

Abstract
Background and Purpose
Inhibition of apoptosis may attenuate the irreversible injury associated with reperfusion. In the current study, we focused on the cytoprotective effects and the underlying mechanism of sodium tanshinone IIA silate (STS) against damage induced by oxygen-glucose deprivation/recovery (OGD/R). in H9c2 cardiomyocytes and the underlying mechanisms.
Experimental Approach
We used a model of cardiac ischaemia/reperfusion, OGD/R in H9c2 cardiomyocytes, to assess the cardioprotective effects of STS. Apoptosis of cells was measured with Hoechst 33342-based fluorescence microscopy, and annexin V-FITC-based flow cytometry. Caspase-3 and caspase-8 activities and mitochondrial membrane potential were also measured using commercial kits. TNF-α in the cell culture supernatant fractions were measured with sandwich elisa, and protein levels assayed using Western blot.
Key Results
STS inhibited OGD/R-induced apoptosis by suppressing JNK-mediated activation of NF-κB, TNF-α expression, activation of caspase-3 and caspase-8 and the Bax/Bcl-2 ratio. Additionally, positive feedback between NF-κB and TNF-α and amplification of TNF-α were inhibited, suggesting that STS plays a protective role against apoptosis in cardiomyocytes, even upon activation of pro-inflammatory cytokines. Interestingly, the cytoprotective effects of STS on OGD/R-induced apoptosis and promotion of cell survival were attenuated after inhibition of PI3K.
Conclusion and Implications
The inhibitory effects of STS on TNF-α and positive feedback signalling of the NF-κB/TNF-α pathways may play important roles in myocardial protection against ischaemia/reperfusion. These protective effects of STS are mediated by suppressing JNK activity through activation of the PI3K-Akt pathway.
Keywords: myocardial cell apoptosis, sodium tanshinone IIA silate, NF-κB/TNF-α, PI3K/Akt
Introduction
Ischaemia and reperfusion (I/R) is a pathological condition characterized by initial restriction of blood supply to an organ, followed by restoration of perfusion and concomitant reoxygenation (Eltzschig and Eckle, 2011). Reperfusion of coronary flow is necessary to resuscitate the ischaemic or hypoxic myocardium. Timely reperfusion facilitates cardiomyocyte salvage and decreases cardiac morbidity and mortality. Unfortunately, reperfusion also elicits a number of adverse reactions, including a process termed I/R injury, which may limit its beneficial actions. A number of studies have shown that myocardial apoptosis occurs during ischaemia and increasing experimental evidence suggests that apoptosis is primarily induced during reperfusion (Fliss and Gattinger, 1996; Zhao et al., 2000). The apoptotic process is initiated shortly after the onset of ischaemia, and becomes markedly enhanced during reperfusion. Therefore, inhibition of apoptosis may attenuate the irreversible injury associated with reperfusion.
Salviae miltiorrhiza, a traditional medicinal herb commonly known as ‘Danshen’, is an important source of a large number of active natural compounds mainly classified as aqueous and lipid-soluble fractions (Zhou et al., 2005). The major lipophilic compounds include tanshinone I, tanshinone IIA, tanshinone IIB and cryptotanshinone and tanshinone IIA is the most abundant of these and the most active diterpenoid quinone compound. In particular, tanshinone IIA exerts protective effects against experimental I/R injury (Fu et al., 2007; Zhang et al., 2010) although the mechanisms involved are still unclear at present. Sodium tanshinone IIA silate (STS) is a water-soluble derivative of tanshinone IIA and STS accepts electrons from complex I and is converted to its semiquinone form, which reduces the oxygen molecules (Jiang et al., 2009). STS administered via injection is marketed as a drug in China for the treatment of coronary heart diseases. However, STS has additionally been associated with dizziness, headaches, nausea, abdominal pain, rash, dermatitis and pruritus. The mechanism underlying its cardioprotective activity is yet to be established.
Reperfusion injury is associated with an inflammatory cascade that perpetuates damage to cardiac tissue after a period of ischaemia. One of the central players in this cascade is the transcription factor NF-κB, known to play a crucial role in regulating inflammatory signal transduction and cytokine production. NF-κB activation by I/R in the myocardium is a very early regulatory event during reperfusion. Many effector genes including those encoding cytokines such as TNF-α and IL-6, are activated by NF-κB (Li et al., 2011a) to enhance expression of pro-inflammatory cytokines. Research on patients undergoing coronary artery bypass graft surgery (Kawamura et al., 2005b) and myocardial biopsy in rats (Li et al., 2011b) has confirmed that myocardial expression of TNF-α is significantly higher after I/R. TNF-α contributes to post-ischaemic myocardial dysfunction via direct suppression of contractility and induction of myocyte apoptosis (Meldrum, 1998). Moreover, a neutralizing antibody against TNF-α has been shown to exert protective anti-apoptotic effects in cardiomyocytes (Minamino et al., 1999; Li et al., 2007). Interestingly, TNF-α acts as both the target gene and inducer of NF-κB (Pahl, 1999) and its production should be suppressed early in ischaemia (Shames et al., 2002). This cytokine has the unique ability to self-amplify through a positive feedback loop targeting NF-κB. Up-regulation of TNF-α in a localized area of the myocardium, for instance, an ischaemic region, readily induces TNF-α in the neighbouring normal myocardium, leading to amplified cytokine effects (Nian et al., 2004). Irwin and co-workers reported that maximal levels of TNF-α mRNA are detectable in the infarct and peri-infarct zones during the initial period after myocardial infarction. In contrast, by day 35 after myocardial infarction, the ‘contralateral normal zone’ in infarcted hearts displayed the highest level of TNF-α expression (Irwin et al., 1999). Thus, in the inflammatory processes following myocardial I/R, cardiomyocytes appear to play an active role in maintaining the inflammatory state, by not only responding to pro-inflammatory cytokines but also releasing several cytokines spontaneously and reacting to stimulation with other cytokines. The myocardium is therefore at the centre of an inflammatory positive feedback loop when hearts are reperfused. Blockade of NF-κB translocation has significant potential for breaking the positive feedback loop of inflammation (Onai et al., 2004).
Earlier studies demonstrated that STS inhibited TNF-α expression (Ren et al., 2010). However, whether this TNF-α decrease contributes to the protective effects of STS against cell injury during myocardial I/R is currently unclear. In the present study, we employed a cellular oxygen-glucose deprivation/recovery (OGD/R) model of I/R to determine whether STS could have cardioprotective effects against I/R injury and to establish the underlying mechanism(s). Taken together, our findings show that STS effectively protects H9c2 cardiomyocytes from OGD/R-induced apoptosis. Moreover, the PI3K-Akt pathway is involved in the inhibitory effects of STS on NF-κB activation, TNF-α expression and positive feedback signalling of the NF-κB/TNF-α pathways.
Methods
Cell culture and treatment
H9c2 rat myocardial cells (ATCC CRL 1446) were cultured in DMEM supplemented with 10% FBS (Hyclone), penicillin G (100 U·mL−1), streptomycin (100 μg·mL−1) and glutamine (2 mM). Before stimulation, cells were seeded in culture dishes or plates and grown to 80% confluency. H9c2 cells were treated with OGD/R or TNF-α in the presence or absence of STS. Cells were exposed to OGD as described previously with minor modifications (Zhu et al., 2010). Briefly, cells were rinsed twice, incubated in glucose-free DMEM and subsequently placed in an anaerobic chamber containing a mixture of 95% N2 and 5% CO2 at 37°C for 6 h. Following OGD, glucose was added to normal levels (final concentration: 4.5 mg·mL−1), and cells were incubated under normal growth conditions (95% air and 5% CO2) for an additional 18 h, as OGD/R, unless otherwise specified.
Hoechst staining for detection of nuclear condensation and fragmentation
H9c2 nuclei were stained with the chromatin dye Hoechst 33342 (Sigma-Aldrich, St. Louis, MO, USA). Briefly, cells were fixed with 4% paraformaldehyde for 10 min at room temperature, washed twice with PBS, and incubated with Hoechst 33342 (2 μg·mL−1) in PBS at room temperature for 30 min, followed by two further washes with PBS. Fluorescent-labelled invasive cells were photographed under a fluorescence microscope.
Measurement of caspase-3 and -8 activities
Caspase-3 and -8 activities were measured with a Caspase Activity Kit from Beyotime Institute of Biotechnology (Shanghai Province, China), using the substrate peptides Ac-DEVD-pNA and Ac-IETD-pNA respectively. Briefly, cells were lysed, and the supernatant was mixed with buffer containing the substrate peptides for caspase attached to p-nitroanilide (pNA). The release of pNA was quantified by determining the absorbance with a SpectraMax M2e Microplate Reader (Molecular Devices, Sunnyvale, CA, USA) at 405 nm. Caspase activities were expressed as a relative percentage of the control value.
Measurement of mitochondrial membrane potential (MMP)
Loss of MMP was assessed using 5,5′,6,6′-tetracholoro-1,1′3,3′-tetraethybenzimidazole-carbocyanide iodine (JC-1; Beyotime). Cells seeded in 60 mm Petri dishes were exposed to OGD/R or TNF-α with or without STS. Next, total cells were collected in 1.5 mL tubes and incubated with JC-1 for 20 min at 37°C. Fluorescence was detected with a SpectraMax M2e Microplate Reader (Molecular Devices) at excitation and emission wavelengths of 490 nm/530 nm and 25 nm/590 nm for monomeric and aggregated JC-1 respectively. The ratios of aggregated JC-1 to monomeric JC-1 fluorescence were calculated to determine the MMP of H9c2 cells. The MMP of STS-treated cells was compared with that of the control group.
Annexin V-FITC/propidium iodide (PI) staining for detecting phosphatidylserine translocation
Cell apoptosis was measured with annexin V-FITC and PI using a detection kit purchased from Beyotime Institute of Biotechnology. Briefly, cells were harvested at the indicated time periods, washed once with cold PBS, and resuspended in 195 μL binding buffer (1 × 105 cells), followed by staining with 5 μL Annexin V-FITC in the dark for 10 min at room temperature. Cells were centrifuged at 1000× g for 5 min, and the pellet was resuspended in 190 μL binding buffer. Next, cells were incubated with 10 μL PI solution on an ice bath in the dark. After filtration (300 apertures), the percentage of cell apoptosis was determined using flow cytometry (BD FACSCalibar, San Jose, CA, USA).
Reverse transcription PCR (RT-PCR)
Total RNA was extracted from H9c2 cells using TRIzol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. RNA (2 μg) was reverse transcribed at 37°C for 60 min in a 10 μL reaction mixture using the Reverse Transcription System from Promega (Fitchburg, WI, USA). PCR SuperMix containing Taq DNA polymerase (Takara, Dalian, China) was used to amplify the cDNA obtained from reverse transcription. Previously described primers (5′-CCTCTTCTCATTCCTGCTCG-3′; 5′-GGTATGAAATGGCAAATCGG-3′) were employed for rat TNF-α amplification (Liu et al., 2009). The reaction was carried out at an annealing temperature of 54°C and monitored to ensure exponential amplification. Amplified products were separated on a 1% agarose gel and visualized with ethidium bromide.
Isolation of cytoplasmic and nuclear proteins
Cytoplasmic and nuclear proteins were extracted using a Nuclear and Cytoplasmic extraction kit from Beyotime Institute of Biotechnology (China). Cells were dissolved with cytoplasmic protein extraction agent A supplemented with a cocktail. After vortexing for 5 s, tubes were incubated for 10–15 min on ice to promote lysis. Following addition of cytoplasmic protein extraction agent B, tubes were vortexed for 5 s and incubated on ice for 1 min. Samples were centrifuged for 5 min at 14 000× g at 4°C, and the supernatant, collected as the cytosolic fraction. The pellet was resuspended in a nuclear protein extraction agent supplemented with a cocktail. After vortexing the tubes 15–20 times for 30 min and centrifuging at 4°C for 10 min at 14 000× g, supernatant fractions containing the nuclear extracts were obtained.
MTT assay for cell viability
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay is based on cleavage of MTT by mitochondrial dehydrogenases, reflecting cell viability. Briefly, cardiomyocytes were treated with STS for 24 h, followed by 0.5 mg·mL−1 MTT, and incubated for 4 h at 37°C. After removal of the MTT-containing medium, DMSO was added, and absorbance was measured at 570 nm.
Electrophoretic mobility shift assay (EMSA)
EMSA was carried out with a commercially available Chemiluminescent EMSA Kit from Beyotime Institute of Biotechnology (China) according to the manufacturer's protocol. An aliquot of 6 μg nuclear protein was used to assess DNA-binding activity using biotin-labelled oligonucleotide probes containing the consensus sequence of NF-κB (5′-AGTTGAGGGGACTTTCCCAGGC-3′ and 3′-TCAACTCCCCTGAAAGGGTCCG-5′). The specificity of binding was assessed by competition with the unlabeled probes.
Western blot analysis
After various treatments, cells were washed twice with PBS and lysed in RIPA buffer containing protease and phosphatase inhibitor cocktails (MERCK, Whitehouse Station, NJ, USA). Whole-cell lysates of cultured cells were prepared by scraping cells into RIPA buffer. The resulting cell lysates were clarified by centrifugation at 12 000× g for 15 min at 4°C. Equal amounts of protein from each sample were separated by electrophoresis on an 8% or 12% polyacrylamide SDS gel followed by transfer to a polyvinylidene difluoride membrane (Bio-Rad, Berkeley, CA, USA) for immunoblotting. Membranes were blocked with 5% non-fat milk in TBST buffer (50 mM Tris, pH 7.5, 250 mM NaCl, 0.1% Tween 20) and probed with the indicated antibodies overnight at 4°C. After five washes in TBST, membranes were exposed to the appropriate secondary antibodies for 2 h at room temperature. Immunoreactive bands were visualized using chemiluminescent detection reagents, according to the manufacturer's instructions.
elisa for TNF-α
The culture medium was collected after treatment and centrifuged at 600× g for 5 min to pellet the cell debris. The supernatant was removed and stored at −80°C prior to analysis. TNF-α levels in the supernatant were determined with sandwich elisa using the dual antibody kits (R&D Systems) according to the manufacturer's instructions and expressed as pg mL−1.
Statistical analysis
Each assay was performed at least three times, and all data are presented as means ± SD. Student's t-test was used to compare the difference between two group means and one-way anova followed by Dunnett's multiple comparison test applied for more than two groups. P < 0.05 was considered statistically significant.
Materials
DMEM cell culture medium was obtained from Gibco Inc. (Carlsbad, CA, USA). Signal pathway inhibitors were procured from Calbiochem Inc. (San Diego, CA, USA). The TNF-α antibody was purchased from Abcam Inc. (San Francisco, CA, USA), and antibodies specific for Akt, phospho-Akt, phospho-IKKα/β, NF-κB p65, phospho-NF-κB p65, IκBα, phospho-IκBα, caspase-3, cleaved caspase-3, Bax, Bcl-2, β-actin and Histone H3 from Cell Signaling Technology Inc. (Danvers, MA, USA). Antibodies for JNK and phospho-JNK were acquired from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). The primers for reverse transcription and PCR were synthesized by Sangon Inc. (Shanghai, China). PCR-related reagents were purchased from Takara Inc., and all other reagents were purchased from Sigma-Aldrich unless otherwise specified.
Results
STS suppresses cardiomyocyte apoptosis induced by OGD/R
Hoechst 33342 staining revealed that the number of cells with condensed or fragmented chromatin increased dramatically after OGD for 6 h followed by 18 h incubation. This process was significantly inhibited in cells treated with STS (Figure 1A). To confirm these results, Annexin V-PI staining followed by flow cytometry, was performed. OGD/R led to an increase in the number of apoptotic cells, compared with vehicle, which was suppressed by STS (Figure 1B). To assess the involvement of caspase activation in the block of OGD/R-induced apoptosis by STS, we examined the activities of the initiating caspase-3 and -8 in the mitochondria-mediated apoptosis pathway. As shown in Figure 1C, after exposure of cells to OGD/R, caspase-3 and -8 activities were enhanced, compared with the control group. STS significantly attenuated this increase in caspase activity in a dose-dependent manner. Moreover, the MMP of H9c2 cells subjected to OGD/R was significantly decreased and this decrease was markedly reversed in the presence of STS, in a dose-dependent manner (Figure 1D). No cell toxicity was observed after STS treatment, even at the maximal dose of STS (10 μM; Figure 1E).
Figure 1.
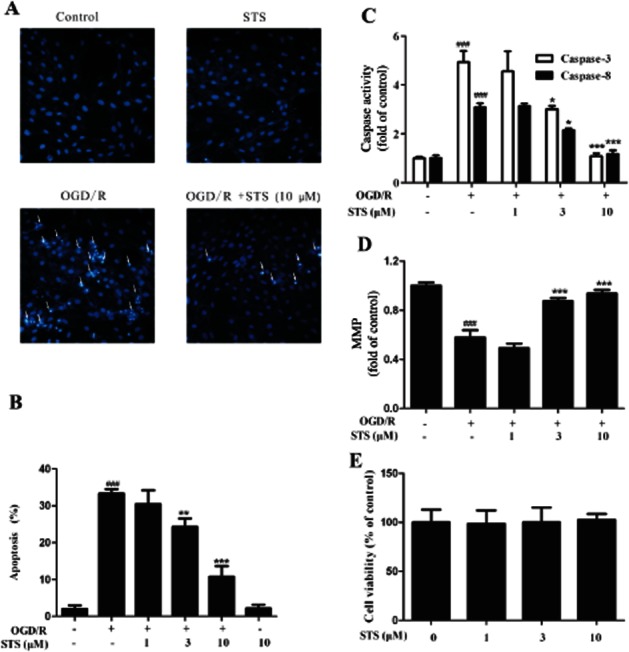
STS attenuates OGD-induced H9c2 cardiomyocyte apoptosis. Cells were subjected to 6 h OGD and 18 h recovery with or without STS treatment. Apoptosis of cells was determined with (A) Hoechst 33342-based fluorescence microscopy. Arrowheads in the pictures indicate the nuclei of apoptotic cells, (B) Annexin V-FITC-based flow cytometry (Annexin V-FITC +/PI- population of gated cells was measured with CellQuest software and presented as an apoptosis ratio). (C) Caspase-3 and caspase-8 activities and (D) MMP were also determined, after OGD/R and treatment with STS. Values are presented as means ± SD from three independent experiments. ###P < 0.001, significantly different from control, *P < 0.05, **P < 0.01, **P < 0.001, significantly different from OGD/R alone. (E) Cell viability was determined using the MTT assay. STS (10 μM) induced no cell toxicity (P > 0.05).
Effects of STS on expression of cleaved caspase-3, Bcl-2 and Bax proteins during OGD/R
Activation of caspase-3 via proteolytic processing of pro-caspase-3 into 17 and 12 kDa subunits serves as an early marker of apoptosis in various cell types. Consistent with the change in caspase-3 activity shown in the Figure 1, the 17 kDa cleaved caspase-3 fragment was detected in cardiomyocytes exposed to OGD/R. Treatment with STS prevented this OGD/R-induced fragmentation of caspase-3 (Figure 2A). The apoptotic process involves a number of regulatory genes mediated by cell death signals. Among these, the Bcl-2 protein family has emerged as a key regulatory component of the cell death process and consists of death antagonists (Bcl-2, Bcl-xL) and death agonists (Bax, Bak), which function primarily to protect or disrupt the integrity of the mitochondrial membrane and control the release of (pro)apoptotic intermembrane proteins(van Empel et al., 2005). Data from the present study revealed significant down-regulation of Bcl-2 expression and up-regulation of Bax following exposure to OGD/R, which was reversed by STS (Figure 2B).
Figure 2.
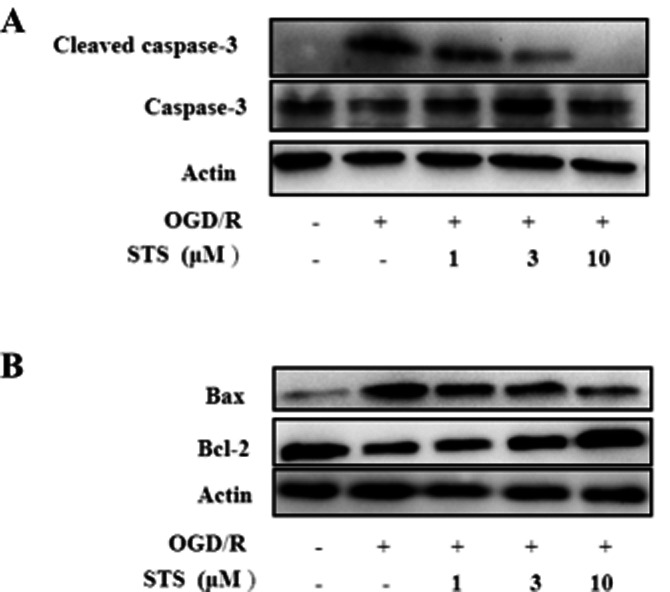
Effects of STS on expression of cleaved caspase-3, Bax and Bcl-2. Cells were subjected to 6 h OGD and 18 h recovery with or without STS treatment. Total protein (30 μg) was assayed for Western blot with antibodies specific for (A) cleaved caspase-3, caspase-3 and (B) Bax and Bcl-2. β-Actin was employed as a loading control. Results are means of three independent experiments.
STS inhibits OGD/R-induced activation of NF-κB and subsequent TNF-α expression, and protects cardiomyocytes from apoptosis
Activation of NF-κB is critical in promoting myocardial inflammation (Kawamura et al., 2005a) and its inhibition may thus underlie the cardioprotective activity of STS. In our experiments, STS inhibited the expression and translocation of NF-κB (Figure 3A). Treatment with OGD/R in the absence or presence of STS affected both the nuclear and cytoplasmic content of p65, which suggested that not all the increased protein was translocated. Activation of NF-κB is controlled by an inhibitory subunit, inhibitor of κB (I-κB), which retains NF-κB in the cytoplasm by physically masking its nuclear translocation signal. Dissociation of the cytoplasmic complex of NF-κB and IκB leads to release of active NF-κB, which translocates into the nucleus to activate target enhancers (Baeuerle and Baltimore, 1988). Active NF-κB has been detected in the cytoplasm under a number of conditions (Baeuerle and Baltimore, 1988; Baeuerle et al., 1988b). These findings, along with the reversibility of NF-κB activation in vivo (Baeuerle et al., 1988b), suggest that the protein translocates freely in and out of the nucleus. Cytoplasmic NF-κB release after OGD/R, which facilitated its translocation into nucleus, was reversed by STS in a dose-dependent manner (Figure 3B).
Figure 3.
TNF-α is involved in OGD/R-mediated apoptotic death. STS inhibits OGD/R-induced NF-κB activation and subsequent TNF-α expression to protect cardiomyocytes against apoptosis. Cells were subjected to 6 h OGD and 18 h recovery with or without the following indicated treatments: (A) Total protein (30 μg) or cytoplasmic or nuclear extracts (15 μg) were analysed by Western blot with antibodies specific for the indicated proteins, with β-actin as a total protein-loading control, Histone H3 (nuclear marker) and β-actin (cytoplasmic marker) as loading controls for nuclear and cytoplasmic extracts respectively. Results are means of three independent experiments. (B) Cytoplasmic extracts were analysed by Western blot with anti-p-NF-κB p65 and β-actin antibodies. Results are means of three independent experiments. (C) The specific DNA-binding activity of nuclear NF-κB was analysed with EMSA using a probe corresponding to the NF-κB binding site. Specificity of binding was confirmed by cold competition experiments with 50-fold molar excess of unlabelled NF-κB duplex oligonucleotide. The arrow indicates the NF-κB-DNA complexes. The graph shows results from one out of three independent experiments. (D) Cells were subjected to 6 h OGD and 4 h recovery with or without STS. Cytoplasmic extracts were analysed by Western blot with antibodies specific for the indicated proteins. Results are means of three independent experiments. (E and G) TNF-α and β-actin mRNA expression levels were determined using RT-PCR. Results shown are means of five independent experiments. (F and H) TNF-α release was measured using elisa. Results are expressed as mean values ± SD of three independent experiments performed in triplicate. ###P < 0.001, significantly different from control, **P < 0.01, **P < 0.001, significantly different from OGD/R alone. (I and J) Apoptosis of cells was determined with Annexin V-FITC-based flow cytometry. The annexin V-FITC +/PI- population of gated cells was measured with CellQuest software, and is presented as the apoptosis ratio. Results are expressed as means ± SD of three independent experiments performed in triplicate. ###P < 0.001, significantly different from control, ***P < 0.001, significantly different from OGD/R alone. (K and L) Total protein (30 μg) was analysed by Western blot with antibodies specific for cleaved caspase-3 and caspase-3. β-actin was used as the loading control. Data are means of three independent experiments.
To further ascertain whether STS is directly involved in inhibition of NF-κB DNA-binding activity, we performed EMSA assays. Exposure of cardiomyocytes to OGD/R resulted in a dramatic increase in binding of nuclear proteins to the NF-κB consensus sequence, while treatment with STS significantly suppressed this NF-κB DNA-binding activity (Figure 3C). Activation of NF-κB requires sequential phosphorylation, ubiquitination and degradation of IκB as well as consequent exposure of the nuclear localization signal on the NF-κB molecule (Li and Verma, 2002). Activation of IKK is the crucial step promoting disruption of the NF-κB/IκBα complex, and IKKβ-dependent IκBα degradation is responsible for stimulating canonical NF-κB signalling (Bonizzi and Karin, 2004). In our experiments, STS inhibited the phosphorylation of IKKβ and phosphorylation and ubiquitination of IκB (Figure 3D), in turn, inducing suppression of NF-κB activation.
STS suppressed TNF-α expression (Figure 3E and F) after OGD/R, in a dose-dependent manner and this suppression was mimicked by BAY 11–7082, a commercial NF-κB inhibitor (Figure 3G and H). To assess the causal role of TNF-α in OGD/R-induced apoptosis, we employed a specific neutralizing antibody (1 μg·mL−1) (Li et al., 2007). Treatment of cardiomyocytes with the anti-TNF-α antibody or BAY 11–7082 during OGD/R, significantly decreased the number of apoptotic cells, as shown by annexin V-FITC-PI staining. Also, the OGD/R-mediated activation of caspase-3 was completely inhibited (Figure 3I–L). These results provide strong evidence that OGD/R stimulated NF-κB activation and subsequent TNF-α expression, which was the primary cause for cardiomyocyte apoptosis. STS inhibited this process, consequently protecting cardiomyocytes from OGD/R-induced apoptosis.
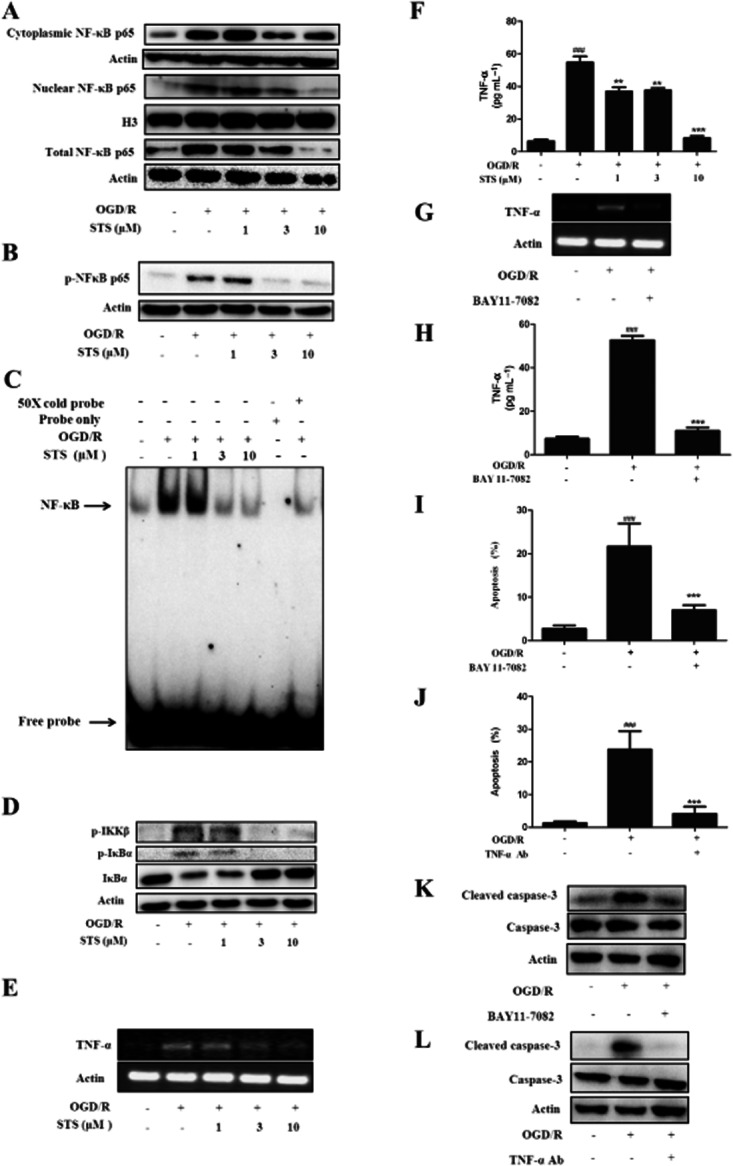
STS suppresses TNF-α-induced apoptosis of cardiomyocytes
The above results suggested that inhibition of TNF-α was involved in the protective effects of STS against OGD/R-induced myocardial cell death. To confirm this hypothesis, we further examined whether STS attenuated apoptosis in H9c2 cardiomyocytes, directly induced by TNF-α. Exposure to TNF-α induced apoptosis in H9c2 cardiomyocytes, as shown with Hoechst 33342 which stained apoptotic nuclei (Figure 4A) The number of apoptotic cells was lower in the cultures treated with STS and TNF-α than in those treated with TNF-α alone, signifying a protective effect against TNF-α-induced apoptosis. Similar results were obtained with annexin V-FITC-based flow cytometry. Data are presented as a percentage of apoptotic cells. Treatment of cells with TNF-α led to significant induction of H9c2 cell death, indicative of apoptotic accumulation, which was concentration-dependently attenuated by STS (Figure 4B). To clarify the mechanisms underlying the effects of STS on signalling pathways in TNF-α-induced apoptosis, we investigated the activation of caspase-8 and -3 in TNF-α-treated cells. As expected, TNF-α induced activation of caspase-8 and -3 in H9c2 cells, which was significantly attenuated by STS (Figure 4C). Moreover, MMP was decreased in cells treated with TNF-α for 24 h, and STS also prevented the TNF-α-induced disruption of mitochondrial integrity (Figure 4D).
Figure 4.
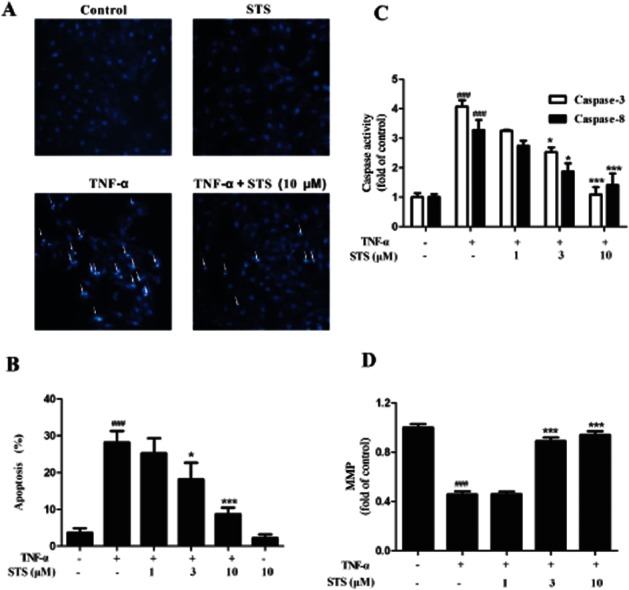
STS suppresses TNF-α-induced H9c2 cardiomyocyte apoptosis. Cells were incubated with 20 ng·mL−1 TNF-α in the presence or absence of the indicated concentrations of STS for 24 h. Apoptosis of cells was determined with (A) Hoechst 33342-based fluorescence microscopy. Arrowheads in the pictures indicate the nuclei of apoptotic cells, and (B) Annexin V-FITC-based flow cytometry (the Annexin V-FITC +/PI- population of gated cells was measured with CellQuest software and presented as an apoptosis ratio). (C) Caspase-3 and caspase-8 activities and (D) MMP were in these cultures were also measured. Values are presented as means ± SD from three independent experiments. ###P < 0.001, significantly different from control, *P < 0.05, ***P < 0.001, significantly different from TNF-α alone.
STS modulates the expression of cleaved caspase-3, Bcl-2 and Bax proteins in TNF-α-treated cardiomyocytes
To further characterize the inhibitory effects of STS on TNF-α-induced apoptosis, we examined whether STS suppresses the cleavage of inactive pre-caspase-3 into its active form. In the present study, cleavage of caspase-3 was triggered by TNF-α, and conversely, inhibited by STS (Figure 5A). We additionally investigated whether STS modulates expression of the Bcl-2 protein family in TNF-α-treated cells. Bcl-2 expression was suppressed, while the Bax content was increased in TNF-α-stimulated cells. Conversely, treatment with STS led to enhanced Bcl-2 and decreased Bax expression. This decrease in Bax/Bcl-2 ratio should lead to modulation of mitochondria apoptosis signalling and attenuate TNF-α cytotoxicity in cardiomyocytes (Figure 5B).
Figure 5.
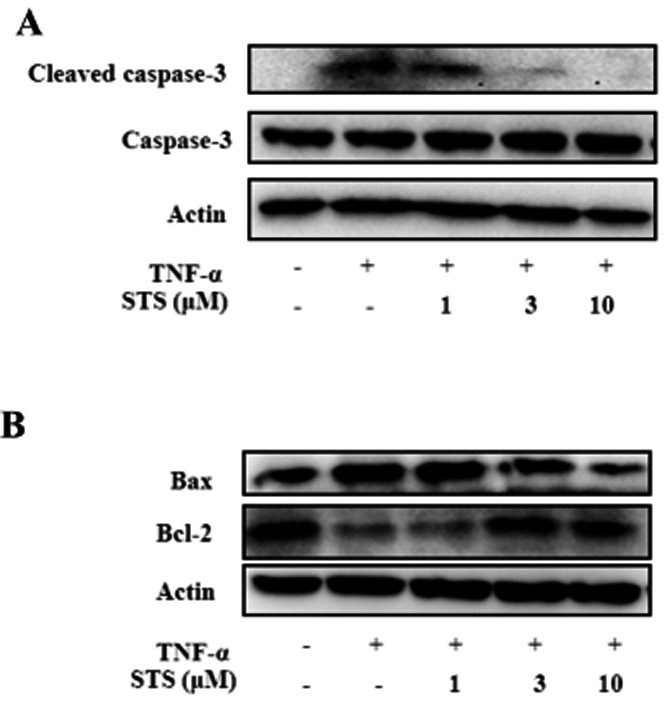
Effects of STS on expression of caspase-3, Bax and Bcl-2. Cells were incubated with 20 ng·ml−1 TNF-α in the presence or absence of the indicated concentrations of STS for 24 h, Total protein (30 μg) was analysed by Western blot with antibodies specific for (A) cleaved caspase-3, caspase-3 (B) Bax and Bcl-2. β-Actin was used as the loading control. Results are representative of three independent experiments.
STS interrupts positive feedback signalling of the NF-κB/TNF-α pathway and TNF-α amplification
NF-κB regulates TNF-α expression and is also activated by the proinflammatory cytokine, leading to positive feedback amplification, with chronic activation of NF-κB in certain cell types (Barnes, 1997). This type of feedforward amplification occurs in HeLa, HMEEC-1 and NHBE cells (Watanabe et al., 2004). To confirm whether a positive feedback amplification loop exists between NF-κB and TNF-α activity in cardiomyocytes that may be affected by STS, H9c2 cells were treated with TNF-α in the absence or presence of STS. The addition of TNF-α to H9c2 cells induced translocation of NF-κB, and triggered a positive feedback loop targeting this transcription factor, leading to the self-amplification of TNF-α. Notably, STS inhibited TNF-α-activated NF-κB translocation in a dose-dependent manner (Figure 6A) and subsequent amplification of TNF-α (Figure 6B). Our results clearly demonstrate that STS inhibits the positive feedback cycle between TNF-α and NF-κB.
Figure 6.
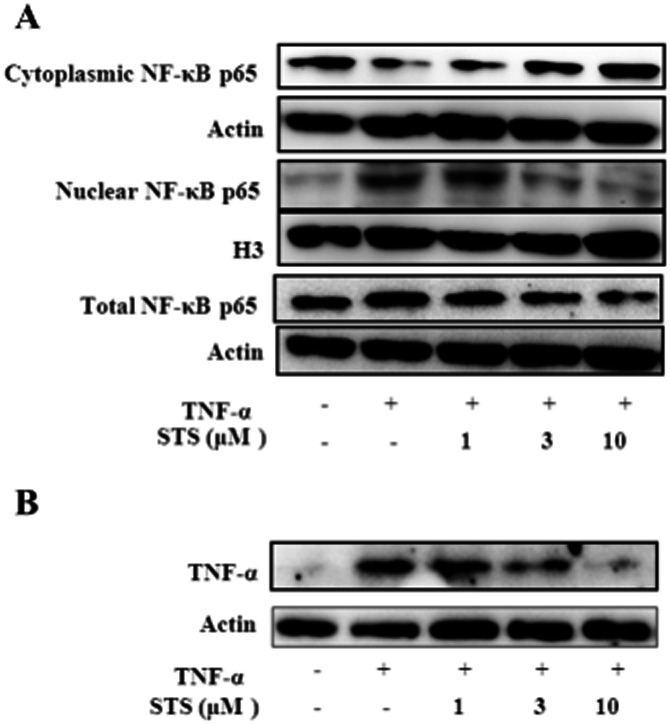
STS disrupts the positive feedback signalling of the NF-κB/TNF-α pathway and amplification of TNF-α. Cells were incubated with 20 ng·ml−1 TNF-α in the presence or absence of the indicated concentrations of STS for 24 h. (A) Cell lysates and nuclear extracts were prepared, and the levels of cytoplasmic, nuclear and total p65 determined using Western blot. (B) TNF-α protein levels was determined via Western blot analysis. Results are representative of three independent experiments.
Effects of PI3K/Akt activation on the STS-attenuated positive NF-κB/TNF-α feedback loop
MAPK and PI3K/Akt signal cascades play important roles in both apoptosis and survival in cardiomyocytes (Bishopric et al., 2001; Sussman et al., 2011). In view of the recent findings that activation of PI3K/Akt negatively regulates the NF-κB activation pathway and improves survival and cardiac function in septic mice (Williams et al., 2006), we investigated the effects of STS on MAPK and Akt activation. STS alone or together with OGD/R (or TNF-α) enhanced phosphorylation of Akt (Figure 7A, B), but had no significant effects on MAP kinases phosphorylation (data not shown) in H9c2 cells. We further assessed the suppressive effects of inhibitors of p38 MAPK (SB203580, 10 μM) and JNK1/2 (SP600125, 10 μM) on OGD/R-induced phosphorylation and degradation of IκB in cardiomyocytes. Pretreatment with SP600125 significantly inhibited OGD/R-induced phosphorylation and degradation of IκB (Figure 7C). Moreover, the inhibition by STS of JNK1/2 phosphorylation, IκB phosphorylation and degradation, and NF-κB p65 phosphorylation were significantly reversed upon inhibition of PI3K (Figure 7D, E). These results suggest that STS protects cardiomyocytes from apoptosis through enhancement of Akt-mediated survival signalling, specifically by suppressing JNK1/2 activity in the PI3K-Akt pathway. To establish whether Akt is involved in the inhibition of NF-κB activation, TNF-α expression and the NF-κB/TNF-α feedback loop, we used LY294002 (20 μM) or wortmannin (500 nM) to inhibit the activation of downstream Akt. Data from Western blot, RT-PCR and elisa assays collectively showed that PI3K inhibitors blocked the suppressive effects of STS by promoting the translocation (Figure 7F) and DNA-binding activity of NF-κB (Figure 7G), TNF-α expression (Figure 7H, I), and the positive feedback loop between NF-κB and TNF-α (Figure 7J, K) in H9c2 myocardial cells. In view of these findings, we suggest that STS inhibits OGD/R-induced JNK activity and subsequent degradation of I-κB and NF-κB activation, TNF-α expression and the NF-κB/TNF-α feedback loop through activation of the PI3K-Akt pathway.
Figure 7.
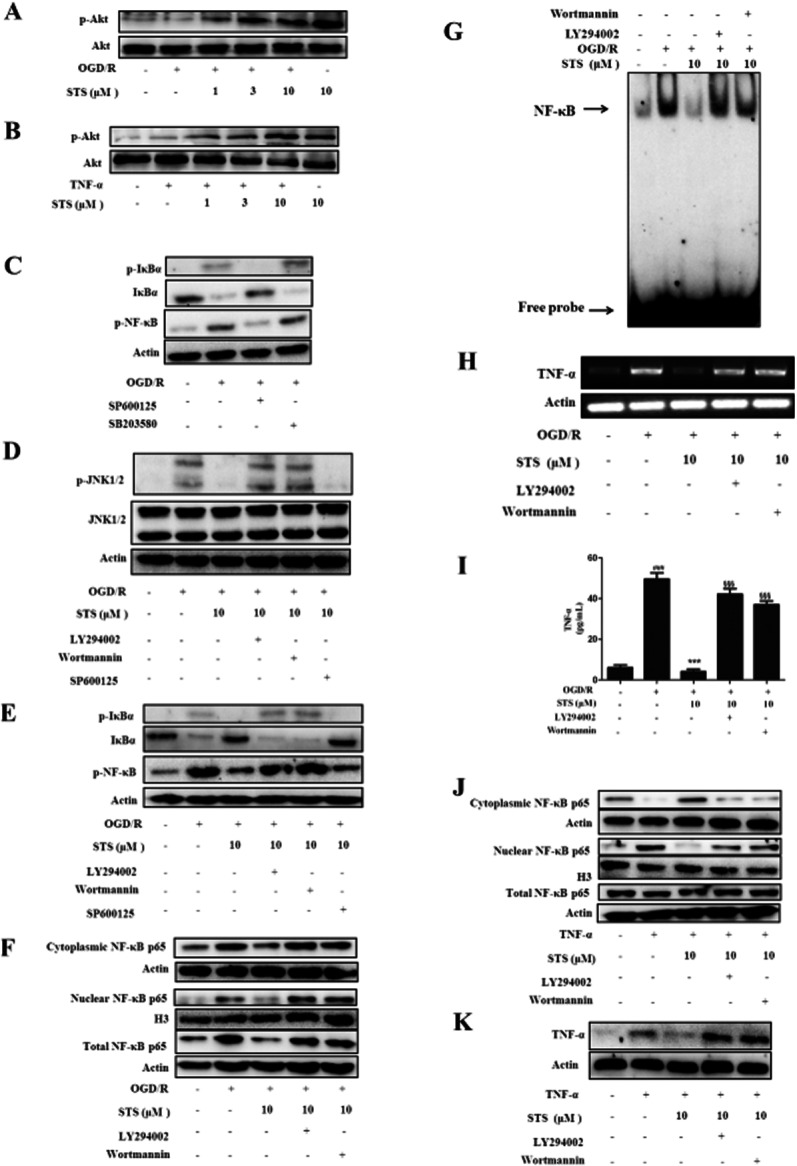
Akt phosphorylation plays a critical role in STS-mediated inhibition of OGD/R-induced JNK1/2 and NF-κB activation, subsequent TNF-α expression, positive NF-κB/TNF-α feedback loop, and TNF-α amplification. (A and B) Cells were exposed to OGD/R or TNF-α with or without STS. Total protein (30 μg) was analysed by Western blot with antibodies specific for p-Akt or Akt, with β-actin as the loading control. Results are representative of three independent experiments. (C–E) Cells were incubated with SP600125 (10 μM), SB203580 (10 μM), LY294002 (20 μM) or wortmannin (500 nM) for 30 min, followed by 6 h OGD and 4 h recovery with or without STS. Whole-cell lysates were examined using Western blot with the indicated antibodies. Results are representative of three independent experiments. (F) Cells were incubated with the LY294002 (20 μM) or wortmannin (500 nM) for 30 min, and subjected to 6 h OGD and 18 h recovery with or without STS. Cell lysates and cytoplasmic and nuclear extracts were prepared, and the levels of cytoplasmic, nuclear and total p65 protein determined using Western blot. Results are representative of three independent experiments. (G) The specific DNA-binding activity for nuclear NF-κB was analysed with EMSA. The graph depicts results from one of three independent experiments. (H) TNF-α and β-actin mRNA expression levels were determined using RT-PCR. Representative results of five independent experiments are shown. (I) TNF-α release was measured with elisa. Results are expressed as mean values ± SD of three independent experiments performed in triplicate. ###P < 0.001, significantly different from control, ***P < 0.001, significantly different from OGD/R alone, §§§P < 0.001, significantly different from OGD/R + STS. (J and K) Translocation of NF-κB and TNF-α expression were examined to determine the effects of STS on the positive feedback loop of NF-κB/TNF-α. Results are representative of three independent experiments.
Role of Akt in the protective effect of STS against OGD/R-induced cardiomyocyte apoptosis
Finally, to further confirm the role of the PI3K-Akt pathway in mediating the protective influence of STS on H9c2 cardiomyocytes, we examined the effects of co-administration of LY294002 or wortmannin and STS on OGD/R-induced cardiomyocyte apoptosis. LY294002 or wortmannin alone had no pro-apoptotic effects in cardiomyocytes (Supporting Information Figure S1). Hoechst 33342 staining, annexin V-FITC-based flow cytometry assay, and Western blot for activated caspase-3 further confirmed that loss of Akt activation via PI3K suppression suppressed the inhibitory effects of STS on OGD/R-induced apoptosis and caspase-3 activation (Figure 8). Our results support the involvement of the Akt signalling pathway in the protective effects of STS against OGD/R-induced apoptosis in cardiomyocytes.
Figure 8.
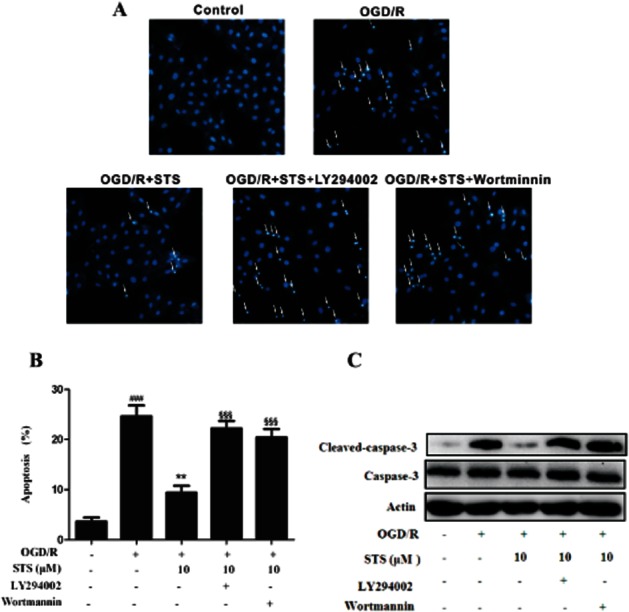
Inhibition of PI3K blocks the cytoprotective effects of STS on OGD/R-treated H9c2 myocardial cells. Cells were incubated with LY294002 (20 μM) or wortmannin (500 nM) for 30 min, and subjected to 6 h OGD and 18 h recovery with or without STS. Apoptosis of cells was determined with (A) Hoechst 33342-based fluorescence microscopy, and (B) Annexin V-FITC-based flow cytometry, The Annexin V-FITC +/PI- population of gated cells was measured with CellQuest software and presented as an apoptosis ratio. Results are expressed as mean values ± SD of three independent experiments performed in triplicate. ###P < 0.001, significantly different from control, ***P < 0.001, significantly different from OGD/R alone, §§§P < 0.001, significantly different from OGD/R + STS. (C) Total protein (30 μg) was analysed by Western blot with antibodies specific for cleaved caspase-3 and caspase-3. β-Actin was used as a protein-loading control. Results are representative of three independent experiments.
Discussion and conclusions
In the present study, we showed that STS prevents apoptosis in cardiomyocytes by inhibiting TNF-α and positive feedback signalling of the NF-κB/TNF-α pathways with no cell toxicity, even at a high concentration of 10 μM. The mechanism appears to involve inhibition of apoptosis-related activation of caspases-3 and -8 as well as reduction of the Bax/Bcl-2 ratio. Interestingly, OGD/R or STS treatment regulated the cleaved caspase-3 level without affecting caspase-3 in cardiomyocytes, consistent with previous findings (Dhingra et al., 2009; Park et al., 2011). The caspase-3 level may be dependent on the context and period of treatment (Zhao et al., 2010; Cuadrado et al., 2011; Kuo et al., 2012; Tsai et al., 2012). We observed no changes in the caspase-3 levels in cardiomyocytes subjected to 6 h OGD followed by 18 h recovery with or without STS treatment. The anti-apoptosis effects were associated with reduction of pro-inflammatory pathways, linked with myocardial infarction. Our results indicate that the inhibitory effects of STS on TNF-α and positive feedback signalling of the NF-κB/TNF-α pathways mediated by JNK are critical for protection of cardiomyocytes against OGD/R, our model of I/R. The protective effects of STS are mediated via up-regulation of Akt phosphorylation, an interesting self-gain signalling that possibly enhances its activity. However, the causal relationship between up-regulation of Akt phosphorylation and STS activity requires further investigation.
NF-κB is a critical regulator of numerous genes implicated in immune and inflammatory responses. NF-κB commonly exists as a heterodimer composed of Rel A (p65) and p50, and is normally located in the cytoplasm bound to its endogenous inhibitor protein, IκB. The upstream IKK promotes IκB phosphorylation, ubiquitination and degradation in the 26S proteasome, liberation of NF-κB and translocation to the nucleus, and binding to κB sites (specific domains within the promoters of downstream genes) to activate their transcription. Many genes involved in inflammation, such as TNF-α and IL-6, contain functional κB sites (Niu et al., 2009). In the inflammatory response, NF-κB is suggested to act on genes for pro-inflammatory cytokines, chemokines and enzymes that generate mediators of inflammation, immune receptors and adhesion molecules, which play key roles in the initial recruitment of leukocytes to inflammation sites. TNF-α is a pro-inflammatory cytokine with potent pro-apoptotic effects, which acts through the cell surface receptor TNFR1 (Krown et al., 1996). The specific role of NF-κB in healing the damage after myocardial infarction (MI) remains controversial (Gordon et al., 2011). Several studies have identified NF-κB as a mediator of the inflammatory response to myocardial ischaemia. Toll-like receptors, which signal through NF-κB, have been shown to play a pivotal role in left ventricular remodelling after MI (Shishido et al., 2003). IKK/NF-κB activation in cardiomyocytes induces reversible inflammatory cardiomyopathy and heart failure (Maier et al., 2012). In the rat myocardial I/R model, a NF-κB inhibitor, BAY 11–7082, significantly reduced infarct size and preserved myocardial function by reducing inflammation and apoptosis (Kim et al., 2010). In vivo administration of the proteasome inhibitor, ALLN, prevented burn-related NF-κB nuclear translocation in the myocardium, inhibited cardiac myocyte secretion of TNF-α, reduced circulating TNF-α levels and improved cardiac contractile function (Maass et al., 2002). NF-κB-dependent gene expression is stimulus and cell type specific (Sen and Smale, 2010). The duration of the NF-κB response (Sen and Smale, 2010) and the cellular context and timing of activation (Gordon et al., 2011) contribute to selective gene activation. Inflammation is a major factor in heart disease, and prolonged inflammatory response promotes cytotoxicity in cardiomyocytes (Gordon et al., 2011). A positive feedback amplification loop is reported between NF-κB and TNF-α activity. Thus, activated NF-κB may act in concert with the TNF-α gene promoter to enhance TNF-α expression. TNF-α, in turn, triggers I-κB phosphorylation and increases NF-κB activity (Watanabe et al., 2004). In the present study, OGD/R-induced ischemic/reperfusion injury in myocardial H9c2 cells (measured using the apoptosis ratio) promoted NF-κB nuclear migration (observed via immunoblotting of the cytosolic and nuclear fractions) and significant TNF-α secretion by cardiomyocytes (estimated using elisa). STS inhibited NF-κB activation and further reduced TNF-α expression. However, the TNF-α concentration measured in whole medium and that required for induction of apoptosis were significantly different. It is possible that the TNF-α concentration around cardiomyocytes is considerably higher than that measured in whole medium. In addition, STS inhibited positive feedback signalling of the NF-κB/TNF-α pathways. We investigated the effects of STS on TNF-α-induced apoptosis and NF-κB activation in cardiomyocytes. Previous experimental and clinical reports demonstrated strong correlations of circulating TNF-α with myocardial necrosis following MI (Li et al., 1999). STS prevented the TNF-α-induced NF-κB nuclear migration and the consequent increase in TNF-α. In our cells exposed to OGD/R, the protective effects of STS were similar to those of ischaemia preconditioning (Xiong et al., 2011). STS inhibited TNF-α-induced apoptosis and NF-κB and TNF-α expression, supporting its use as post-conditioning upon activation of pro-inflammatory cytokines. NF-κB is the main factor in the positive feedback loop of inflammation, and its inhibition may be effective therapy for myocardial reperfusion injury from the viewpoint of preventing inflammation. However, inhibition of NF-κB also carries a significant risk of adverse consequences, such as potentiation of apoptosis or unintentional disruption of signalling in non-target tissues. Drugs commonly used in cardiovascular medicine, such as aspirin and statins, often possess intrinsic anti-NF-κB properties (Baldwin, 2001; Planavila et al., 2005). These drugs generally act as weak antagonists and inhibit central kinases in the classic NF-κB activation pathway.
Akt plays an important role in myocardial biology (Sussman et al., 2011). Earlier research has shown that the PI3K-Akt pathway facilitates cardiomyocyte survival during I/R (Matsui and Rosenzweig, 2005) and adenoviral-mediated expression of myr-AKT protects cultured neonatal cardiomyocytes from hypoxia-induced apoptosis (Matsui et al., 1999). To determine whether the protective effects of STS were associated with the PI3K-Akt pathway, the specific PI3K inhibitors, LY294002 and wortmannin, were employed, and the effects of co-administration of PI3K inhibitor and STS examined, compared with those of STS alone. Treatment with both the PI3K inhibitor and STS lead to NF-κB activation and increased production of TNF-α, recovery of the positive feedback loop of NF-κB/TNF-α, and enhancement of cardiomyocyte apoptosis. Thus, LY294002 or wortmannin blocked the cytoprotective effects of STS, suggesting that STS exerted its cytoprotective effects through activation of the PI3K/Akt pathway. To our knowledge, the current study has highlighted the novel finding that inhibition of the NF-κB/TNF-α pathway mediated by PI3K-Akt phosphorylation played a critical role in cytoprotection by STS. Moreover, our data support and extend previous work by colleagues reporting that STS protects against apoptosis in cardiomyocytes resulting from up-regulation of Akt phosphorylation (Zhang et al., 2010; Hong et al., 2012).
In conclusion, STS inhibited the activation of JNK1/2, the degradation of IκB, the translocation and DNA-binding activity of NF-κB, and subsequently, TNF-α expression, as well as the positive feedback signalling of NF-κB/TNF-α pathways. These inhibitory effects could play important roles in the mechanism of STS-mediated myocardial protection against damage induced by I/R. The protective effects of STS were mediated by the PI3K-Akt pathway. While our understanding of the contribution of NF-κB activation and TNF-α secretion to myocardial injury and dysfunction after I/R is incomplete, it is likely that therapeutic strategies that modulate the main factor in the positive feedback loop of inflammation and NF-κB-dependent inflammatory gene transcription and translation could have clinical utility for treatment of myocardial reperfusion injury, in terms of preventing inflammation.
Glossary
- I/R
ischaemia and reperfusion
- MMP
mitochondrial membrane potential
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- OGD/R
oxygen-glucose deprivation/recovery
- PI
propidium iodide
- STS
sodium tanshinone IIA silate
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 LY294002 or wortmannin alone showed no pro-apoptosis effect on cardiomyocytes. Cells were incubated with LY294002 (20 μM) or wortmannin (500 nM) for 24 h. Apoptosis was determined with Hoechst 33342-based fluorescence microscopy.
References
- Baeuerle PA, Baltimore D. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science. 1988;242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Lenardo M, Pierce JW, Baltimore D. Phorbol-ester-induced activation of the NF-kappa B transcription factor involves dissociation of an apparently cytoplasmic NF-kappa B/inhibitor complex. Cold Spring Harb Symp Quant Biol. 1988b;53(Pt 2):789–798. doi: 10.1101/sqb.1988.053.01.089. [DOI] [PubMed] [Google Scholar]
- Baldwin AS., Jr Series introduction: the transcription factor NF-kappaB and human disease. J Clin Invest. 2001;107:3–6. doi: 10.1172/JCI11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ. Nuclear factor-kappa B. Int J Biochem Cell Biol. 1997;29:867–870. doi: 10.1016/s1357-2725(96)00159-8. [DOI] [PubMed] [Google Scholar]
- Bishopric NH, Andreka P, Slepak T, Webster KA. Molecular mechanisms of apoptosis in the cardiac myocyte. Curr Opin Pharmacol. 2001;1:141–150. doi: 10.1016/s1471-4892(01)00032-7. [DOI] [PubMed] [Google Scholar]
- Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Cuadrado I, Fernandez-Velasco M, Bosca L, de Las Heras B. Labdane diterpenes protect against anoxia/reperfusion injury in cardiomyocytes: involvement of AKT activation. Cell Death Dis. 2011;2:e229. doi: 10.1038/cddis.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhingra S, Sharma AK, Arora RC, Slezak J, Singal PK. IL-10 attenuates TNF-alpha-induced NF kappaB pathway activation and cardiomyocyte apoptosis. Cardiovasc Res. 2009;82:59–66. doi: 10.1093/cvr/cvp040. [DOI] [PubMed] [Google Scholar]
- Eltzschig HK, Eckle T. Ischemia and reperfusion[mdash]from mechanism to translation. Nat Med. 2011;17:1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Empel VP, Bertrand AT, Hofstra L, Crijns HJ, Doevendans PA, De Windt LJ. Myocyte apoptosis in heart failure. Cardiovasc Res. 2005;67:21–29. doi: 10.1016/j.cardiores.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Fliss H, Gattinger D. Apoptosis in ischemic and reperfused rat myocardium. Circ Res. 1996;79:949–956. doi: 10.1161/01.res.79.5.949. [DOI] [PubMed] [Google Scholar]
- Fu J, Huang H, Liu J, Pi R, Chen J, Liu P. Tanshinone IIA protects cardiac myocytes against oxidative stress-triggered damage and apoptosis. Eur J Pharmacol. 2007;568:213–221. doi: 10.1016/j.ejphar.2007.04.031. [DOI] [PubMed] [Google Scholar]
- Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-kappaB in the heart: to be or not to NF-kappaB. Circ Res. 2011;108:1122–1132. doi: 10.1161/CIRCRESAHA.110.226928. [DOI] [PubMed] [Google Scholar]
- Hong HJ, Liu JC, Chen PY, Chen JJ, Chan P, Cheng TH. Tanshinone IIA prevents doxorubicin-induced cardiomyocyte apoptosis through Akt-dependent pathway. Int J Cardiol. 2012;157:174–179. doi: 10.1016/j.ijcard.2010.12.012. [DOI] [PubMed] [Google Scholar]
- Irwin MW, Mak S, Mann DL, Qu R, Penninger JM, Yan A, et al. Tissue expression and immunolocalization of tumor necrosis factor-alpha in postinfarction dysfunctional myocardium. Circulation. 1999;99:1492–1498. doi: 10.1161/01.cir.99.11.1492. [DOI] [PubMed] [Google Scholar]
- Jiang B, Zhang L, Wang Y, Li M, Wu W, Guan S, et al. Tanshinone IIA sodium sulfonate protects against cardiotoxicity induced by doxorubicin in vitro and in vivo. Food Chem Toxicol. 2009;47:1538–1544. doi: 10.1016/j.fct.2009.03.038. [DOI] [PubMed] [Google Scholar]
- Kawamura N, Kubota T, Kawano S, Monden Y, Feldman AM, Tsutsui H, et al. Blockade of NF-kappaB improves cardiac function and survival without affecting inflammation in TNF-alpha-induced cardiomyopathy. Cardiovasc Res. 2005a;66:520–529. doi: 10.1016/j.cardiores.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Kawamura T, Kadosaki M, Nara N, Wei J, Endo S, Inada K. Nicorandil attenuates NF-kappaB activation, adhesion molecule expression, and cytokine production in patients with coronary artery bypass surgery. Shock. 2005b;24:103–108. doi: 10.1097/01.shk.0000168874.83401.3f. [DOI] [PubMed] [Google Scholar]
- Kim YS, Kim JS, Kwon JS, Jeong MH, Cho JG, Park JC, et al. BAY 11-7082, a nuclear factor-kappaB inhibitor, reduces inflammation and apoptosis in a rat cardiac ischemia-reperfusion injury model. Int Heart J. 2010;51:348–353. doi: 10.1536/ihj.51.348. [DOI] [PubMed] [Google Scholar]
- Krown KA, Page MT, Nguyen C, Zechner D, Gutierrez V, Comstock KL, et al. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J Clin Invest. 1996;98:2854–2865. doi: 10.1172/JCI119114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo WW, Wang WJ, Tsai CY, Way CL, Hsu HH, Chen LM. Diallyl trisufide (DATS) suppresses high glucose-induced cardiomyocyte apoptosis by inhibiting JNK/NFkappaB signaling via attenuating ROS generation. Int J Cardiol. 2012;S0167-5273:15257–15268. doi: 10.1016/j.ijcard.2012.09.080. [DOI] [PubMed] [Google Scholar]
- Li C, Gao Y, Tian J, Shen J, Xing Y, Liu Z. Sophocarpine administration preserves myocardial function from ischemia-reperfusion in rats via NF-kappaB inactivation. J Ethnopharmacol. 2011a;135:620–625. doi: 10.1016/j.jep.2011.03.052. [DOI] [PubMed] [Google Scholar]
- Li C, Gao Y, Xing Y, Zhu H, Shen J, Tian J. Fucoidan, a sulfated polysaccharide from brown algae, against myocardial ischemia-reperfusion injury in rats via regulating the inflammation response. Food Chem Toxicol. 2011b;49:2090–2095. doi: 10.1016/j.fct.2011.05.022. [DOI] [PubMed] [Google Scholar]
- Li D, Zhao L, Liu M, Du X, Ding W, Zhang J, et al. Kinetics of tumor necrosis factor alpha in plasma and the cardioprotective effect of a monoclonal antibody to tumor necrosis factor alpha in acute myocardial infarction. Am Heart J. 1999;137:1145–1152. doi: 10.1016/s0002-8703(99)70375-3. [DOI] [PubMed] [Google Scholar]
- Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- Li S, Jiao X, Tao L, Liu H, Cao Y, Lopez BL, et al. Tumor necrosis factor-alpha in mechanic trauma plasma mediates cardiomyocyte apoptosis. Am J Physiol Heart Circ Physiol. 2007;293:H1847–H1852. doi: 10.1152/ajpheart.00578.2007. [DOI] [PubMed] [Google Scholar]
- Liu CJ, Lo JF, Kuo CH, Chu CH, Chen LM, Tsai FJ, et al. Akt mediates 17beta-estradiol and/or estrogen receptor-alpha inhibition of LPS-induced tumor necresis factor-alpha expression and myocardial cell apoptosis by suppressing the JNK1/2-NFkappaB pathway. J Cell Mol Med. 2009;13(9B):3655–3667. doi: 10.1111/j.1582-4934.2009.00669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass DL, Hybki DP, White J, Horton JW. The time course of cardiac NF-kappaB activation and TNF-alpha secretion by cardiac myocytes after burn injury: contribution to burn-related cardiac contractile dysfunction. Shock. 2002;17:293–299. doi: 10.1097/00024382-200204000-00009. [DOI] [PubMed] [Google Scholar]
- Maier HJ, Schips TG, Wietelmann A, Kruger M, Brunner C, Sauter M, et al. Cardiomyocyte-specific IkappaB kinase (IKK)/NF-kappaB activation induces reversible inflammatory cardiomyopathy and heart failure. Proc Natl Acad Sci U S A. 2012;109:11794–11799. doi: 10.1073/pnas.1116584109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Rosenzweig A. Convergent signal transduction pathways controlling cardiomyocyte survival and function: the role of PI 3-kinase and Akt. J Mol Cell Cardiol. 2005;38:63–71. doi: 10.1016/j.yjmcc.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Matsui T, Li L, del Monte F, Fukui Y, Franke TF, Hajjar RJ, et al. Adenoviral gene transfer of activated phosphatidylinositol 3′-kinase and Akt inhibits apoptosis of hypoxic cardiomyocytes in vitro. Circulation. 1999;100:2373–2379. doi: 10.1161/01.cir.100.23.2373. [DOI] [PubMed] [Google Scholar]
- Meldrum DR. Tumor necrosis factor in the heart. Am J Physiol. 1998;274(3 Pt 2):R577–R595. doi: 10.1152/ajpregu.1998.274.3.R577. [DOI] [PubMed] [Google Scholar]
- Minamino T, Yujiri T, Papst PJ, Chan ED, Johnson GL, Terada N. MEKK1 suppresses oxidative stress-induced apoptosis of embryonic stem cell-derived cardiac myocytes. Proc Natl Acad Sci U S A. 1999;96:15127–15132. doi: 10.1073/pnas.96.26.15127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. 2004;94:1543–1553. doi: 10.1161/01.RES.0000130526.20854.fa. [DOI] [PubMed] [Google Scholar]
- Niu F, Zhang X, Chang L, Wu J, Yu Y, Chen J, et al. Trichostatin A enhances OGD-astrocyte viability by inhibiting inflammatory reaction mediated by NF-kappaB. Brain Res Bull. 2009;78:342–346. doi: 10.1016/j.brainresbull.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Onai Y, Suzuki J, Kakuta T, Maejima Y, Haraguchi G, Fukasawa H, et al. Inhibition of IkappaB phosphorylation in cardiomyocytes attenuates myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004;63:51–59. doi: 10.1016/j.cardiores.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- Park M, Youn B, Zheng XL, Wu D, Xu A, Sweeney G. Globular adiponectin, acting via AdipoR1/APPL1, protects H9c2 cells from hypoxia/reoxygenation-induced apoptosis. Plos One. 2011;6:e19143. doi: 10.1371/journal.pone.0019143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planavila A, Laguna JC, Vazquez-Carrera M. Atorvastatin improves peroxisome proliferator-activated receptor signaling in cardiac hypertrophy by preventing nuclear factor-kappa B activation. Biochim Biophys Acta. 2005;1687:76–83. doi: 10.1016/j.bbalip.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Ren ZH, Tong YH, Xu W, Ma J, Chen Y. Tanshinone II A attenuates inflammatory responses of rats with myocardial infarction by reducing MCP-1 expression. Phytomedicine. 2010;17:212–218. doi: 10.1016/j.phymed.2009.08.010. [DOI] [PubMed] [Google Scholar]
- Sen R, Smale ST. Selectivity of the NF-{kappa}B response. Cold Spring Harb Perspect in Biol. 2010;2:a000257. doi: 10.1101/cshperspect.a000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shames BD, Barton HH, Reznikov LL, Cairns CB, Banerjee A, Harken AH, et al. Ischemia alone is sufficient to induce TNF-alpha mRNA and peptide in the myocardium. Shock. 2002;17:114–119. doi: 10.1097/00024382-200202000-00006. [DOI] [PubMed] [Google Scholar]
- Shishido T, Nozaki N, Yamaguchi S, Shibata Y, Nitobe J, Miyamoto T, et al. Toll-like receptor-2 modulates ventricular remodeling after myocardial infarction. Circulation. 2003;108:2905–2910. doi: 10.1161/01.CIR.0000101921.93016.1C. [DOI] [PubMed] [Google Scholar]
- Sussman MA, Volkers M, Fischer K, Bailey B, Cottage CT, Din S, et al. Myocardial AKT: the omnipresent nexus. Physiol Rev. 2011;91:1023–1070. doi: 10.1152/physrev.00024.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai KH, Wang WJ, Lin CW, Pai P, Lai TY, Tsai CY, et al. NADPH oxidase-derived superoxide anion-induced apoptosis is mediated via the JNK-dependent activation of NF-kappaB in cardiomyocytes exposed to high glucose. J Cell Physiol. 2012;227:1347–1357. doi: 10.1002/jcp.22847. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Jono H, Han J, Lim DJ, Li JD. Synergistic activation of NF-kappaB by nontypeable Haemophilus influenzae and tumor necrosis factor alpha. Proc Natl Acad Sci U S A. 2004;101:3563–3568. doi: 10.1073/pnas.0400557101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DL, Ozment-Skelton T, Li C. Modulation of the phosphoinositide 3-kinase signaling pathway alters host response to sepsis, inflammation, and ischemia/reperfusion injury. Shock. 2006;25:432–439. doi: 10.1097/01.shk.0000209542.76305.55. [DOI] [PubMed] [Google Scholar]
- Xiong J, Wang Q, Xue FS, Yuan YJ, Li S, Liu JH, et al. Comparison of cardioprotective and anti-inflammatory effects of ischemia pre- and postconditioning in rats with myocardial ischemia-reperfusion injury. Inflamm Res. 2011;60:547–554. doi: 10.1007/s00011-010-0303-4. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wei L, Sun D, Cao F, Gao H, Zhao L, et al. Tanshinone IIA pretreatment protects myocardium against ischaemia/reperfusion injury through the phosphatidylinositol 3-kinase/Akt-dependent pathway in diabetic rats. Diabetes Obes Metab. 2010;12:316–322. doi: 10.1111/j.1463-1326.2009.01166.x. [DOI] [PubMed] [Google Scholar]
- Zhao J, Li J, Li W, Li Y, Shan H, Gong Y, et al. Effects of spironolactone on atrial structural remodelling in a canine model of atrial fibrillation produced by prolonged atrial pacing. Br J Pharmacol. 2010;159:1584–1594. doi: 10.1111/j.1476-5381.2009.00551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZQ, Nakamura M, Wang NP, Wilcox JN, Shearer S, Ronson RS, et al. Reperfusion induces myocardial apoptotic cell death. Cardiovasc Res. 2000;45:651–660. doi: 10.1016/s0008-6363(99)00354-5. [DOI] [PubMed] [Google Scholar]
- Zhou L, Zuo Z, Chow MS. Danshen: an overview of its chemistry, pharmacology, pharmacokinetics, and clinical use. J Clin Pharmacol. 2005;45:1345–1359. doi: 10.1177/0091270005282630. [DOI] [PubMed] [Google Scholar]
- Zhu JR, Tao YF, Lou S, Wu ZM. Protective effects of ginsenoside Rb(3) on oxygen and glucose deprivation-induced ischemic injury in PC12 cells. Acta Pharmacol Sin. 2010;31:273–280. doi: 10.1038/aps.2010.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.