | 0.801 ± 0.039 |
| N00236460 | 121.0 ± 20.3 | 295.8 ± 78.0 | 1.180 ± 0.030 | 0.708 ± 0.015 |
| S 50131a | 126.8 ± 14.1 | 216.0 ± 43.7 | 1.274 ± 0.046 | 0.740 ± 0.025 |
| S 50131b | 125.9 ± 13.7 | 314.2 ± 48.1 | 1.376 ± 0.049 | 0.812 ± 0.023 |
| Vehicle | 106.1 ± 14.0 | 260.5 ± 75.2 | 1.385 ± 0.058 | 0.824 ± 0.029 |
| GKA50 | 129.2 ± 32.0 | 275.1 ± 77.2 | 1.292 ± 0.055* | 0.737 ± 0.028 |
| S 51434a | 92.8 ± 9.0 | 174.4 ± 43.6 | 1.376 ± 0.041 | 0.827 ± 0.017 |
| S 51434b | 176.4 ± 60.2 | 443.4 ± 117.2 | 1.313 ± 0.049 | 0.836 ± 0.027 |
Results are means ± SEM of at least nine mice per group.
P < 0.05 significantly different from vehicle.
Low-dose group.
High-dose group.
We then asked whether the lipid profile changes observed with S 50131 and S 51434 occurred earlier than 4 weeks. Therefore, plasma FFAs and hepatic TGs were examined after a 4-day treatment with both GKAs in comparison to GKA50 (Figure 6). Contrary to the 4-week study, plasma FFAs were unaffected by GKA treatment at 4 days. However, hepatic TGs were increased by S 50131 (mean 58%, P < 0.01) and S 51434 (mean 108%, P < 0.01).
Figure 6.
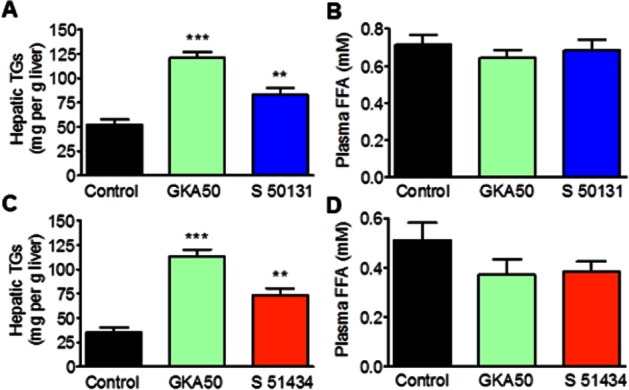
S 50131 and S 51434 increase the hepatic triglyceride (TG) content without any effect on plasma free fatty acids (FFAs) after 4 days of daily oral treatment in db/db mice. (A,C), Hepatic TG content. (B,D), Plasma FFA levels. S 50131 (A,B) and S 51434 (C,D) were evaluated in independent experiments with GKA50 being used as positive control. All compounds were administered once daily at 130 μmol·kg−1, corresponding to 60.4 mg·kg−1 for GKA 50, 40.0 mg·kg−1 for S 50131 and 43.9 mg·kg−1 for S 51434. Data are means ± SEM of 10 animals. **P < 0.01, ***P < 0.001, significantly different from vehicle; Student's t-test.
The main findings observed with all four GKAS are summarized in Table 3.
Table 3.
Effects of N00236460, GKA50, S 50131 and S 51434 on hepatic triglycerides (TGs), plasma free fatty acids (FFAs) and HbA1c after a 4-day or 4-week treatment, and acute antihyperglycaemic activity 6 h after oral administration in db/db mice
Discussion
Because of its critical role in the regulation of glucose homeostasis, pharmacological activation of GK has for long been viewed as an attractive target to treat type 2 diabetes. Activating GK mutations in humans and rodents, while causing hypoglycaemia, did not alter plasma lipid profiles (Gloyn et al., 2003; Pino et al., 2007; Wabitsch et al., 2007), suggesting that manipulating GK could have a positive balance on the regulation of glucose metabolism. In contrast, naturally occurring genetic variations in GCKR, a liver-specific GK inhibitory protein and transgenic animal models of hepatic GK overexpression were associated with altered hepatic and/or plasma lipid profiles (O'Doherty et al., 1999; Jackerott et al., 2002; Ferre et al., 2003; Saxena et al., 2007; Santoro et al., 2012), casting doubt on the safety of the pharmacological approach. We recently demonstrated that GKAs belonging to three distinct structural families effectively decreased plasma HbA1c levels but concomitantly increased hepatic TGs after chronic administration in hyperglycaemic db/db mice (De Ceuninck et al., 2013). Syntheses with a new chemical scaffold resulted in S 50131 and S 51434, two GKAs with potent antihyperglycaemic activity after acute oral administration in diabetic mice, but surprisingly no effect on HbA1c levels after 4 weeks of oral treatment at pharmacological doses. In the 4-week study, these two GKAs markedly increased the plasma FFA levels, which for S 50131 was also associated with increased hepatic TGs. In contrast, no FFA increase could be seen with either compound after a 4-day treatment, although the hepatic TGs were clearly increased. These findings, together with those previously reported with different structural classes of GKAs, prompt serious reflection regarding the safety aspects of GKAs as treatment for patients with type 2 diabetes.
Although we did not investigate the cause-effect relationship between the increase of plasma FFAs and the absence of efficacy on antidiabetic endpoints in the present study, current knowledge suggests that those events are linked. Among other effects, elevated plasma FFA levels can lead to hepatic and muscle insulin resistance by decreasing insulin-dependent glucose uptake in the muscle and glycogen stores in the liver (Ferrannini et al., 1983; Kovacs and Stumvoll, 2005). Because of the very high basal plasma insulin levels in db/db mice, S 50131 in the present study did not further increase basal insulinaemia (results not shown). This was in agreement with results previously reported in this model with GKA50, N00236460 and other GKAs from distinct chemical families (De Ceuninck et al., 2013). However, the finding that glycogen stores were decreased in the livers of GKA-treated mice suggested that all of them increased hepatic insulin resistance. The efficacy and safety profiles of GKAs used in the present study further suggested that elevated circulating FFAs rather than hepatic TGs were responsible for the lack of antidiabetic efficacy of S 50131 and S 51434. Indeed, the increase of plasma FFAs observed with S 50131 and S 51434 was not observed with GKA50 and N00236460. Furthermore, the two latter were able to decrease HbA1c despite increased hepatic TGs. Finally, S 51434 enhanced circulating FFAs but did not alter hepatic TGs. The reason for the increase in plasma FFAs with S 50131 and S 51434 in db/db mice is unknown. It is unlikely to be due to enhanced de novo lipogenesis, occurring as a consequence of increased hepatic carbohydrate flux, as hepatic lipids are secreted from the liver essentially as very low-density lipoproteins (Postic and Girard, 2008). Another possibility would be enhanced adipose tissue lipolysis. However, the weight of epididymal fat was unchanged in mice treated for 4 weeks with S 51434 (not shown); therefore, this is probably not the explanation. Finally, decreased hepatic fatty acid uptake could also contribute to elevated plasma FFAs, but this was not measured in the present study. At this stage, this question deserves further investigation.
Whatever mechanisms are involved, the fact that circulating FFA levels were increased with these two novel GKAs is an additional safety warning for the use of this pharmacological class in humans, especially in patients with type 2 diabetes for whom lipid-associated complications require special attention (Valensi and Picard, 2011). In contrast to the increased hepatic TG content induced by GKA treatment, reported previously (De Ceuninck et al., 2013) and in the present study, circulating FFAs, however, can easily be monitored in clinical practice. It is interesting to note that recent publications and press releases still claim that GKAs represent a promising new class of drugs for the treatment of type 2 diabetes. In contradiction with these assertions however, a large number of GKAs did not reach clinical development despite good efficacy in preclinical research. Very often, the reasons for failure were not disclosed. However, treatment with MK-0941 was reported to be associated with an increased incidence of hypoglycaemia, a lack of sustained efficacy, and elevations in plasma TG levels and blood pressure (Meininger et al., 2011). RO0281675, a GKA showing potent glucose-lowering and insulin-releasing activity in mice (Grimsby et al., 2003), was recently disclosed as inducing hepatic steatosis in toxicology studies in rats and dogs. However, it was suggested that the formation of a thiourea metabolite was the causative agent (Sarabu et al., 2012). Piragliatin, a structurally related compound that reached phase 2 clinical development, revealed no evidence of hepatic steatosis in subchronic or chronic toxicological studies in rats and dogs, but its development was stopped because of an unfavourable risk-benefit profile (Sarabu et al., 2012). In our hands, piragliatin showed an upward trend in hepatic steatosis when administered orally for 4 days in db/db mice (De Ceuninck et al., 2013). However, after a 4-week treatment at pharmacological doses in db/db mice, piragliatin did not increase hepatic or plasma TGs, but did dose-dependently increase plasma FFA levels with no effect on HbA1c (De Ceuninck et al., unpublished experiments). This profile strikingly resembling that of the tetrahydrocyclopropa[a]indene compounds tested in the present study, further supported a relationship between elevated plasma FFAs and the lack of antidiabetic efficacy of S 50131 and S 51434 observed after 4 weeks.
One critical question is whether GKAs can still be considered as a viable and safe option for the treatment of type 2 diabetes. As hepatic and pancreatic GK are very similar in protein sequence and enzyme activity (with only few differences found in the N-terminal amino acid sequence), most GKAs stimulate glycogen synthesis in hepatocytes and insulin secretion in islets of Langerhans in vitro. However, probably due to pharmacodynamic issues, GKAs do not often reach the pancreas and rather concentrate in the liver when administered orally in animal models. On the one hand, these ‘liver-selective’ GKAs were generally safe regarding hypoglycaemic events, but induced hepatic steatosis and/or dyslipidaemia, possibly associated with a lack of sustained efficacy. On the other hand, dual GKAs (reaching the pancreas and the liver), while similarly being associated with disturbed lipid homeostasis, also caused hypoglycaemia. They did this by increasing the affinity of GK for glucose, which in turn, induced insulin secretion at inappropriate glucose levels. Based on the current knowledge and on the safety issues uncovered recently, succeeding with this particular class of compounds, if feasible, will remain a challenge. At the present time, we believe that the manipulation of GK activity to offer a safe, long-term antidiabetic benefit should be reconsidered.
Acknowledgments
We thank Michel Wierzbicki and Jean-Marie Fourquez for the chemical synthesis of GKA50 and N00236460.
Glossary
- ALAT
alanine aminotransferase
- ASAT
aspartyl aminotransferase
- FFA
free fatty acid
- GCKR
glucokinase regulatory protein
- GK
glucokinase
- GKA
glucokinase activator
- TG
triglyceride
Conflict of interest
All authors are employees of Servier group subsidiaries and declare no competing financial interests.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Effect of S 50131 and S 51434 on glycaemia by comparison with GKA50 and N00236460 after 2 and 4 weeks of daily oral administration in db/db mice.
References
- Agius L. Targeting hepatic glucokinase in type 2 diabetes: weighing the benefits and risks. Diabetes. 2009;58:18–20. doi: 10.2337/db08-1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebernitz GR, Beaulieu V, Dale BA, Deacon R, Duttaroy A, Gao J, et al. Investigation of functionally liver selective glucokinase activators for the treatment of type 2 diabetes. J Med Chem. 2009;52:6142–6152. doi: 10.1021/jm900839k. [DOI] [PubMed] [Google Scholar]
- Bertrand M, Jackson P, Walther B. Rapid assessment of drug metabolism in the drug discovery process. Eur J Pharm Sci. 2000;11(Suppl. 2):S61–S72. doi: 10.1016/s0928-0987(00)00165-2. [DOI] [PubMed] [Google Scholar]
- Bontemps F, Hue L, Hers HG. Phosphorylation of glucose in isolated rat hepatocytes. Sigmoidal kinetics explained by the activity of glucokinase alone. Biochem J. 1978;15:603–611. doi: 10.1042/bj1740603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocklehurst KJ, Payne VA, Davies RA, Carroll D, Vertigan HL, Wightman HJ, et al. Stimulation of hepatocyte glucose metabolism by novel small molecule glucokinase activators. Diabetes. 2004;53:535–541. doi: 10.2337/diabetes.53.3.535. [DOI] [PubMed] [Google Scholar]
- Chahil TJ, Ginsberg HN. Diabetic dyslipidemia. Endocrinol Metab Clin North Am. 2006;25:491–510. doi: 10.1016/j.ecl.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Coope GJ, Atkinson AM, Allott C, McKerrecher D, Johnstone C, Pike KG, et al. Predictive blood glucose lowering efficacy by glucokinase activators in high fat fed female Zucker rats. Br J Pharmacol. 2006;149:328–335. doi: 10.1038/sj.bjp.0706848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ceuninck F, Kargar C, Ilic C, Caliez A, Rolin JO, Umbdenstock T, et al. Small molecule glucokinase activators disturb lipid homeostasis and induce fatty liver in rodents: a warning for therapeutic applications in humans. Br J Pharmacol. 2013;168:339–353. doi: 10.1111/j.1476-5381.2012.02184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efanov AM, Barrett DG, Brenner MB, Briggs SL, Delaunois A, Durbin JD, et al. A novel glucokinase activator modulates pancreatic islet and hepatocyte function. Endocrinology. 2005;146:3696–3701. doi: 10.1210/en.2005-0377. [DOI] [PubMed] [Google Scholar]
- Ferrannini E, Barrett EJ, Bevilacqua S, DeFronzo RA. Effect of fatty acids on glucose production and utilization in man. J Clin Invest. 1983;72:1737–1747. doi: 10.1172/JCI111133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre T, Riu E, Franckhauser S, Agudo J, Bosch F. Long-term overexpression of glucokinase in the liver of transgenic mice leads to insulin resistance. Diabetologia. 2003;46:1662–1668. doi: 10.1007/s00125-003-1244-z. [DOI] [PubMed] [Google Scholar]
- Fuller H, Stevens LK. Prevalence of hypertension among diabetic patients and its relation to vascular risk. Diabetes Hypertension Study Group. J Hum Hypertens. 1991;5:237–243. [PubMed] [Google Scholar]
- Fyfe MC, White JR, Taylor A, Chatfield R, Wargent E, Printz RL, et al. Glucokinase activator PSN-GK1 displays enhanced antihyperglycaemic and insulinotropic actions. Diabetologia. 2007;50:1277–1287. doi: 10.1007/s00125-007-0646-8. [DOI] [PubMed] [Google Scholar]
- Gloyn AL, Noordam K, Willemsen MA, Ellard S, Lam WW, Campbell IW, et al. Insights into the biochemical and genetic basis of glucokinase activation from naturally occurring hypoglycemia mutations. Diabetes. 2003;52:2433–2440. doi: 10.2337/diabetes.52.9.2433. [DOI] [PubMed] [Google Scholar]
- Grimsby J, Sarabu R, Corbett WL, Haynes NE, Bizzarro FT, Coffey JW, et al. Allosteric activators of glucokinase: potential role in diabetes therapy. Science. 2003;301:370–373. doi: 10.1126/science.1084073. [DOI] [PubMed] [Google Scholar]
- Jackerott M, Baudry A, Bucchini D, Jami J, Joshi RL. Improved metabolic disorders of insulin receptor-deficient mice by transgenic overexpression of glucokinase in the liver. Diabetologia. 2002;45:1292–1297. doi: 10.1007/s00125-002-0881-y. [DOI] [PubMed] [Google Scholar]
- Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia. 2003;46:3–19. doi: 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- Kamata K, Mitsuya M, Nishimura T, Eiki J, Nagata Y. Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure. 2004;12:429–438. doi: 10.1016/j.str.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Keppler D, Decker K. Glycogen: determination with amyloglucosidase. In: Bergmeyer HU, editor. Methods of Enzymatic Analysis. Weinheim/New York: Verlag Chemie/Academic Press Inc; 1974. pp. 1127–1131. [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs P, Stumvoll M. Fatty acids and insulin resistance in muscle and liver. Best Pract Res Clin Endocrinol Metab. 2005;19:625–635. doi: 10.1016/j.beem.2005.07.003. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKerrecher D, Allen JV, Caulkett PW, Donald CS, Fenwick ML, Grange E, et al. Design of a potent, soluble glucokinase activator with excellent in vivo efficacy. Bioorg Med Chem Lett. 2006;16:2705–2709. doi: 10.1016/j.bmcl.2006.02.022. [DOI] [PubMed] [Google Scholar]
- Matschinsky FM. Glucokinase as glucose sensor and metabolic signal generator in pancreatic beta-cells and hepatocytes. Diabetes. 1990;39:647–652. doi: 10.2337/diab.39.6.647. [DOI] [PubMed] [Google Scholar]
- Meglasson MD, Matschinsky FM. Pancreatic islet glucose metabolism and regulation of insulin secretion. Diabetes Metab Rev. 1986;2:163–214. doi: 10.1002/dmr.5610020301. [DOI] [PubMed] [Google Scholar]
- Meininger GE, Scott R, Alba M, Shentu Y, Luo E, Amin H, et al. Effects of MK-0941, a novel glucokinase activator, on glycemic control in insulin-treated patients with type 2 diabetes. Diabetes Care. 2011;34:2560–2566. doi: 10.2337/dc11-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Doherty R, Lehman DL, Télémaque-Potts S, Newgard CB. Metabolic impact of glucokinase overexpression in liver: lowering of blood glucose in fed rats is accompanied by hyperlipidemia. Diabetes. 1999;48:2022–2027. doi: 10.2337/diabetes.48.10.2022. [DOI] [PubMed] [Google Scholar]
- Ohyama S, Takano H, Lino T, Nishimura T, Zhou YP, Langdon RB, et al. Small-molecule glucokinase activator lowers blood glucose in the sulfonylurea-desensitized rat. Eur J Pharmacol. 2010;640:250–256. doi: 10.1016/j.ejphar.2010.04.054. [DOI] [PubMed] [Google Scholar]
- Peter A, Stefan N, Cegan A, Walenta M, Wagner S, Königsrainer A, et al. Hepatic glucokinase expression is associated with lipogenesis and fatty liver in humans. J Clin Endocrinol Metab. 2011;96:E1126–E1130. doi: 10.1210/jc.2010-2017. [DOI] [PubMed] [Google Scholar]
- Pfefferkorn JA, Guzman-Perez A, Litchfield J, Aiello R, Treadway JL, Pettersen J, et al. Discovery of (S)-6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido) nicotinic acid as a hepatoselective glucokinase activator clinical candidate for treating type 2 diabetes mellitus. J Med Chem. 2012;55:1318–1333. doi: 10.1021/jm2014887. [DOI] [PubMed] [Google Scholar]
- Pino MF, Kim KA, Shelton KD, Lindner J, Odili S, Li C, et al. Glucokinase thermolability and hepatic regulatory protein binding are essential factors for predicting the blood glucose phenotype of missense mutations. J Biol Chem. 2007;282:13906–13916. doi: 10.1074/jbc.M610094200. [DOI] [PubMed] [Google Scholar]
- Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. 2008;118:829–837. doi: 10.1172/JCI34275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees MG, Gloyn AL. Small molecular glucokinase activators: has another new anti-diabetic therapeutic lost favour? Br J Pharmacol. 2013;168:335–338. doi: 10.1111/j.1476-5381.2012.02201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro N, Zhang CK, Zhao H, Pakstis AJ, Kim G, Kursawe R, et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology. 2012;55:781–789. doi: 10.1002/hep.24806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarabu R, Bizzarro FT, Corbett WL, Dvorozniak MT, Geng W, Grippo JF, et al. Discovery of piragliatin–first glucokinase activator studied in type 2 diabetic patients. J Med Chem. 2012;55:7021–7036. doi: 10.1021/jm3008689. [DOI] [PubMed] [Google Scholar]
- Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- Schuit FC, Huypens P, Heimberg H, Pipeleers DG. Glucose sensing in pancreatic beta-cells: a model for the study of other glucose-regulated cells in gut, pancreas, and hypothalamus. Diabetes. 2001;50:1–11. doi: 10.2337/diabetes.50.1.1. [DOI] [PubMed] [Google Scholar]
- Seglen PO. Preparation of isolated rat liver cells. In: Prescott DM, editor. Methods in Cell Biology. XIII. New York: Academic Press; 1976. pp. 29–83. Ch. 4. [DOI] [PubMed] [Google Scholar]
- Stamler J, Vaccaro O, Neaton JD, Wentworth D. Diabetes, other risk factors, and 12-yr cardiovascular mortality for men screened in the Multiple Risk Factor Intervention Trial. Diabetes Care. 1993;16:434–444. doi: 10.2337/diacare.16.2.434. [DOI] [PubMed] [Google Scholar]
- Tahrani AA, Bailey CJ, Del Prato S, Barnett AH. Management of type 2 diabetes: new and future developments in treatment. Lancet. 2011;378:182–197. doi: 10.1016/S0140-6736(11)60207-9. [DOI] [PubMed] [Google Scholar]
- Ursø B, Cope DL, Kalloo-Hosein HE, Hayward AC, Whitehead JP, O'Rahilly S, et al. Differences in signaling properties of the cytoplasmic domains of the insulin receptor and insulin-like growth factor Receptor in 3T3-L1 adipocytes. J Biol Chem. 1999;274:30864–30873. doi: 10.1074/jbc.274.43.30864. [DOI] [PubMed] [Google Scholar]
- Valensi P, Picard S. Lipids, lipid-lowering therapy and diabetes complications. Diabetes Metab. 2011;37:15–24. doi: 10.1016/j.diabet.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Verspohl EJ. Novel pharmacological approaches to the treatment of type 2 diabetes. Pharmacol Rev. 2012;64:188–237. doi: 10.1124/pr.110.003319. [DOI] [PubMed] [Google Scholar]
- Wabitsch M, Lahr G, Van de Bunt M, Marchant C, Lindner M, von Puttkamer J, et al. Heterogeneity in disease severity in a family with a novel G68V GCK activating mutation causing persistent hyperinsulinaemic hypoglycemia of infancy. Diabet Med. 2007;24:1393–1399. doi: 10.1111/j.1464-5491.2007.02285.x. [DOI] [PubMed] [Google Scholar]
- Winzell MS, Coghlan M, Leighton B, Frangioudakis G, Smith DM, Storlien LH, et al. Chronic glucokinase activation reduces glycemia and improves glucose tolerance in high-fat diet fed mice. Eur J Pharmacol. 2011;663:80–86. doi: 10.1016/j.ejphar.2011.05.009. [DOI] [PubMed] [Google Scholar]
- Yee S. In vitro permeability across Caco-2 cells (colonic) can predict in vivo (small intestinal) absorption in man – fact or myth. Pharm Res. 1997;14:763–766. doi: 10.1023/a:1012102522787. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.