

Abstract
Background and Purpose
High-altitude pulmonary oedema (HAPE) experienced under high-altitude conditions is attributed to mitochondrial redox distress. Hence, hypobaric hypoxia (HH)-induced alteration in expression of mitochondrial biogenesis and dynamics genes was determined in rat lung. Further, such alteration was correlated with expression of mitochondrial DNA (mtDNA)-encoded oxidative phosphorylation (mtOXPHOS) genes. The prophylactic effect of dexamethasone (DEX) in counteracting the HH-induced mitochondrial distress was used as control to understand adaptation to high-altitude exposure.
Experimental Approach
Rats pretreated with DEX were exposed to normobaric normoxia (NN) or HH. HH-induced injury was assessed as an increase in lung water content, tissue damage and oxidant generation. Mitochondrial number, mtDNA content and mtOXPHOS activities were measured to determine mitochondrial function. The expression of mitochondrial biogenesis, dynamics and mtOXPHOS genes was studied.
Key Results
HH-induced lung injury was associated with decreased mitochondrial number, mtDNA content and mtOXPHOS activities. HH exposure decreased the nuclear gene oestrogen-related receptor-α (ERRα), which interacts with PPAR-γ coactivator-1α (PGC-1α) in controlling mitochondrial metabolism. Consequently, mtOXPHOS transcripts are repressed under HH. Further, HH modulated mitochondrial dynamics by decreasing mitofusin 2 (Mfn2) and augmenting fission 1 (Fis1) and dynamin-related protein 1 (Drp1) expression. Nevertheless, DEX treatment under NN (i.e. adaptation to HH) did not affect mitochondrial biogenesis and dynamics, but increased mtOXPHOS transcripts. Further, mtOXPHOS activities increased together with reduced oxidant generation. Also, DEX pretreatment normalized ERRα along with mitochondrial dynamics genes and increased mtOXPHOS transcripts to elicit the mitochondrial function under HH.
Conclusions and Implications
HH stress (HAPE)-mediated mitochondrial dysfunction is due to repressed ERRα and mtOXPHOS transcripts. Thus, ERRα-mediated protection of mitochondrial bioenergetics might be the likely candidate required for lung adaptation to HH.
Keywords: high-altitude pulmonary oedema, dexamethasone, oestrogen-related receptor-α, hypobaric hypoxia, mitofusin-2, mitochondrial fission, mitochondrial biogenesis, oxidative phosphorylation
Introduction
Rapid ascent to high altitude (>2500 m) by humans without prior acclimatization protocol leads to the development of high-altitude pulmonary oedema (HAPE) (Hultgren, 1996). Several aetiological factors, like pulmonary hypertension, hypoxic pulmonary vasoconstriction, elevated capillary pressure, injury to the blood–gas barrier and increased pulmonary vascular permeability are attributed to the pathophysiology of HAPE (West et al., 1991; Maggiorini et al., 2001; Hopkins et al., 2005). However, at the molecular level, an increased generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS) was observed under severe hypobaric hypoxic (HH) conditions (Lin et al., 2011; Shukla et al., 2011). Such oxidative stress is due to mitochondrial disturbances causing protein(s) oxidation and DNA cleavages, ultimately culminating in mitochondrial dysfunction. As a consequence, reduced ATP is produced to disrupt cellular energy homeostasis (Paradies et al., 2002; Magalhaes et al., 2005). Hence, an adaptive mechanism under severe HH conditions must operate at the mitochondrial level as a first line of defence in order to meet cellular energy demand. This counteracting phenomenon (i.e. HH adaptation) could be achieved by augmenting mitochondrial biogenesis as well as its dynamics and oxidative phosphorylation (mtOXPHOS) gene expression, which are in turn controlled by both nuclear DNA (nDNA) and mitochondrial DNA (mtDNA). A schematic diagram showing the nuclear and mitochondrial cross-talk reported so far is depicted in Figure 1.
Figure 1.
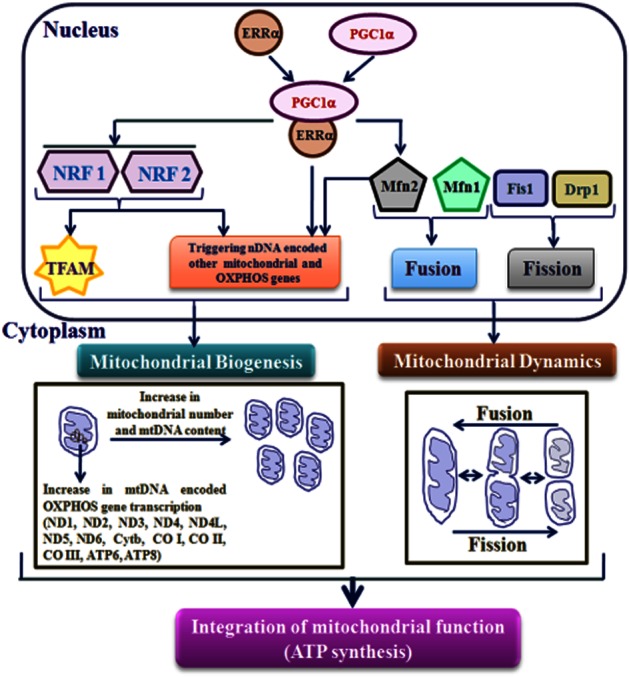
Orchestration of mitochondrial function by PGC-1α and ERRα. Interaction among genes (considered in the present study) encoded by nDNA and mtDNA, which participates in transcriptional network regulating mitochondrial biogenesis, dynamics and mtOXPHOS are depicted schematically. The nDNA-encoded PGC-1α interacts with ERRα to form ‘PGC-1α-ERRα’ complex and coordinately control the transcription of genes involved in mitochondrial biogenesis and mtOXPHOS. ‘PGC-1α-ERRα’ complex directly triggers the genes encoding mitochondrial structural components and mtOXPHOS enzymes, as well as indirectly by enhancing the expression of downstream transcriptional regulators such as NRF1 and NRF2. Subsequently, NRFs also enhance the TFAM expression, which directly activates the transcription and replication of mtDNA. In turn, mtDNA-encoded 13 subunits along with nDNA-encoded subunits forms multi-subunit mtOXPHOS complexes required for ATP synthesis. Mitochondrial function is also regulated by the processes of fusion (Mfn1 and Mfn2) and fission (Fis1 and Drp1) that lead to proper organization of the mitochondrial network. Along with mitochondrial biogenesis, ‘PGC-1α-ERRα’ complex also regulates the Mfn2 expression. Apart from fusion process, Mfn2 drives the mitochondrial metabolism through regulation of nDNA-encoded subunits of OXPHOS.
The nDNA-encoded ‘transcription co-regulator’ called PPAR-γ coactivator-1α (PGC-1α) plays a key role in the overall expression of genes involved in mitochondrial biogenesis and metabolism (Wu et al., 1999). PGC-1α, through inducing the expression of nDNA-binding transcription factors such as nuclear respiratory factors (NRF1 and NRF2) and co-activation of NRF1, in turn activates the transcription of mitochondrial transcription factor A (TFAM). TFAM further regulates mtDNA for its replication and transcription (Wu et al., 1999; Ekstrand et al., 2004). Thus, TFAM along with other mitochondrial transcription factors control the mtDNA expression of 13 proteins: NADH-dehydrogenase-ubiquinone reductase (ND)1 to ND6 and ND4L of complex I (seven protein subunits of ND); cytochrome b (Cyt b) of complex III (one protein subunit of ubiquinol-cytochrome c reductase); cytochrome c oxidase I/II/III (CO I/II/III) of complex IV (three protein subunits of CO); and ATP6 and 8 of complex V (two protein subunits of ATP synthetase) of the mtOXPHOS system (Fernandez-Silva et al., 2003). The nuclear receptor oestrogen-related receptor-α (ERRα), along with PGC-1α is instrumental in regulating cellular energy homeostasis and mitochondrial biogenesis. The inhibition of ERRα diminishes the effect of PGC-1α in inducing mitochondrial biogenesis and cellular respiration (Mootha et al., 2004; Schreiber et al., 2004). In addition, ERRα occupies the extended region of the promoters of all gene-encoding enzyme/protein(s) participating in the citric acid cycle and OXPHOS functions (Charest-Marcotte et al., 2010). Along with mitochondrial biogenesis, the relationship between mitochondrial functions and its dynamics (fusion/fission) under various physiological and pathological conditions are gaining significance. Generally, in mammalian cells, the mitochondrial fusion process is largely controlled by mitofusin 1 and 2 (Mfn1 and 2), the two highly conserved GTPases that are localized in the mitochondrial outer membrane (Hales and Fuller, 1997). Mitochondrial fission 1 (Fis1) and the dynamin-related protein 1 (Drp1) are core components of the mammalian mitochondrial fission machinery. Fis1 protein is located throughout the surface of the outer membrane and is involved in recruiting Drp1, which identifies fission sites (Smirnova et al., 1998; James et al., 2003) leading to an increased number of mitochondria.
Several drugs (nifedipine, salmeterol) or increased oxygen availability through ventilation are in vogue essentially to treat the HH stress-induced lung dysfunction. However, prophylactic administration of synthetic glucocorticoid dexamethasone (DEX) a day prior to high-altitude ascent by humans are shown to prevent the onset of HAPE, even in susceptible individuals (Maggiorini et al., 2006). Yet, in the individuals experiencing acute mountain sickness, treatments with DEX started after the episode could not prevent the onset of symptoms of HAPE (Bartsch et al., 1990). Such observations suggest that the genomic effects exerted by DEX at the cellular level (which require days rather than hours) are critical in counteracting the HAPE (i.e. inducing adaptation) at HH conditions. Various mechanisms detailing the mode of action of DEX in preventing HAPE have been postulated. They include an increase in surfactant production (Young and Silbajoris, 1986), strengthening of cell-to-cell tight junctions (Stelzner et al., 1988) and up-regulation of alveolar sodium (Na) transporters (Guney et al., 2007). All these effects of DEX are high-energy demanding biological activities that require optimal mitochondrial function for the uninterrupted supply of ATP under HH conditions. In this context, it is worth mentioning that DEX has been shown to increase the transcription of mtDNA-encoded OXPHOS genes as well, even under normoxic conditions (Scheller and Sekeris, 2003); this may play a predominant role in the maintenance of cellular energy status especially under HH stress conditions. However, the molecular mechanisms by which DEX brings about these effects are not enumerated.
Although the effect of hypoxia-induced oxidative stress on mitochondrial dysfunction has been studied in other tissues (Magalhaes et al., 2005; Zungu et al., 2007; Gutsaeva et al., 2008), to the best of our knowledge, no data is available regarding the effect of in vivo acute and severe simulated high-altitude hypoxia exposure on rodent lung mitochondrial function. Additionally, the cross-talk between nDNA (involved in mitochondrial biogenesis/dynamics), and mtDNA (OXPHOS) gene expression, that may have an effect on mitochondrial function in cellular energy homeostasis has not been well-characterized. Therefore, in the present study, acute but severe HH conditions are employed to impose high-altitude stress in rats; and their lung tissue were then examined for the expression of genes involved in mitochondrial biogenesis (PGC-1α, ERRα, NRF1, NRF2 and TFAM), dynamics (Mfn1, Mfn2, Drp1, Fis1) and mtOXPHOS (ND1 to 6, ND4L, Cyt b, CO I/II/III, ATP6 and ATP8). Essentially, this study also deciphers whether DEX prophylaxis-mediated adaptation to HH conditions is accomplished by the maintenance of mitochondrial functions.
Methods
Animals
Male Sprague-Dawley rats (200–225 g) were used in the study. The animals were maintained in the Animal house of the institute at 28 ± 2°C and subjected to a 12–12 h light–dark cycle with free access to food and water. The experiments were carried out in accordance with the guidelines of the ethics committee of the institute Bharathiar University, Coimbatore, India. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Hypobaric hypoxic exposure
The rats were randomly assigned to four experimental groups (n = 6): saline-treated normobaric normoxia (NN); saline-treated HH; DEX-treated NN; and DEX-treated HH. DEX (2 mg·kg−1·day−1) or an equivalent volume of saline was administered through i.p. for 3 consecutive days (Guney et al., 2007). From the third day of treatment onwards, the rats continued to be exposed to NN or exposed to a simulated altitude of 9142 m in an animal decompression chamber (Seven Star, Delhi, India) coupled to a mercury barometer at 28°C for 6 h (Shukla et al., 2011). The airflow and relative humidity in the chamber were maintained at 2 L min−1 and 50–55%, respectively. The rats were provided with food and water ad libitum during the experimental protocol. All the animals survived the high-altitude exposure. After exposure to HH, the animals were killed immediately by cervical dislocation; and the lungs were dissected en bloc after perfusion with ice-cold PBS to remove blood. One portion of fresh lung tissue from the various experimental groups was used for extraction of total RNA and isolation of mitochondria. The second portion was snap frozen and stored at −80°C for biochemical and enzyme assay.
Determination of HH-induced toxicity in lung
Analysis of oedema formation in lung
Lung water content was used as an index of oedema formation. The water content of the lung tissue was calculated as the difference between wet weight and dry weight and expressed as milligrams of water per milligrams of dry tissue (Shukla et al., 2011).
Preparation of lung tissue
All the lung tissue obtained for biochemical measurements was washed in 0.9% sodium chloride and kept in ice. The tissue was homogenized with cold 1.15% potassium chloride fortified with a protease inhibitor cocktail to make a 10% homogenate (w/v) and centrifuged at 400× g for 10 min at 4°C. The protein concentrations of the tissue homogenates were determined using BSA as standard (Lowry et al., 1951).
Analysis of lactate dehydrogenase (LDH) activity
LDH activity in the lung homogenate was quantified using reduction of 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide dye (Abe and Matsuki, 2000).
Analysis of oxidative stress markers
Measurement of RNS
The total of NO metabolites nitrate (after reduction to nitrite with 20 mM vanadium chloride) and nitrite present in the supernatant of lung homogenate was assayed using Griess reagent (Sigma Chemical Company, St. Louis, MO, USA) in accordance with the manufacturer's instructions (Boyaci et al., 2006).
Measurement of ROS
About 5 mg of lung tissue in 1 mL of Hank's balanced salt solution was taken, and ROS production measured spectrophotometrically by the quantitative nitro blue tretazolium (NBT) reduction assay as reported previously (Mookerjee et al., 2006).
Determination of gene expression using real-time PCR
Total RNA from lung tissue was extracted using the RNA extraction kit (RBC Real Genomics, Real Biotech Corporation, Taipei, Taiwan) following the manufacturer's protocols. The concentration and purity of the RNA was determined using a UV spectrophotometer (Implen GMbH, Munchen, Germany) by measuring the absorbance at 260 and 280 nm. RNA integrity was assessed in a randomly chosen subset of samples using agarose gel electrophoresis and ethidium bromide staining. RNA isolated from each two different animals per group was pooled proportionately (i.e. to make three biological replicates per group). About 1 μg of total RNA was reverse-transcribed with first-strand cDNA synthesis kit (Fermentas, Thermo Scientific, Waltham, MA, USA) in accordance with the manufacturer's instructions. Quantitative real-time PCR amplification reactions were performed in 20 μL final volume via SYBR Green chemistry (Roche Diagnostics India Pvt. Ltd., Maharashtra, India) according to the manufacturer's instructions on LightCycler® 480 system (Roche Diagnostics Asia Pacific Pte Ltd., Singapore). The β-actin gene amplification acted as an internal control. The details of the primer sequences (Mikula et al., 2005; de Cavanagh et al., 2008; Ding et al., 2010) for the various genes studied are given in Supporting Information Table S1. Amplification specificity was controlled by melting curve analysis. The ‘REST-2009’ application was used to understand the relative expression profile with the input of Cp value and relative efficiency of each primer pair (Pfaffl, 2009).
Determination of mtDNA/nDNA ratio
MtDNA/nDNA ratio was quantified by the method described earlier using the d-loop region of mtDNA and GAPDH gene of nDNA. The mtDNA/nDNA ratio was reported as 2–ΔCp (de Cavanagh et al., 2008).
Determination of mitochondrial content and function
Isolation of mitochondria
Mitochondria of the lung tissue were isolated using MITOISO1 kit (Sigma Chemical Company) following the manufacturer's instructions. The functional specificities of citrate synthase (CS) and different mitochondrial complexes (I, III, IV and V), in terms of their specific enzyme activities, were measured using isolated mitochondria. All the assays were performed at 30°C.
Analysis of CS activity
CS, a mitochondrial matrix enzyme, is commonly used as a marker for the contents of intact mitochondria. About 50 μg of mitochondrial protein preparation was used for activity measurement. Enzyme activity was assayed following the acetylation of 5, 5′-dithio-bis-(2-nitrobenzoic acid) at 412 nm (Craig, 1973).
Analysis of mitochondrial OXPHOS complex activity
The activity of complex I was measured by monitoring the reduction of 2, 6-dichlorophenol indophenol in the presence of 200 mM NADH at 600 nm (Janssen et al., 2007). The activity of complex III was measured as an increase in absorbance because of the reduction of cytochrome c at 550 nm (Krahenbuhl et al., 1994). The activity of complex IV was measured as a decrease in absorbance because of the oxidation of cytochrome c at 550 nm (Capaldi et al., 1995). Complex V activity was measured as oligomycin-sensitive Mg2+-ATPase activity by measuring the increasing oxidation of NADH at 340 nm in the presence of LDH, pyruvate kinase and ATP (Zheng and Ramirez, 2000).
Statistical analysis
For relative expression studies, REST 2009 uses randomization and bootstrapping methods to test the statistical significance of gene expression ratios and calculates 95% confidence intervals for relative fold changes. The hypothesis test P(H1) obtained represents the probability of the alternate hypothesis that the difference between the sample and control groups is due only to chance. The significance level was set at P(H1) < 0.05. (Pfaffl, 2009). The gene expression ratio was reported in the form of a box and whisker plot showing median expression (horizontal line), surrounded by the first and third quartiles of those values (box) and the extreme values (whiskers). Results of the biochemical experiments were reported as mean values ± SD. Data was analysed with one-way anova. When a significant effect was detected, the Bonferroni's post hoc test was used to compare among groups as applicable. The significance level was set at P < 0.05.
Materials
RNA extraction kit was procured from RBC Real Genomics, Real Biotech Corporation. DEX, NBT, MITOISO1 kit and all the other chemicals used in the mtOXPHOS complex assay were procured from Sigma Chemical Company. First-strand cDNA synthesis kit was procured from Fermentas, Thermo Scientific. LightCycler 480 SYBR Green I Master was procured from Roche Diagnostics India Pvt. Ltd.. The primers were procured from Integrated DNA Technologies (Coralville, IA, USA). All the other chemicals used were indigenous in nature and of the highest purity.
Results
HH condition-induced lung injury
Animals exposed to acute, but severe HH conditions were evaluated for the lung injury by measuring (i) pulmonary oedema and (ii) increase in LDH activity of lung tissue. The severity of pulmonary oedema was determined in terms of its water content. There was no significant change in oedema either in the control (i.e. saline-treated) or in the DEX pretreated animals kept in NN conditions. However, in animals exposed to HH, there was a significant increase in oedema (∼120%; P < 0.01), in comparison with NN animals (Figure 2A). The increase in oedema observed in HH conditions was brought back (P < 0.01) to NN levels in animals subjected with DEX pretreatment (i.e. adaptation).
Figure 2.
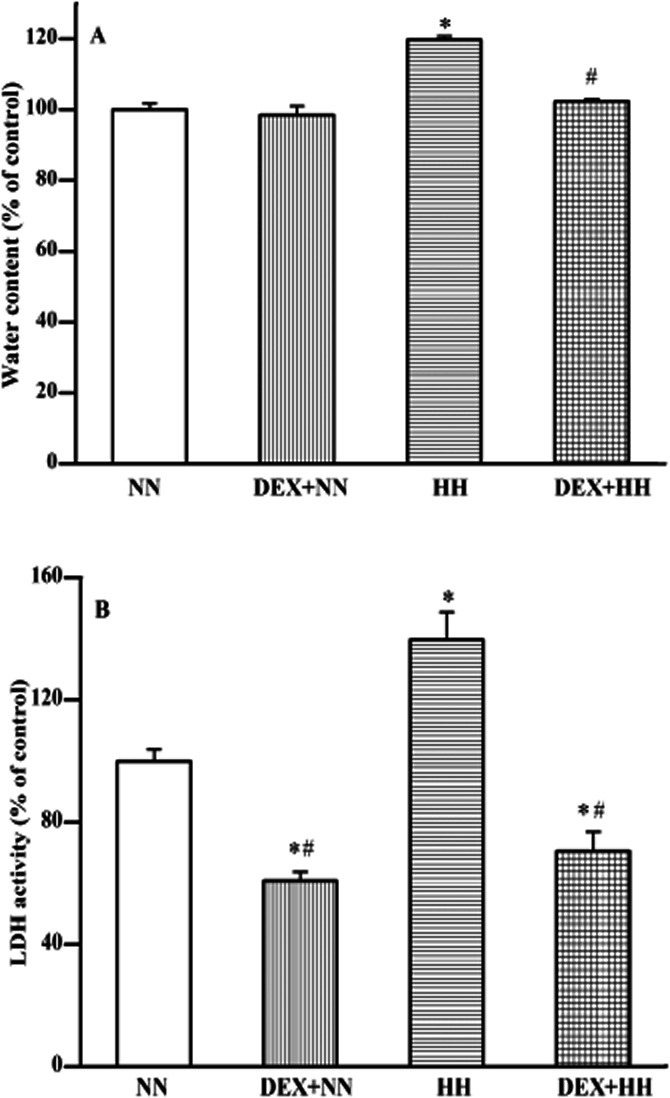
Effect of DEX on HH-induced pulmonary oedema (A) and LDH activity (B) in rat lung. The pulmonary oedema (i.e. increase in water content) of lung tissue of animals exposed to NN or HH conditions with or without DEX pretreatment was determined (A). Similarly, lung tissue damage was assessed as an increase in LDH activity (B). The extent of lung oedema/damage under NN ( ) condition is compared with DEX plus NN (
) condition is compared with DEX plus NN ( ), HH (
), HH ( ) and DEX plus HH (
) and DEX plus HH ( ) conditions. DEX prophylaxis did not alter the water content but decreased LDH activity in the lung tissue of animals maintained under NN conditions. Increase in water content and LDH activity observed under HH conditions reverted to NN levels by DEX pretreatment. This shows the adaptation of lung to HH stress by DEX pretreatment. Results are expressed in % variation as mean ± SD (n = 6 per group). *P < 0.05 versus NN; #P < 0.05 versus HH. The 100% value in absolute quantity of oedema and LDH activity, respectively, corresponds to 3.85 ± 0.067 mg mg−1 dry tissue and 0.62 ± 0.023 U mg−1 of protein. Determination of oedema and LDH activity are described in Materials and Methods.
) conditions. DEX prophylaxis did not alter the water content but decreased LDH activity in the lung tissue of animals maintained under NN conditions. Increase in water content and LDH activity observed under HH conditions reverted to NN levels by DEX pretreatment. This shows the adaptation of lung to HH stress by DEX pretreatment. Results are expressed in % variation as mean ± SD (n = 6 per group). *P < 0.05 versus NN; #P < 0.05 versus HH. The 100% value in absolute quantity of oedema and LDH activity, respectively, corresponds to 3.85 ± 0.067 mg mg−1 dry tissue and 0.62 ± 0.023 U mg−1 of protein. Determination of oedema and LDH activity are described in Materials and Methods.
LDH activity was significantly reduced (∼60%; P < 0.05) in animals kept under NN conditions, but pretreated with DEX, as compared with untreated animals. On the other hand, animals exposed to HH recorded a significant increase (∼130%; P < 0.05) in their lung LDH activity. Also, DEX pretreatment followed by HH exposure protected the lung as observed by the maintenance of LDH activity. It is almost equal to the animals subjected with DEX pretreatment (Figure 2B).
HH-induced lung injury is due to oxidative stress
ROS and RNS generation in lung tissues were measured as markers of oxidative stress. ROS generation was increased approximately twofold under HH in comparison with NN animals (Figure 3A). An interesting observation is that DEX pretreatment decreased ROS generation significantly (P < 0.05), even in the animals maintained under NN conditions. Also, pretreatment of animals with DEX prevented the effect of HH-induced ROS generation. It almost maintained to the level of NN animals subjected with DEX pretreatment (P < 0.05).
Figure 3.
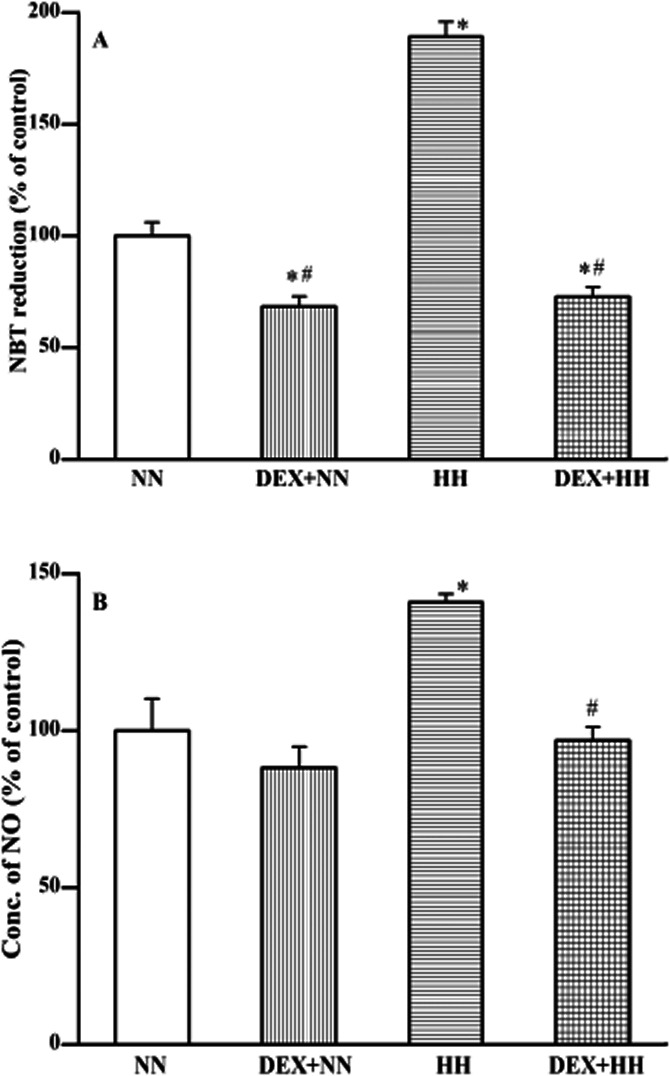
DEX pretreatment (or HH adaptation) protects against HH-induced ROS (A) and RNS (B) production. The ROS generation (i.e. increase in NBT reduction) in lung tissues of animals exposed to NN or HH conditions with or without DEX pretreatment were determined (A). Similarly, RNS generation was assessed as an increase in NO content (B). The generation of ROS/RNS under NN ( ) condition is compared with DEX plus NN (
) condition is compared with DEX plus NN ( ), HH (
), HH ( ) and DEX plus HH (
) and DEX plus HH ( ) conditions. DEX prophylaxis decreased ROS but did not alter the RNS generation in lung tissue of animals maintained under NN condition. However, profound increase in ROS and RNS generation observed under HH condition were reverted to NN levels by DEX pretreatment. Results are expressed in % variation as mean ± SD (n = 6 per group). *P < 0.05 versus NN; #P < 0.05 versus HH. The 100% value in absolute quantity of ROS and NO, respectively, corresponds to 0.355 ± 0.021 OD at 630 nm mg−1 tissues and 799 ± 101.27 nmoles mg−1 of protein. Determination of ROS and RNS generation are described in Materials and Methods.
) conditions. DEX prophylaxis decreased ROS but did not alter the RNS generation in lung tissue of animals maintained under NN condition. However, profound increase in ROS and RNS generation observed under HH condition were reverted to NN levels by DEX pretreatment. Results are expressed in % variation as mean ± SD (n = 6 per group). *P < 0.05 versus NN; #P < 0.05 versus HH. The 100% value in absolute quantity of ROS and NO, respectively, corresponds to 0.355 ± 0.021 OD at 630 nm mg−1 tissues and 799 ± 101.27 nmoles mg−1 of protein. Determination of ROS and RNS generation are described in Materials and Methods.
Similarly, RNS generation was increased (∼140%; P < 0.01) under HH conditions when compared with NN animals (Figure 3B). Pretreatment with DEX significantly inhibited HH-induced RNS generation and maintained the level similar to that of NN animals (P < 0.05). Thus, DEX contributes (i.e. during adaptation) in protecting to the level of ROS and RNS formation under HH conditions.
HH-induced oxidative stress affected mitochondrial complexes
HH-induced pulmonary oxidative stress was monitored further, in terms of lung tissue mtOXPHOS enzyme function (Figure 4). The diverse functional activity unique to each mitochondrial complex in animals exposed to HH recorded a substantial decrease (∼45 to 65%; P < 0.01) in comparison with NN animals. However, in animals pretreated with DEX exhibited a significant increase (∼130 to 150%; P < 0.01) in all respiratory complexes activities. DEX pretreatment followed by HH exposure in animals, registered stabilized functional properties of all mitochondrial complexes at their NN levels. Thus, during adaptation, the contributory effect of DEX in reducing ROS/RNS was the manifestation of stabilized functional properties of all mitochondrial complexes under NN and HH conditions.
Figure 4.

DEX pretreatment (or HH adaptation) protects against HH-induced mitochondrial complexes dysfunction. The integrity of mitochondrial complex I (A), complex III (B), complex IV (C) and complex V (D) of lung tissues of animals exposed to NN or HH conditions with or without DEX pretreatment was determined in terms of their protein specificity (i.e. functional activity). The respective functional activity under NN ( ) condition is compared with DEX plus NN (
) condition is compared with DEX plus NN ( ), HH (
), HH ( ) and DEX plus HH (
) and DEX plus HH ( ) conditions. A profound increase in all complex activities was observed in DEX-treated animals maintained under NN conditions. The decreased functional activities observed under HH conditions were restored to their NN levels in the presence of DEX. Results are expressed in % variation as mean ± SD (n = 6 per group). *P < 0.05 versus NN; #P < 0.05 versus HH. The 100% value in absolute quantity of complex I, III, IV and V, respectively, corresponds to 189 ± 16.54, 886 ± 85.27, 1493 ± 230.96 and 183.6 ± 10.41 nmoles min−1 mg−1 of protein. Determination of mtOXPHOS activities is described in Materials and Methods.
) conditions. A profound increase in all complex activities was observed in DEX-treated animals maintained under NN conditions. The decreased functional activities observed under HH conditions were restored to their NN levels in the presence of DEX. Results are expressed in % variation as mean ± SD (n = 6 per group). *P < 0.05 versus NN; #P < 0.05 versus HH. The 100% value in absolute quantity of complex I, III, IV and V, respectively, corresponds to 189 ± 16.54, 886 ± 85.27, 1493 ± 230.96 and 183.6 ± 10.41 nmoles min−1 mg−1 of protein. Determination of mtOXPHOS activities is described in Materials and Methods.
HH-induced oxidative stress also affected mitochondrial content and mtDNA
The CS activity (Figure 5A) to determine number of mitochondria and the mtDNA content, in terms of the ratio of mtDNA (D-loop region) to nDNA (GAPDH gene) (Figure 5B) were monitored. The CS activity and mtDNA/nDNA ratio of lung tissues were not altered significantly in animals maintained under NN conditions either with or without DEX pretreatment. However, in animals exposed to HH, a drastic fall in CS activity was observed concomitant with the decreased content (∼50 to 60%; P < 0.01) of mtDNA relative to nDNA. DEX pretreatment, however, restored CS activity and mtDNA content to the NN level (P < 0.01) even under HH conditions. Altogether, these results demonstrate the plausible role of DEX in counteracting the HH stress, involving the stabilization of mitochondrial content and its DNA integrity under HH.
Figure 5.
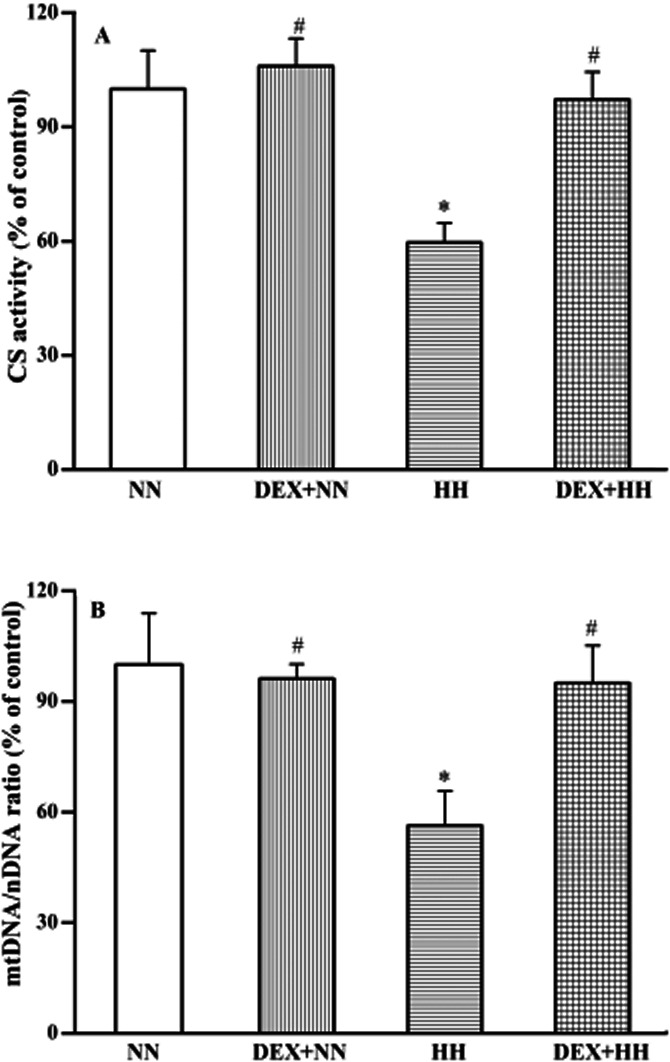
DEX pretreatment (or HH adaptation) protects against HH-induced mitochondrial disintegrity. The mitochondrial number (i.e. CS activity) of lung tissues of animals exposed to NN or HH conditions with or without DEX pretreatment were determined (A). Similarly, mtDNA content was calculated as the mtDNA/nDNA ratio (B). The mitochondrial and mtDNA content under NN ( ) condition is compared with DEX plus NN (
) condition is compared with DEX plus NN ( ), HH (
), HH ( ) and DEX plus HH (
) and DEX plus HH ( ) conditions. DEX prophylaxis did not alter either the CS or mtDNA content of animals maintained under NN conditions. A profound decrease in both the CS activity and mtDNA content was observed with HH treatment. However, the presence of DEX in HH restores both the CS as well as mtDNA content significantly to NN levels. Results are expressed in % variation as mean ± SD (n = 6 per group). *P < 0.05 versus NN; #P < 0.05 versus HH. The 100% value in absolute quantity of CS activity and mtDNA/nDNA ratio (2–ΔCp), respectively, corresponds to 1.41 ± 0.14 nmoles min−1 mg−1 of protein and 300.56 ± 41.78. Determination of CS activity and mtDNA/nDNA ratio is described in Materials and Methods.
) conditions. DEX prophylaxis did not alter either the CS or mtDNA content of animals maintained under NN conditions. A profound decrease in both the CS activity and mtDNA content was observed with HH treatment. However, the presence of DEX in HH restores both the CS as well as mtDNA content significantly to NN levels. Results are expressed in % variation as mean ± SD (n = 6 per group). *P < 0.05 versus NN; #P < 0.05 versus HH. The 100% value in absolute quantity of CS activity and mtDNA/nDNA ratio (2–ΔCp), respectively, corresponds to 1.41 ± 0.14 nmoles min−1 mg−1 of protein and 300.56 ± 41.78. Determination of CS activity and mtDNA/nDNA ratio is described in Materials and Methods.
Effect of HH on mitochondrial functional gene expression
In order to elucidate the involvement of nDNA in the regulation of mtDNA/mitochondrial content under HH, the levels of nDNA-encoded gene regulators involved in mitochondrial biogenesis and dynamics (Figure 6A) were measured using QRT-PCR. Under HH, PGC-1α mRNA was increased [∼2-fold; P(H1) < 0.05]; however, selective and profound repression of ERRα was observed. Despite the abundance of PGC-1α mRNA, the expression of downstream regulatory genes involved in mitochondrial biogenesis such as NRF1, NRF2 and TFAM remained unchanged compared with NN animals. In contrast, the expression of genes involved in mitochondrial dynamics was significantly altered. Out of the two genes involved in mitofusion, only Mfn2 expression was significantly decreased whereas, Mfn1 was not altered under HH conditions. Conversely, expression of mitochondrial fission genes: Fis1 and Drp1 were significantly increased [∼2.5- to 4-fold; P(H1) < 0.05] in HH-exposed animals. This modulation in levels of nDNA-encoded regulators especially the ERRα was manifested into a significant decrease [P(H1) < 0.05; Figure 6B] of all mtDNA-encoded genes involved in OXPHOS. The relative gene expression profile obtained for different genes using ‘REST-2009’ software enumerates the statistical significance of each reaction (Supporting Information Table S2).
Figure 6.
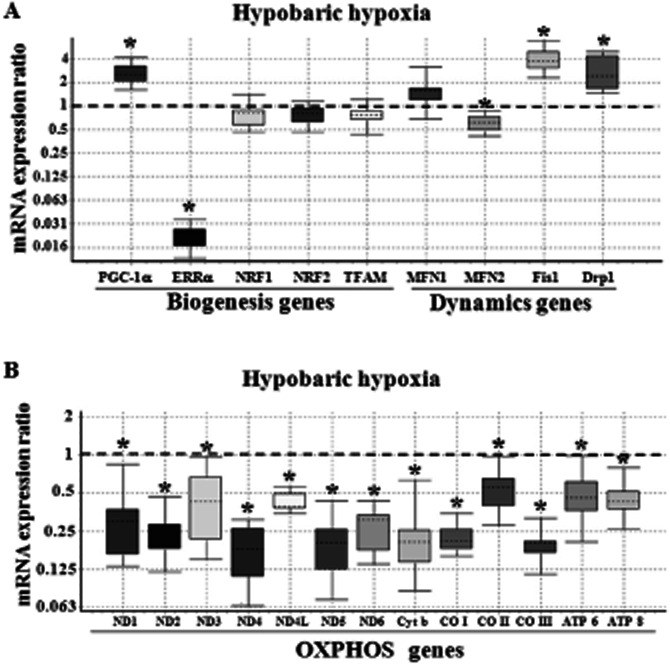
Effect of HH on the relative expression of mitochondrial biogenesis/dynamics genes (A) and mtDNA-encoded OXPHOS genes (B). The relative expression of genes in lung tissue of rat exposed to HH condition was measured using QRT-PCR as detailed under Materials and Methods. The Box and whisker plot reports the mRNA expression ratio on the y-axis of different genes denoted in the x-axis. The gene expression ratio of rat lung tissue exposed to HH was calculated relative to the control group (i.e. without DEX treatment) maintained in NN conditions (fixed at 1), using β-actin gene expression for normalization. Note that although PGC-1α mRNA transcript level was increased (∼2.5-fold), a selective repression of ERRα (∼0.022) was observed. Also, expression of fusion gene Mfn2 was decreased along with increased fission genes (Fis1 and Drp1) transcript level. The decrease in expression of all 13 mtOXPHOS genes under HH conditions was highly significant (*P(H1) < 0.05 in comparison to control group; n = 3 per group). The name of the genes as abbreviated in x-axis is given in expansion under abbreviations section.
Adaptation to HH condition is mediated through mitochondrial functional genes
Prophylactic treatment of animals with DEX under NN conditions (i.e. adaptation to HH) increased the expression of PGC-1α mRNA, but other genes involved in mitochondrial biogenesis and dynamics were not altered (Figure 7A). Contrary to this effect, treatment of animals with DEX facilitated a 2- to 12-fold [P(H1) < 0.05; Figure 7B] increase in all 13 genes of mitochondrial origin in comparison with NN animals (Supporting Information Table S3). The gene expression profiles of DEX pretreated animals subjected to HH conditions are depicted in Figure 8. PGC-1α mRNA increased by DEX pretreatment was upheld even in HH-exposed animals. Also, ERRα transcripts were maintained at the NN level by DEX pretreatment (Figure 8A). The other downstream regulators of nDNA origin involved in mitochondrial biogenesis (NRF1, NRF2, TFAM) were maintained at the NN level. A close look at Figures 6A and 8A shows that under HH conditions, decreased Mfn2 and increased mitochondrial fission genes (Fis1 and Drp1) are restored to the NN level by DEX pretreatment. Similarly, genes involved in mtOXPHOS are increased in DEX pretreated animals, even under HH conditions (Figure 8B, Supporting Information Table S4). Thus, adaptability to HH stress (i.e. DEX pretreatment) is mainly favoured by the increased level of mtOXPHOS gene product.
Figure 7.
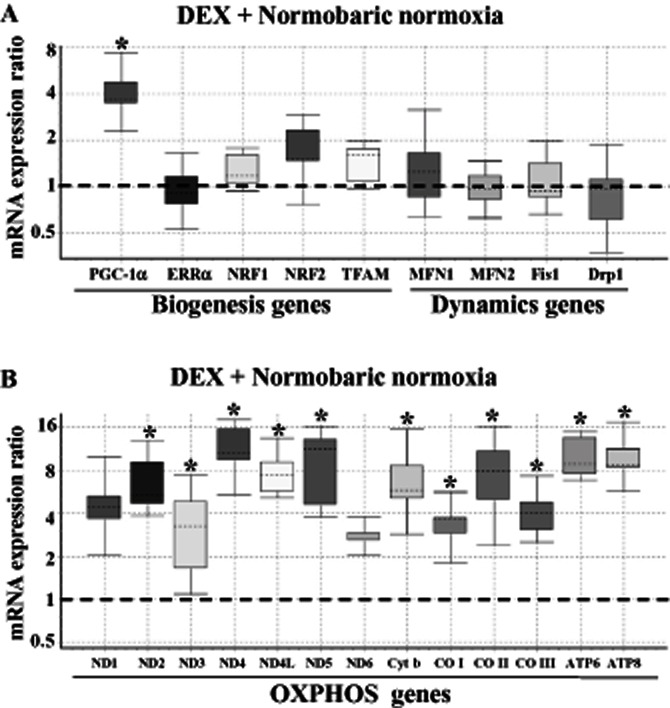
Effect of DEX (or HH adaptation) on the relative expression of mitochondrial biogenesis/dynamics genes (A) and mtDNA-encoded OXPHOS genes (B). The relative expression of genes in the lung tissue derived from DEX pretreated rats under NN conditions was measured using QRT-PCR as detailed under Materials and Methods. The Box and whisker plot reports the mRNA expression ratio on the y-axis of different genes denoted on the x-axis. The expression ratio of DEX pretreated rat lung was calculated relative to the control group (i.e. without DEX treatment) maintained in NN conditions (fixed at 1) using β-actin gene expression for normalization. Note that DEX selectively increased (∼3.5-fold) the PGC-1α mRNA transcript. However, other genes involved in mitochondrial biogenesis/dynamics were maintained at NN levels only. Also, DEX treatment augmented (∼2- to 12-fold) the expression of mtDNA-encoded OXPHOS genes, which is highly significant (*P(H1) < 0.05 in comparison with the control group; n = 3 per group). The name of the genes as abbreviated in x-axis is given in expansion under abbreviations section.
Figure 8.
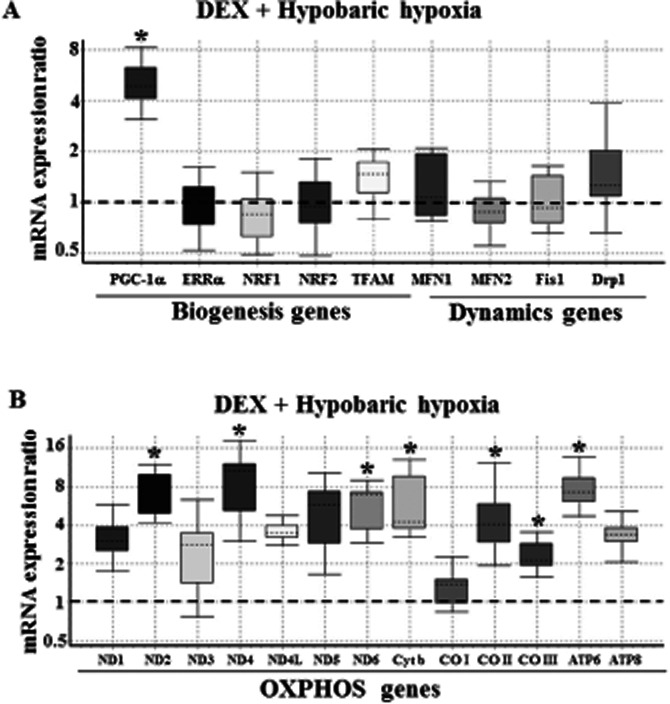
Protective effect of DEX pretreatment (or HH adaptation) against HH on the expression of mitochondrial biogenesis/dynamics genes (A) and mtDNA-encoded OXPHOS genes (B). The relative expression of genes in lung tissue derived from DEX pretreated rat but subjected to HH condition was measured using QRT-PCR as detailed under Materials and Methods. The Box and whisker plot reports the mRNA expression ratio on y-axis of different genes denoted in x-axis. The expression ratio of DEX pretreated rat lung subjected to HH were calculated relative to the control group maintained in NN conditions (fixed at 1) using β-actin gene expression for normalization. Note that DEX pretreatment has selectively increased (5-fold) the PGC-1α mRNA transcript. Also, other genes involved in mitochondrial biogenesis/dynamics are normalized to NN levels. Further, DEX pretreatment upheld the increased expression of some mtDNA-encoded OXPHOS genes by 2- to 8-fold even under HH condition, which are highly significant (*P(H1) < 0.05 in comparison with the control group; n = 3 per group). The name of the genes as abbreviated in x-axis is given in expansion under abbreviations section.
Discussion
The pulmonary oedema (HAPE) experienced under high-altitude hypoxic (i.e. HH) stress conditions is very similar in rats and humans (Dempsey and Forster, 1982; Boyaci et al., 2006). In the present rat model study, an attempt has been made in identifying the dysregulation of mitochondrial functional genomics of lung as an early signalling event in response to acute but severe HH stress. Further, DEX, the prophylactic corticosteroid drug widely used for high-altitude acclimatization, is employed in the present study as positive control to see its effect on the molecular event of mitochondria in counteracting the HH stress and restoring the normalcy (i.e. HAPE adaptation). Secondly, DEX pre-administered to HAPE-susceptible human individuals were protected from HAPE during HH stress conditions and, hence, the effect of DEX in NN conditions in all likelihood would mimic adaptive mechanisms essential to get rid off HAPE under HH stress.
Adaptation to HH stress is mediated through mitochondrial functions
Exposure of rats to ‘in vitro-stimulus’ of 9142 m altitude at 28°C for 6 h resulted in pulmonary oedema. Concomitantly, an increase in LDH activity was also observed, which might have triggered the cytotoxic effects observed at this simulated high altitude (Shukla et al., 2011). However, pretreatment with DEX significantly reduced the LDH activity and, consequently, a reduction in oedema (Figure 2) was also observed. Further, an increase in ROS and RNS generation (Figure 3) was observed in rats under stimulated HH stress as an index of oxidative stress. However, DEX pretreatment substantially decreased not only the excessive ROS generations under HH, but also decreased the ROS formation under NN (Figure 3). DEX, the widely acclaimed regulator of ROS homeostasis is reported to be involved in promoting ROS scavenging systems at the transcriptional level (Jose et al., 1997). Thus, the HAPE experienced by the rats due to ‘in vitro-stimulus’ is possibly mediated through oxidative stress. Such oxidative stress has caused mitochondrial dysfunction due to decreased activities of mtOXPHOS complexes (Figure 4). Further, DEX was shown to regulate all mtOXPHOS complexes activities by enhancing its functions not only at NN, but also under HH condition (Figure 4). Thus, during HAPE acclimatization (i.e. DEX pretreatment) maintenance of mitochondrial complex functions to keep the ROS/RNS formation at bay could be the molecular event, which must happen in the lung. It may be also inferred that mitochondria are the immediate targets in oxidative stress, especially under severe HH conditions. Additional evidence like decreased mitochondrial number (i.e. CS activity) and mtDNA (i.e. mtDNA/nDNA ratio) observed during stimulated HH stress (Figure 5) support our hypothesis of maintaining the mitochondrial functions to NN condition is the prerequisite for HAPE acclimatization. These observations give an insight into coordinated regulatory alteration of mitochondrial-related genes under HH stress. Also, while bioenergetics homeostasis in mitochondria is achieved through post-translation modification of several metabolic enzymes, long-term adaptation is achieved by the regulation of mitochondrial metabolic genes at the transcriptional level through co-regulators and transcription factors (Feige and Auwerx, 2007; Charest-Marcotte et al., 2010).
Deregulation of nDNA-encoded mitochondrial regulators impairs mitochondrial function under HH stress
The nDNA-encoded mitochondrial regulators, PGC-1α and ERRα complementarily brings about mitochondrial biogenesis and dynamics functions. However, HH caused a selective repression of ERRα while the level of PGC-1α was increased. Surprisingly, the other further downstream nDNA regulators like NRF1, NRF2 and TFAM involved in mitochondrial biogenesis were not altered (Figure 6A). Thus, under HH stress, at a restricted level of ERRα, one cannot expect its interaction with PGC-1α towards forming an active complex ‘ERRα–PGC-1α’ (as shown in Figure 1) and, hence, decreased the expression of mtDNA-encoded OXPHOS genes (Figure 6B). Also, the role of PGC-1α (through induction of NRFs and interaction with NRF1) in influencing mtOXPHOS genes proposed (Wu et al., 1999) are not operating in lung tissue under stimulated HH condition. In other words, although a phenomenal increase in the levels of PGC-1α was observed, in the absence of ERRα (as observed in the present study), the mitochondrial biogenesis could not be effected under HH conditions, which is in perfect resonance with the earlier reports (Mootha et al., 2004; Schreiber et al., 2004). Interestingly, in parallel to this observation, in ERRα null rats and human heart failure cases, a decreased ERRα gene expression along with increased levels of PGC-1α and NRF2 were reported. Such gene expression was then characterized as a compensatory mechanism (Sihag et al., 2009; Karamanlidis et al., 2010). Taking these earlier inputs together with the results in Figures 5 and 6, we pinpoint that mitochondrial biogenesis and mtOXPHOS are impaired due to decreased ERRα along with depleted mtDNA (Sihag et al., 2009; Karamanlidis et al., 2010) under HH condition.
Another significant finding is that stimulated HH conditions induced mitochondrial fission (Fis1 and Drp1) genes along with decreased Mfn2 expression (Figure 6A). This would result in an imbalance of mitochondrial dynamics (i.e. fusion and fission) that will eventually lead to mitochondrial fragmentation. Also, reduced ATP synthesis along with elevated oxidative stress under HH conditions would create a favourable condition for initiation of mitochondrial fragmentation (Benard et al., 2007; Chen et al., 2010; Sauvanet et al., 2010). Similar to HH exposure, various other stresses that involves elevated hydrostatic pressure like external elevated pressure, ischaemia/reperfusion and shear stress has also resulted in significant mitochondrial fragmentation (Ju et al., 2007; Giedt et al., 2012). Apart from such induced disturbances on mitochondrial dynamics, down-regulated Mfn2 (Figure 6A) could also trigger mitochondrial de-energization, in part, by repressing nDNA-encoded OXPHOS complex genes expression, which is again PGC–1α-independent (Pich et al., 2005). In fact, decreased ERRα (Figure 6A), which controls the expression of Mfn2 (Cartoni et al., 2005), independent of the increased PGC-1α observed in the present study, is singularly responsible for mtOXPHOS dysregulation. The decreased expression of ERRα under HH stress (i.e. HAPE) is the forerunner of triggering a cascade of events interlinking transcriptional dysfunction in mitochondria as it is the ‘bioenergetics regulon’ that concurrently regulates the genes involved in mitochondrial biogenesis, dynamics and mtOXPHOS (energy production; Charest-Marcotte et al., 2010). In this context, it is worth mentioning several reports (Huss et al., 2007; Karamanlidis et al., 2010; Hu et al., 2011) in which the role of ERRα in bioenergetics homeostasis and mitochondrial functional adaptation has been emphasized.
mtOXPHOS genes as molecular markers in HH adaptation
DEX has been administered before venturing into high altitude for better adaptability or to prevent the onset of HAPE. In order to understand the role of DEX on nDNA and mtDNA in bringing about acclimatization, its effect under NN conditions were studied. DEX-treated animals kept under NN conditions favoured an increase of the PGC-1α gene only among all the mitochondrial biogenesis/dynamics genes (Figure 7A). Contrary to this, all the mtOXPHOS complex genes were increased (Figure 7B) by DEX under NN conditions. This is attributed to direct effects of DEX on mtDNA-encoded genes, possibly through the glucocorticoid receptor localized in mitochondria (Psarra and Sekeris, 2011). This effect need not be through PGC-1α induction of NRFs and its interaction with NRF1 as reported (Wu et al., 1999) because there is no augmentation of these genes proportional to the increased expression of mtOXPHOS genes (Figure 7B). Secondly, DEX under NN conditions did not influence the expression of TFAM gene (Figure 7A) individually; this again explains the reasons for not having any variation either in mtDNA content or mitochondrial density as observed in Figure 5. Thus, we lay emphasis on the plausible direct action of DEX on mitochondrial transcription (Scheller and Sekeris, 2003). To put it differently, molecular adaptation to HH stress (i.e. DEX treatment of animals, but maintained under NN conditions) invariably facilitated increased mtOXPHOS genes expression and, hence, increased mtOXPHOS complex functions. Thus, one can comfortably conclude that under NN condition, the molecular event triggered for adaptation to HAPE is the augmentation of mtOXPHOS genes.
HH adaptability is regulated by ERRα
Prophylactic treatment of DEX normalized the levels of ERRα together with other biogenesis genes besides elevating PGC-1α level under HH conditions (Figure 8A). Also, the increased transcripts level of mtOXPHOS genes by DEX pretreatment was maintained even under HH condition (Figure 8B). Elevated level of PGC-1α in DEX-treated animals, nevertheless, could not have independent influence on mitochondrial function per se as corroborated by the results from Figures 5, 6 and 8. However, ERRα is significantly decreased under HH stress conditions, but is restored to the NN level in the DEX pretreated animal. This effect correlates well with decreased mitochondrial biogenesis (i.e. mitochondrial number and mtDNA content) and expression of mtOXPHOS genes under HH, which is either normalized or augmented by DEX pretreatment under HH conditions. Yet, another significant interpretation is that only the normoxic level of ERRα is quite sufficient (irrespective of increased PGC-1α observed) in eliciting mitochondrial function because HH stress down-regulated the ERRα function. In addition, DEX pretreatment under HH conditions resulted in the maintenance of the mitochondrial fusion/fission gene expression to NN levels (Figure 8A). Along with normalized expression of mitochondrial dynamics genes, the effect of DEX treatment (i.e. adaptation) on metabolic augmentation to preserve ATP synthesis and reduce oxidative stress would avert mitochondrial fragmentation under HH conditions. This is possible as the relationship between mitochondrial bioenergetics and mitochondrial network organization is bidirectional (Benard et al., 2007).
Conclusion
The scheme showing the HH exposure-induced mitochondrial dysfunction in rat lung and mechanism of HH adaptation rendered by DEX pre-administration is depicted in Figure 9. In conclusion, the present study demonstrates that the underlying molecular mechanism involved in lung mitochondrial dysfunction under high-altitude (HH) stress are well-correlated with the decreased expression of ERRα. This has resulted in decreased mitochondrial biogenesis and expression of mtOXPHOS genes. In addition, HH has resulted in the aberration of mitochondrial dynamics as is evidenced by increased fission (Fis1 and Drp1)- and decreased fusion (Mfn2)-related gene expression. The prophylactic administration of DEX (i.e. HH adaptation) overcomes this dysfunction through the preservation of mitochondrial bioenergetics possibly by normalizing ERRα and elevating mtOXPHOS genes expression. These observations made in the present study indicate that (i) HAPE susceptibility under HH stress is due to mitochondrial dysfunction; and (ii) HAPE adaptation might be due to protective effects on mitochondrial function through mtOXPHOS genes and its major nDNA regulators like ERRα.
Figure 9.
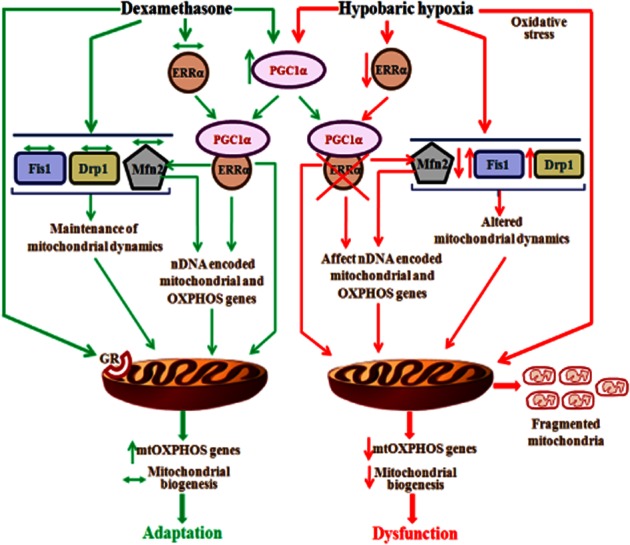
Scheme showing molecular mechanism of HH exposure-induced mitochondrial dysfunction and DEX pre-administration (or HH adaptation)-mediated protection in rat lung. Under HH condition, decreased ERRα-mediated repression of ‘ERRα-PGC-1α’ active form has eventually resulted in loss of mitochondrial biogenesis-mediated adaptation and decreased Mfn2 transcripts leading to mitochondrial de-energization. Also, decreased ERRα along with oxidative stress-mediated mtDNA damage has resulted in decreased expression of genes involved in mtOXPHOS complex to further affect its function. In addition, increased expression of genes involved in fission (Fis1 and Drp1) along with decreased Mfn2 will result in mitochondrial fragmentation under HH condition. On the other hand, prophylactic treatment of DEX (i.e. adaptation to HH) has normalized the expression of ERRα and increased the PGC-1α transcripts under HH conditions. Hence, the formation of ‘ERRα-PGC-1α’ active form would maintain the Mfn2 transcripts and mitochondrial biogenesis. Also, DEX increased the transcripts of mtOXPHOS genes through the interaction with mitochondrial localized glucocorticoid receptor (GR) to augment the mtOXPHOS functions. In addition, mitochondrial dynamics genes are restored to NN level. Thus, the protection of ERRα along with elevated expression of mtOXPHOS genes are the likely candidates prerequisite for lung adaptation to HH stress.
Acknowledgments
This work was supported by grants from DRDO-BU Center for Life Sciences, Bharathiar University, Coimbatore, Tamil Nadu, India. One of the authors, Ms. C. Loganathan, acknowledges DRDO-BU Center for Life Sciences for the award of Senior Research Fellowship.
Glossary
- CO I/II/III
cytochrome c oxidase I/II/III
- CS
citrate synthase
- Cyt b
cytochrome b
- DEX
dexamethasone
- Drp1
dynamin-related protein 1
- ERRα
oestrogen-related receptor-α
- Fis1
fission 1
- HAPE
high-altitude pulmonary oedema
- Mfn1/2
mitofusin1/2
- mtDNA
mitochondrial DNA
- ND1 to 6, 4L
NADH-dehydrogenase-ubiquinone reductase1 to 6, 4L
- nDNA
nuclear DNA
- NRF1/2
nuclear respiratory factor 1/2
- OXPHOS
oxidative phosphorylation
- PGC-1α
PPAR-γ coactivator-1α
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- TFAM
mitochondrial transcription factor A
Conflicts of interest
All the authors declare no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Table S1 Details of primer sequences.
Table S2 Effect of hypobaric hypoxia on relative expression of genes involved in mitochondrial functions of lung tissue.
Table S3 Effect of DEX treatment on relative expression of genes involved in mitochondrial functions of lung tissue.
Table S4 Effect of dexamethasone pretreatment followed by hypobaric hypoxia on relative expression of genes involved in mitochondrial functions of lung tissue.
References
- Abe K, Matsuki N. Measurement of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction activity and lactate dehydrogenase release using MTT. Neurosci Res. 2000;38:325–329. doi: 10.1016/s0168-0102(00)00188-7. [DOI] [PubMed] [Google Scholar]
- Bartsch P, Vock P, Franciolli M. High altitude pulmonary edema after successful treatment of acute mountain sickness with dexamethasone. Wilder Med. 1990;1:162–164. [Google Scholar]
- Benard G, Bellance N, James D, Parrone P, Fernandez H, Letellier T, et al. Mitochondrial bioenergetics and structural network organization. J Cell Sci. 2007;120:838–848. doi: 10.1242/jcs.03381. [DOI] [PubMed] [Google Scholar]
- Boyaci H, Maral H, Turan G, Basyigit I, Dillioglugil MO, Yildiz F, et al. Effects of erdosteine on bleomycin-induced lung fibrosis in rats. Mol Cell Biochem. 2006;281:129–137. doi: 10.1007/s11010-006-0640-3. [DOI] [PubMed] [Google Scholar]
- Capaldi RA, Marusich MF, Taanman JW. Mammalian cytochrome-c oxidase: characterization of enzyme and immunological detection of subunits in tissue extracts and whole cells. Methods Enzymol. 1995;260:117–132. doi: 10.1016/0076-6879(95)60134-1. [DOI] [PubMed] [Google Scholar]
- Cartoni R, Leger B, Hock MB, Praz M, Crettenand A, Pich S, et al. Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise. J Physiol. 2005;567:349–358. doi: 10.1113/jphysiol.2005.092031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Cavanagh EM, Flores I, Ferder M, Inserra F, Ferder L. Renin-angiotensin system inhibitors protect against age-related changes in rat liver mitochondrial DNA content and gene expression. Exp Gerontol. 2008;43:919–928. doi: 10.1016/j.exger.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charest-Marcotte A, Dufour CR, Wilson BJ, Tremblay AM, Eichner LJ, Arlow DH, et al. The homeobox protein Prox1 is a negative modulator of ERR{alpha}/PGC-1{alpha} bioenergetic functions. Genes Dev. 2010;24:537–542. doi: 10.1101/gad.1871610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig I. A procedure for the analysis of citrate synthase (E.C. 4.1.3.7) in somatic cell hybrids. Biochem Genet. 1973;9:351–358. doi: 10.1007/BF00486070. [DOI] [PubMed] [Google Scholar]
- Dempsey JA, Forster HV. Mediation of ventilatory alterations. Physiol Rev. 1982;62:262–346. doi: 10.1152/physrev.1982.62.1.262. [DOI] [PubMed] [Google Scholar]
- Ding H, Jiang N, Liu H, Liu X, Liu D, Zhao F, et al. Response of mitochondrial fusion and fission protein gene expression to exercise in rat skeletal muscle. Biochim Biophys Acta. 2010;1800:250–256. doi: 10.1016/j.bbagen.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, et al. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet. 2004;13:935–944. doi: 10.1093/hmg/ddh109. [DOI] [PubMed] [Google Scholar]
- Feige JN, Auwerx J. Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol. 2007;17:292–301. doi: 10.1016/j.tcb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Fernandez-Silva P, Enriquez JA, Montoya J. Replication and transcription of mammalian mitochondrial DNA. Exp Physiol. 2003;88:41–56. doi: 10.1113/eph8802514. [DOI] [PubMed] [Google Scholar]
- Giedt RJ, Yang C, Zweier JL, Matzavinos A, Alevriadou BR. Mitochondrial fission in endothelial cells after simulated ischemia/reperfusion: role of nitric oxide and reactive oxygen species. Free Radic Biol Med. 2012;52:348–356. doi: 10.1016/j.freeradbiomed.2011.10.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guney S, Schuler A, Ott A, Hoschele S, Zugel S, Baloglu E, et al. Dexamethasone prevents transport inhibition by hypoxia in rat lung and alveolar epithelial cells by stimulating activity and expression of Na+-K+-ATPase and epithelial Na+ channels. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1332–L1338. doi: 10.1152/ajplung.00338.2006. [DOI] [PubMed] [Google Scholar]
- Gutsaeva DR, Carraway MS, Suliman HB, Demchenko IT, Shitara H, et al. Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J Neurosci. 2008;28:2015–2024. doi: 10.1523/JNEUROSCI.5654-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales KG, Fuller MT. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell. 1997;90:121–129. doi: 10.1016/s0092-8674(00)80319-0. [DOI] [PubMed] [Google Scholar]
- Hopkins SR, Garg J, Bolar DS, Balouch J, Levin DL. Pulmonary blood flow heterogeneity during hypoxia and high-altitude pulmonary edema. Am J Respir Crit Care Med. 2005;171:83–87. doi: 10.1164/rccm.200406-707OC. [DOI] [PubMed] [Google Scholar]
- Hu X, Xu X, Lu Z, Zhang P, Fassett J, Zhang Y, et al. AMP activated protein kinase-alpha2 regulates expression of estrogen-related receptor-alpha, a metabolic transcription factor related to heart failure development. Hypertension. 2011;58:696–703. doi: 10.1161/HYPERTENSIONAHA.111.174128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultgren HN. High-altitude pulmonary edema: current concepts. Ann Rev Med. 1996;47:267–284. doi: 10.1146/annurev.med.47.1.267. [DOI] [PubMed] [Google Scholar]
- Huss JM, Imahashi K, Dufour CR, Weinheimer CJ, Courtois M, Kovacs A, et al. The nuclear receptor ERRalpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Met. 2007;6:25–37. doi: 10.1016/j.cmet.2007.06.005. [DOI] [PubMed] [Google Scholar]
- James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003;278:36373–36379. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- Janssen AJ, Trijbels FJ, Sengers RC, Smeitink JA, van den Heuvel LP, Wintjes LT. Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin Chem. 2007;53:729–734. doi: 10.1373/clinchem.2006.078873. [DOI] [PubMed] [Google Scholar]
- Jose HJ, Berenice SG, Cecilia VR. Induction of antioxidant enzymes by dexamethasone in the adult rat lung. Life Sci. 1997;60:2059–2067. doi: 10.1016/s0024-3205(97)00193-8. [DOI] [PubMed] [Google Scholar]
- Ju WK, Liu Q, Kim KY, Crowston JG, Lindsey JD, Agarwal N. Elevated hydrostatic pressure triggers mitochondrial fission and decreases cellular ATP in differentiated RGC-5 cells. Invest Ophthal Vis Sci. 2007;48:2145–2151. doi: 10.1167/iovs.06-0573. [DOI] [PubMed] [Google Scholar]
- Karamanlidis G, Nascimben L, Couper GS, Shekar PS, del Monte F, Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res. 2010;106:1541–1548. doi: 10.1161/CIRCRESAHA.109.212753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahenbuhl S, Talos C, Wiesmann U, Hoppel CL. Development and evaluation of a spectrophotometric assay for complex III in isolated mitochondria, tissues and fibroblasts from rats and humans. Clin Chim Acta. 1994;230:177–187. doi: 10.1016/0009-8981(94)90270-4. [DOI] [PubMed] [Google Scholar]
- Lin HJ, Wang CT, Niu KC, Gao C, Li Z, Lin MT, et al. Hypobaric hypoxia preconditioning attenuates acute lung injury during high-altitude exposure in rats via up-regulating heat-shock protein 70. Clin Sci (Lond) 2011;121:223–231. doi: 10.1042/CS20100596. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhaes J, Ascensao A, Soares JM, Ferreira R, Neuparth MJ, Marques F. Acute and severe hypobaric hypoxia increases oxidative stress and impairs mitochondrial function in mouse skeletal muscle. J Appl Physiol. 2005;99:1247–1253. doi: 10.1152/japplphysiol.01324.2004. [DOI] [PubMed] [Google Scholar]
- Maggiorini M, Melot C, Pierre S, Pfeiffer F, Greve I, Sartori C, et al. High-altitude pulmonary edema is initially caused by an increase in capillary pressure. Circulation. 2001;103:2078–2083. doi: 10.1161/01.cir.103.16.2078. [DOI] [PubMed] [Google Scholar]
- Maggiorini M, Brunner-La Rocca HP, Peth S, Fischler M, Bohm T, Bernheim A, et al. Both tadalafil and dexamethasone may reduce the incidence of high-altitude pulmonary edema: a randomized trial. Ann Intern Med. 2006;145:497–506. doi: 10.7326/0003-4819-145-7-200610030-00007. [DOI] [PubMed] [Google Scholar]
- Mikula M, Dzwonek A, Hennig EE, Ostrowski J. Increased mitochondrial gene expression during L6 cell myogenesis is accelerated by insulin. Int J Biochem Cell Biol. 2005;37:1815–1828. doi: 10.1016/j.biocel.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Mookerjee A, Basu JM, Majumde S, Chatterjee S, Panda GS, Dutta P, et al. A novel copper complex induces ROS generation in doxorubicin resistant Ehrlich ascitis carcinoma cells and increases activity of antioxidant enzymes in vital organs in vivo. BMC Cancer. 2006;6:267–271. doi: 10.1186/1471-2407-6-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Handschin C, Arlow D, Xie X, St Pierre J, Sihag S, et al. Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci U S A. 2004;101:6570–6575. doi: 10.1073/pnas.0401401101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradies G, Petrosillo G, Pistolese M, Ruggiero FM. Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene. 2002;286:135–141. doi: 10.1016/s0378-1119(01)00814-9. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. 2009. Rest 2009 Software User Guide. Qiagen, Hilden, Germany. Available at: http://b2b.qiagen.com/products/rest2009software.aspx (accessed 14/12/2009)
- Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, et al. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet. 2005;14:1405–1415. doi: 10.1093/hmg/ddi149. [DOI] [PubMed] [Google Scholar]
- Psarra AM, Sekeris CE. Glucocorticoids induce mitochondrial gene transcription in HepG2 cells: role of the mitochondrial glucocorticoid receptor. Biochim Biophys Acta. 2011;1813:1814–1821. doi: 10.1016/j.bbamcr.2011.05.014. [DOI] [PubMed] [Google Scholar]
- Sauvanet C, Duvezin-Caubet S, di Rago JP, Rojo M. Energetic requirements and bioenergetic modulation of mitochondrial morphology and dynamics. Semin Cell Dev Biol. 2010;21:558–565. doi: 10.1016/j.semcdb.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Scheller K, Sekeris CE. The effects of steroid hormones on the transcription of genes encoding enzymes of oxidative phosphorylation. Exp Physiol. 2003;88:129–140. doi: 10.1113/eph8802507. [DOI] [PubMed] [Google Scholar]
- Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc Natl Acad Sci U S A. 2004;101:6472–6477. doi: 10.1073/pnas.0308686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla D, Saxena S, Purushothaman J, Shrivastava K, Singh M, Shukla S. Hypoxic preconditioning with cobalt ameliorates hypobaric hypoxia induced pulmonary edema in rat. Eur J Pharmacol. 2011;656:101–109. doi: 10.1016/j.ejphar.2011.01.038. [DOI] [PubMed] [Google Scholar]
- Sihag S, Cresci S, Li AY, Sucharov CC, Lehman JJ. PGC-1alpha and ERRalpha target gene downregulation is a signature of the failing human heart. J Mol Cell Cardiol. 2009;46:201–212. doi: 10.1016/j.yjmcc.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Shurland DL, Ryazantsev SN, van der Bliek AM. A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol. 1998;143:351–358. doi: 10.1083/jcb.143.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzner TJ, O'Brien RF, Sato K, Weil JV. Hypoxia-induced increases in pulmonary transvascular protein escape in rats. Modulation by glucocorticoids. J Clin Invest. 1988;82:1840–1847. doi: 10.1172/JCI113800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JB, Tsukimoto K, Mathieu-Costello O, Prediletto R. Stress failure in pulmonary capillaries. J Appl Physiol. 1991;70:1731–1742. doi: 10.1152/jappl.1991.70.4.1731. [DOI] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Young SL, Silbajoris R. Dexamethasone increases adult rat lung surfactant lipids. J Appl Physiol. 1986;60:1665–1672. doi: 10.1152/jappl.1986.60.5.1665. [DOI] [PubMed] [Google Scholar]
- Zheng J, Ramirez VD. Inhibition of mitochondrial proton F0F1-ATPase/ATP synthase by polyphenolic phytochemicals. Br J Pharmacol. 2000;130:1115–1123. doi: 10.1038/sj.bjp.0703397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zungu M, Alcolea MP, Garcia-Palmer FJ, Young ME, Essop MF. Genomic modulation of mitochondrial respiratory genes in the hypertrophied heart reflects adaptive changes in mitochondrial and contractile function. Am J Physiol Heart Circ Physiol. 2007;293:H2819–H2825. doi: 10.1152/ajpheart.00806.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.