

Abstract
Background and Purpose
Mitochondria are a drug target in mitochondrial dysfunction diseases and in antiparasitic chemotherapy. While zebrafish is increasingly used as a biomedical model, its potential for mitochondrial research remains relatively unexplored. Here, we perform the first systematic analysis of how mitochondrial respiratory chain inhibitors affect zebrafish development and cardiovascular function, and assess multiple quinones, including ubiquinone mimetics idebenone and decylubiquinone, and the antimalarial atovaquone.
Experimental Approach
Zebrafish (Danio rerio) embryos were chronically and acutely exposed to mitochondrial inhibitors and quinone analogues. Concentration-response curves, developmental and cardiovascular phenotyping were performed together with sequence analysis of inhibitor-binding mitochondrial subunits in zebrafish versus mouse, human and parasites. Phenotype rescuing was assessed in co-exposure assays.
Key Results
Complex I and II inhibitors induced developmental abnormalities, but their submaximal toxicity was not additive, suggesting active alternative pathways for complex III feeding. Complex III inhibitors evoked a direct normal-to-dead transition. ATP synthase inhibition arrested gastrulation. Menadione induced hypochromic anaemia when transiently present following primitive erythropoiesis. Atovaquone was over 1000-fold less lethal in zebrafish than reported for Plasmodium falciparum, and its toxicity partly rescued by the ubiquinone precursor 4-hydroxybenzoate. Idebenone and decylubiquinone delayed rotenone- but not myxothiazol- or antimycin-evoked cardiac dysfunction.
Conclusion and Implications
This study characterizes pharmacologically induced mitochondrial dysfunction phenotypes in zebrafish, laying the foundation for comparison with future studies addressing mitochondrial dysfunction in this model organism. It has relevant implications for interpreting zebrafish disease models linked to complex I/II inhibition. Further, it evidences zebrafish's potential for in vivo efficacy or toxicity screening of ubiquinone analogues or antiparasitic mitochondria-targeted drugs.
Keywords: mitochondria, zebrafish, Danio rerio, idebenone, decylubiquinone, atovaquone, menadione
Introduction
Mitochondrial dysfunction is a common feature in multiple human diseases. Mitochondrial diseases present diverse clinical symptoms with the most common being neurological and cardiological manifestations, including cardiomyopathies and heart conduction defects (Tuppen et al., 2010; Berardo et al., 2011). Mutations in mitochondrial DNA (mDNA) may affect respiratory chain elements or ribosomal and transfer RNAs required for mitochondrial gene expression, causing diseases such as Leber's hereditary optic neuropathy or mitochondrial myopathy, encephalopathy, lactic acidosis and stroke syndrome (Tuppen et al., 2010). Also, mutations in nuclear encoded mitochondrial proteins are associated with human diseases (e.g. optic atrophy or Charcot-Marie-Tooth 2A). Furthermore, key neurodegenerative disorders such as Alzheimer, Parkinson and Huntington diseases, among others, have been associated with mitochondrial dysfunction (Oliveira, 2010). Current treatment options for mitochondrial dysfunction disorders are scarce and mostly symptomatic (Finsterer, 2010). Further treatment development is partly hindered by limited in vivo models of mitochondrial disease. Notably, strong purifying selection in the mammalian female germ line imposes difficulties in establishing mouse models of mDNA mutations (Stewart et al., 2008).
Zebrafish (Danio rerio) is increasingly used for modelling human diseases (Ingham, 2009). Its mitochondrial genome is sequenced (Broughton et al., 2001) and zebrafish is emerging as a model for studying mitochondria-linked disorders. Indeed, silencing mitofusin-2 in zebrafish generated an in vivo model of Charcot-Marie-Tooth 2A neuropathy (Vettori et al., 2011). Similarly, depletion of cytochrome c oxidase (COX), optic atrophy 3 protein and electron transfer flavoprotein dehydrogenase (ETFDH) were respectively used to model COX deficiency (Baden et al., 2007), Costeff syndrome (Pei et al., 2010) and multiple acyl-CoA dehydrogenase deficiency in zebrafish (Song et al., 2009). Moreover, zebrafish was reported sensitive to mitochondrial toxins linked to Parkinson's disease (Bretaud et al., 2004), and parkin knockdown in embryos led to mitochondrial complex I deficiency (Flinn et al., 2009). Currently, few studies addressed normal zebrafish mitochondrial physiology and bioenergetics. These include analysis of mDNA metabolism (Artuso et al., 2012), oxygen consumption (Stackley et al., 2011), calcium buffering and mitochondrial permeability transition (Azzolin et al., 2010) in zebrafish embryos. Still, further studies are required to validate zebrafish as a model to study mitochondrial dysfunction and its experimental treatment, including the identification of similarities and specific limitations when extrapolating towards mammalian physiology.
Here, we model mitochondrial dysfunction in zebrafish and use it to test mitochondria-targeted drugs. We characterize the effects of mitochondrial inhibitors and quinone analogues on zebrafish embryonic development and cardiovascular function, identifying morphological and functional phenotypes associated with impaired respiratory complexes and ubiquinone deficiency. Among several quinone analogues, we test the ubiquinone mimetics idebenone and decylubiquinone. Also, we test the antimalarial atovaquone, appraising zebrafish's potential for differential toxicity screening of the growing class of mitochondria-targeted antiparasitic drugs (Mather et al., 2007).
Methods
Drugs, solvents and solutions
Mitochondrial inhibitors [rotenone, 3-nitropropionic acid (3NP), myxothiazol, antimycin and oligomycin] and all other drugs/chemicals were from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise stated. Quinones analogues were: natural monomeric naphthoquinones [menadione (MND), juglone (JGL), naphthazarin (NTZ), plumbagin (PLB), lapachol (LPC) and β-lapachone (βLPC)]; natural dimeric naphthoquinones [diospyrin (DPR) and diosquinone (DQN)]; synthetic naphthoquinone [atovaquone (ATV)]; and synthetic benzoquinones [idebenone (IDB) and decylubiquinone (DCB)]. Other drugs include the ubiquinone synthesis precursor [4-hydroxybenzoic acid (4HB)] and respective inhibitor [4-nitrobenzoic acid (4NB)]; the NAD(P)H:quinone oxidoreductase (NQO1) inhibitor [dicoumarol (DCM)]; and the control abnormality inducer [valproic acid (VPA)]. DPR and DQN were isolated from root barks of Diospyros chamaethamnus Dinter ex Mildbr (Costa et al., 1998). 4HB was from Extrasynthese (Genay Cedex, France). For all drugs, chemical structures are in Supporting Information Figure S1, summarized data in Table 2 and Figure 5C, and their mechanism/sites of action depicted in Figure 8. Stock drug solutions were prepared in water, DMSO or methanol, according to their solubility. Prior to assays, stocks were diluted in dechlorinated autoclaved water (egg water). Non-water drug solvents, DMSO or methanol, were kept constant at all drug dilutions and always below 0.5 or 0.05% respectively. These solvent concentrations induced no significant effects (Figure 1B).
Table 2.
Comparative toxicity of mitochondrial inhibitors, quinone analogues and other drugs in zebrafish vs. other species
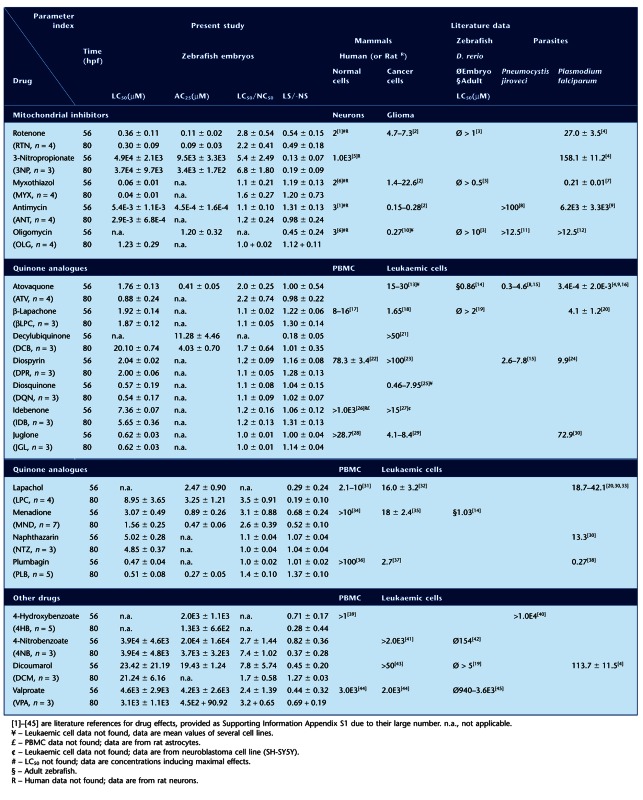 |
Figure 5.

Detailed heartbeat analysis and multiple-abnormalities profiling. A, Concentration-dependent changes in heart rate at 80hpf, showing total weighted average (i, see Methods), and separating embryos without (ii) from those with (iii) cardiac oedema; embryos in asystole were excluded from calculations. Horizontal rectangles are the 95% confidence interval (CI) for control means without (light grey) and with (dark grey, iii) spontaneous cardiac oedema, n = 33 experiments. Data for drugs are mean ± SEM from n = 3–14 experiments. B, Detailed heartbeat analysis at 56 hpf showing examples of normal atrioventricular coordination and frequency (i), bradicardia (ii), atrioventricular block (iii) and supraventricular arrhythmia (iv). C, Multi-dimensional scaling and clustering of abnormality profiles at 80 hpf. Axes are the sum of mean abnormalities for the respective subdomains (Table 1). Clusters: 1 (3NP); 2 (DCB); 3 (MND and 4HB); 4 (VPA, LPC and 4NB); 5 (all other drugs); k-means clustering.
Figure 8.

Mitochondrial and cytosolic biochemical pathways with sites of drug action. A, Mitochondrial respiratory chain, depicting electron flow (dashed-line arrows) from substrates to oxygen, and proton (H+) pumping across the inner mitochondrial membrane (IMM). Drugs (blue text) and inhibitory sites (grey arrows). Note that antimycin (ANT) binds Qi, downstream of a potential electron exit site promoting superoxide (O2–) formation, whereas both myxothiazol (MYX) and atovaquone (ATV) bind the upstream Qo in complex III. B, Mitochondrial and cytosolic dehydrogenases, capable of reducing ubiquinone (UQ) or its analogues idebenone (IDB) and decylubiquinone (DCB). Complex I, NADH dehydrogenase; Complex II, succinate dehydrogenase; ETFDH, electron-transferring-flavoprotein (ETF) dehydrogenase; GPDHm, glycerol-3-phosphate dehydrogenase (mitochondrial); DHODH, dihydroorotate dehydrogenase; NQO1, NAD(P)H: quinone oxidoreductase. C, Lipid metabolism and other metabolic pathways feeding dehydrogenases (ETFDH, GPDHm and DHDOH) that can reduce ubiquinone, thus being alternatives to Complex I and II. D, Ubiquinone, melanin and haemoglobin biosynthesis. Note that: 4-hidroxibenzoate (4HB) is a precursor for UQ synthesis, inhibited by ATV and 4-nitrobenzoate (4NB); NQO1 promotes (white arrows) melanin synthesis, increases and decreases β-lapachone (βLPC) and menadione (MND) toxicity, respectively. Dicoumarol (DCM) and lapachol (LPC) inhibit NQO1; Haem synthesis is critically dependent on mitochondria and its membrane potential (Δψm). [a]–[r] are literature references for the biochemical pathways and drug effects: [a] (Earley et al., 1987); [b] (Krungkrai, 2004); [c] (Dong et al., 2009); [d] (Brière et al., 2004); [e] (Huang et al., 2006); [f] (Gonzalez-Aragon et al., 2007); [g] (Mather et al., 2007); [h] (Devenish et al., 2000); [i] (Haefeli et al., 2011); [j] (Höltta-Vuori et al., 2010); [k] (Knecht et al., 2000); [l] (Bentinger et al., 2010); [m] (Kaneshiro et al., 2000); [n] (Forsman et al., 2010); [o]- (Li et al., 2011); [p] (Kishi et al., 2002); [q] (Choi et al., 2010); [r] (Preusch, 1986).
Figure 1.

Mitochondrial inhibitors and zebrafish development. A, Protocol and images of normal development. B–G, Changes in normal (white circles), abnormal (grey triangles), dead (black circles) and hatched (white diamonds) embryos (% total) across time and drug concentrations. B, Controls – egg water (i), with 0.5% DMSO (ii) and 0.05% methanol (iii, MeOH). C–G, Mitochondrial inhibitors. Data are mean ± SEM of n independent experiments: control: n = 34, 4 and 4, for egg water, DMSO and MeOH, respectively; rotenone, n = 4–14; 3NP, n = 3–9; myxothiazol, n = 4–12; antimycin, n = 5–6; oligomycin, n = 3–5. C-Giii, Arrows – peak abnormalities at 80 hpf.
Egg production
Adult wild-type zebrafish (Da. rerio), were maintained at 28 ± 1°C on a 14 h:10 h light:dark cycle, and handled for egg production as we previously detailed (Soares et al., 2009). Briefly, adults (14:7 male:female) were placed in a 30 L breeding tank on the day before egg collection. Ninety minutes after starting the light period, eggs were collected and cleaned. This time point was recorded as 0 h post fertilization (hours per fertilization, hpf; Figure 1A).
Chronic drug exposure assays
Embryo maintenance and drug treatment
Embryos were randomly distributed in 12-well plates (10 embryos/well; 2 mL egg water/well) and kept at 28 ± 1°C on a 14 h:10 h light:dark cycle throughout the assay. Embryos were continuously exposed to drugs from 4 to 80 hpf. VPA, a known teratogen in zebrafish, was used as positive control for developmental abnormalities and also cardiovascular abnormalities such as bradycardia (Gurvich et al., 2005). Dead embryos were removed during the readings (Figure 1A) and two out of three of each well content was renewed with freshly prepared drug solution at 32 and 56 hpf. All drug concentrations were assayed in duplicate wells (10 + 10 eggs), in at least three independent experiments (≥60 eggs).
Monitoring of zebrafish development and cardiac function
At regular time points (8, 32, 56 and 80 hpf: Figure 1A) embryos were scored as normal, abnormal or dead; hached/unhatched; using a stereomicroscope (Stemi DV4, Zeiss, Göttinger, Germany) or an inverted microscope (Eclipse TS 100, Nikon, Tokyo, Japan) with colour camera (Nikon 8400; Tokyo, Japan) for digital recording. Normal development was as previously described (Kimmel et al., 1995). Abnormalities were organized as: pigmentation, cardiac and structural – for subdivisions description and photographs see Table 1 and Figure 2A. Heart rates (Figure 5A) were measured in two embryos per phenotype per well, during 20 s, at 80 hpf. Weighted averages (Figure 5Ai) consider the embryos with/without cardiac oedema among the live population per well. For detailed heartbeat analyses (Figure 5B), videos were recorded at 20× magnification in an inverted microscope (Eclipse TE300, Nikon), with CCD camera (C6790; Hamamatsu Photonics, Hamamatsu, Japan) and Aquacosmos software (version 2.5; Hamamatsu Photonics). Videos were captured at 28 Hz during 10 s and processed with ImageJ (NIH) for assessing atrioventricular coordination.
Table 1.
Description of zebrafish abnormalities. Criteria for scoring zebrafish as abnormal or dead, and respective reading times
| Pigmentation | Cardiac abnormalities | Structural abnormalities | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Melanin | Anaemia | Oedema | Asystole | Lesions | Tail | Head | Gastrula | Dead | |
| Description | Decreased pigmentation | Hypochromic anaemia | Swelling in cardiac region and/or in yolk sac | Without heart beat and circulation | Necrosis – dark cell tissues Local clots or bleeding – accumulation of erythrocytes | Skeletal abnormalities – lordosis, kyphosis and other which cause tail curvature Abnormal caudal fins | Abnormal craniofacial structure Abnormal eyes Abnormal otoliths | Gastrulation arrest, without tissue decomposition | Generalized necrosis and disintegrated tissues |
| Reading time | ≥32 hpf | ≥56 hpf | ≥32 hpf | ≥56 hpf | ≥32 hpf | ≥32 hpf (except fins: ≥80 hpf) | ≥32 hpf (except otoliths: ≥80 hpf) | ≥32 hpf | ≥8 hpf |
Figure 2.

Mitochondrial inhibitors and abnormalities in zebrafish. A, Representative abnormalities in comparison with post- and pre-hatched control embryos (top and bottom left, respectively) at 80 hpf. B, Spontaneous abnormalities under control conditions; C–G, Mitochondrial inhibitor induced abnormalities at the indicated concentrations (arrows in Figure 1C-Giii); Boxplots (specific abnormalities, % total embryos) show median, mean (+), interquartile distances, maximum and minimum; Symbols (normal, white circles; abnormal, grey triangles; and dead; black circles) show mean ± SEM (% total, embryos). Data are from n independent experiments: control: n = 34; rotenone (RTN), n = 14; 3-nitropropionic acid (3NP), n = 9; myxothiazol (MYX), n = 12; antimycin (ANT), n = 5; oligomycin (OLG), n = 5. *P < 0.05 in boxplots, Kruskal–Wallis ANOVA with Dunn's post hoc (vs. control). *P < 0.05 in grey triangles (mean abnormalities), One-way ANOVA with Bonferroni post hoc (vs. control).
Parameters and abnormality indexes
NC50, AC25 and LC50 signify concentrations required for, respectively, 50% decrease in normal embryos, 25% of embryos with abnormalities and 50% lethality. LS and NS are slopes from concentration-response curves depicting dead and normal embryos respectively. Abnormality indexes were expressed by LC50/NC50 and LS/-NS ratios. Both are ∼1 when increasing drug concentrations convert normal embryos directly into dead embryos. When abnormal embryos survive, the LC50/NC50 ratio is >1. The LS/-NS can still be ∼1, but otherwise is inversely proportional to the resilience of abnormal embryos to increasing drug concentrations. Indeed, an LS steeper or shallower than -NS signifies a more or less abrupt abnormal-to-dead conversion respectively.
Acute drug exposure assays
Hatched embryos at 56 hpf were randomly distributed in 12-well plates (10 embryos in 2 mL egg water/well). Following acute exposure to isolated or combined drugs, embryos were scored for presence/absence of circulation and heartbeat at 10 min intervals. For each treatment, 40–60 embryos, from two to three independent clutches, were assayed in two to three independent experiments, before data aggregation for Kaplan–Meier analysis.
Data sources and comparison of protein sequences
Protein sequence data of NADH dehydrogenase subunit 1 (ND1, complex I), succinate dehydrogenase complex subunit A (complex II), cytochrome b (complex III) and ATPase subunit 6 and 9 (complex V) were obtained from NCBI (http://ncbi.nlm.nih.gov/protein). Percentages of identity were calculated using BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). COBALT (http://www.ncbi.nlm.nih.gov/tools/cobalt) was used to align cytochrome b sequences (Figure 3).
Figure 3.
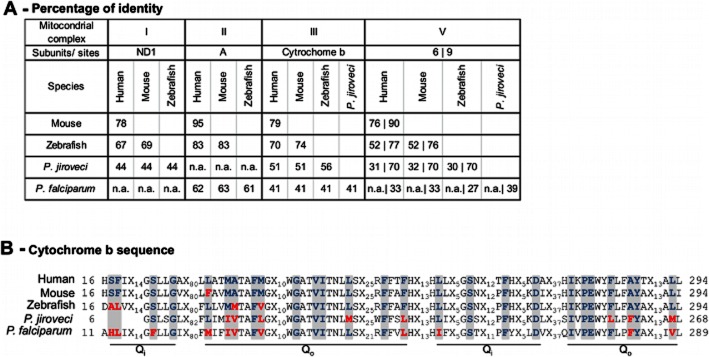
Amino acid sequence comparison of mitochondrial inhibitor-binding subunits. A, Percentage of identity among inhibitor-binding subunits of mitochondrial complexes I, II, III and V (NADH dehydrogenase subunit 1, ND1; succinate dehydrogenase subunit A, cytochrome b, and ATPase subunits 6 and 9), from human, mouse, zebrafish and parasites targetable with quinone analogues (P. jiroveci and P. falciparum; ‘n.a.’, not applicable – no homologue subunit in this species). B, Comparison of Qo and Qi sites (grey highlight). Xn summarises non-Qo/Qi amino acids.
Statistics
Values are mean ± SEM from n independent experiment, unless otherwise stated. Conventional ‘E notation’ is used when appropriate. For normally distributed data, t-test or ANOVA were used, respectively, to compare two or more groups. The Mann–Whitney test and Kruskal–Wallis ANOVA were used for non-normally distributed data. Changes in proportion of embryos with/without circulation or heartbeat were analysed via Kaplan–Meier. Cluster analysis was performed by k-means clustering, preceded by exploratory hierarchical clustering. Linear regressions (for computing NC50, AC25, LC50, and slopes – NS, LS) and non-parametric tests were performed with GraphPad Prism version 5.0 (San Diego, CA, USA). All other statistical analyses were performed with SPSS 20.0 (SPSS Inc., Chicago, IL, USA). Differences from control were considered statistically significant when P < 0.05. Simultaneous biological relevance was assumed only for differences larger than 10% (Δ > 10%).
Results
Mitochondrial inhibitors induce different abnormalities in zebrafish
To assess how mitochondrial dysfunction affects zebrafish development, fertilized eggs were chronically exposed to different mitochondrial inhibitors. Protocol and concentration-response curves are in Figure 1 and representative abnormalities (Figure 2A) described in Table 1.
Complex I and II inhibitors (rotenone and 3NP, respectively) induced more abnormalities in surviving embryos than complex III inhibitors (myxothiazol and antimycin), which primarily induced a direct transition from normal to dead. The ATP synthase inhibitor (oligomycin) arrested surviving embryos at the gastrula stage, followed by a time-dependent death (Figure 1C-G and Figure 2B-G). The order of potency for lethality was antimycin > myxothiazol > rotenone > oligomycin > 3NP (LC50 in Table 2). Comparing LC50/NC50 and LS/-NS ratios at 80 hpf (Table 2) highlights 3NP as the most frequent inducer of abnormalities among the mitochondrial inhibitors (highest LC50/NC50 and lowest LS/-NS; AC25 = 3.4E3 ± 1.7E2μM, n = 4), followed by rotenone, myxothiazol, antimycin and oligomycin. At 56 hpf, oligomycin stands out as an abnormality inducer, but the corresponding embryos do not survive until 80 hpf (Table 2, Figure 2G).
Types and frequency of abnormalities varied across mitochondrial inhibitors (Figure 2B-G). Rotenone and 3NP significantly decreased pigmentation (melanin) at 56 hpf suggesting developmental delay, and induced cardiovascular abnormalities (oedema and asystole); 3NP also caused necrotic lesions and head abnormalities (abnormal otoliths); Figure 2C,D. Oligomycin-induced abnormalities were primarily arrests at the gastrula stage, lethal at 80 hpf (Figure 2G). Myxothiazol and antimycin evoked no statistically significant abnormalities (Figure 2E,F), consistent with direct transition from normal to dead (Figure 1E,F).
Heart rate analysis showed that bradycardia and cardiac oedema were strongly associated (Figure 5Aii vs. Aiii; with few exceptions). Control heart rate was 179 ± 3 beats per minute (bpm) and 163 ± 5 bpm, for embryos without and with spontaneous oedema respectively (Figure 5Aiii). Embryos with oedema caused by rotenone, 3NP, myxothiazol or oligomycin, all displayed concentration-dependent bradycardia, more pronounced than that caused by spontaneous oedema in controls (Figure 5Aiii). In weighted average, however, only 3NP caused relevant bradycardia among the mitochondrial inhibitors (Figure 5Ai), consistent with its higher proportion of oedema (Figure 2Diii). Also, cluster analysis and scaling across three domains (cardiac, structural and pigmentation) clearly separated 3NP from all other mitochondrial inhibitors (Cluster 5; Figure 5C).
Comparison of mitochondrial complex subunits between zebrafish, human, mouse and parasites targetable with quinone analogues
To appraise zebrafish for mitochondrial dysfunction modelling and drug testing, we compared inhibitor-binding subunits for rotenone (Earley et al., 1987), 3NP (Huang et al., 2006), myxothiazol, antimycin, atovaquone (Mather et al., 2007) and oligomycin (Devenish et al., 2000) – (Figure 3A,B). Pneumocystis jiroveci (Hughes et al., 1990) and Plasmodium falciparum (Fry and Pudney, 1992) are included because of relevant cytochrome b differences allowing selective targeting with atovaquone (Fry and Pudney, 1992; Mather et al., 2007). Zebrafish identity with human subunits is only 9–13% lower than mouse versus human, except for ATPase subunit 6 (Figure 3A). Our LC50 for mitochondrial inhibitors in zebrafish were lower than LC50 or concentrations fully inhibiting mammalian mitochondrial complexes, except 3NP where only its AC25 approximated the LC50 reported for mammalian cells (Table 2). The genetic and functional correlation is evidenced by ND1 and complex V subunit 6 absence in Pl. falciparum versus zebrafish/mammals, and the associated 10-fold higher concentrations required for rotenone or oligomycin to kill Pl. falciparum versus zebrafish embryos in this study (Table 2). Differences in cytochrome b Qo (binds myxothiazol/atovaquone) and Qi (binds antimycin) (Figure 3B) are functionally relevant since antimycin displays >1000-fold lower LC50 towards zebrafish/mammals versus parasites. Therapeutically more relevant, atovaquone displays much higher toxicity towards Pl. falciparum versus zebrafish/mammals (Table 2).
Quinone analogues and other ubiquinone-related compounds induce different abnormalities in zebrafish
Aiming to validate zebrafish as in vivo model for exploring the therapeutic potential of quinone analogues, we tested their effects in chronically (4–80 hpf) exposed embryos, along with other drugs for mechanistic studies (Supporting Information Figure S1). All 11 quinone analogues were lethal within the tested range of concentrations (Table 2). The LC50 interval 0.5–1 μM includes PLB, DQN and JGL; 1–10 μM includes ATV, βLPC, DPR, MND, NTZ, IDB and LPC. The least lethal of all quinone analogues was decylubiquinone (DCB; LC50 at 80 hpf = 20.10 ± 0.74 μM, n = 3). Other drugs exhibited an LC50 comprised between 21 μM and 4 mM (DCM, VPA and 4NB); The LC50 for 4HB was >5 mM, its AC25 at 80 hpf was 1.3 ± 0.66 mM, n = 5. Relatively few (4:11) quinone analogues induced a significant proportion of structural/functional abnormalities (ATV, LPC, MND and DCB; Table 2, LC50/NC50>1; Figure 4). Among the ‘other drugs’, 4NB, DCM and VPA exhibited an LC50/NC50>1 at either 56 or 80 hpf (Table 2 and Figure 4).
Figure 4.

Influence of quinone analogues and ubiquinone related compounds on zebrafish development. A-I, i, iii, Mean ± SEM of changes in normal, abnormal, dead and hatched embryos (% total) for the indicated times (56 and 80 hpf) and drug concentrations; ii, iv, Boxplots (specific abnormalities, % total embryos) show median, mean (+), interquartile distances, maximum and minimum, for the indicated concentrations (arrow; peak abnormalities at 80 hpf). Data are from n independent experiments with: A, atovaquone, n = 3–12; B, decylubiquinone, n = 4–8 C, idebenone, n = 3–8; D, 4NB, n = 3–11; E, 4HB, n = 4–16; F, lapachol, n = 3–5; G, dicoumarol, n = 3–8; H, menadione, n = 3–15; and I, β-lapachone, n = 3–10. *P < 0.05, Mann–Whitney (vs. control) if difference in medians >10%. J, Effect of dicoumarol [DCM; NQO1 inhibitor, (Preusch et al., 1991)] upon abnormalities induced by menadione (MND) or β-lapachone (βLPC) at 80 hpf. P > 0.05 for differences between MND/βLPC alone versus combined with 50 or 500 nM DCM, one-way ANOVA with Bonferroni post hoc, n = 5–6. K, Periods of menadione (1 μM) exposure and resulting mean ± SEM of embryos with hypochromic anaemia (% live, 80 hpf). *P < 0.05 versus 48–80hpf exposure, ANOVA with Bonferroni post hoc, n = 3–4 independent experiments. The periods of primitive, transient and definitive haematopoiesis were derived from (Chen and Zon, 2009).
Atovaquone induced abnormalities were primarily cardiac oedema and focal lesions (e.g. bleedings/clots), most noticeable at 1 μM and 56 hpf (some lethal at 80 hpf; Figure 4A). Meaningfully, although atovaquone shares complex III inhibition with myxothiazol and antimycin (Figure 8A), it induces different abnormalities (Figure 2E,F vs. Figure 4A), which may be better explained by atovaquone's inhibition of ubiquinone synthesis (Figure 8D). Consistently, the ubiquinone synthesis inhibitor 4NB replicated distinguishing atovaquone features, such as focal lesions and pronounced cardiac oedema without relevant bradycardia (Figure 4A vs. Figure 4D; Figure 5Aiii).
Decylubiquinone's lethality was strikingly time-dependent increasing about 90% from 56 to 80 hpf (Figure 4B). Decylubiquinone caused a tail fin anomaly (Figure 2A; ∼50% embryos at 10 μM; Figure 4Eiv, 80 hpf), also observed within 90 min of exposure in hatched embryos (experiments in Figure 7A), suggesting altered neuromuscular tonus and not necessarily developmental defects. Interestingly, the structurally related idebenone (terminal –OH in decylubiquinone's aliphatic chain; Supporting Information Figure 1) displayed higher lethality (∼3× lower LC50; Table 2) and no significant abnormalities other than delayed pigmentation (melanin; Figure 4C).
Figure 7.
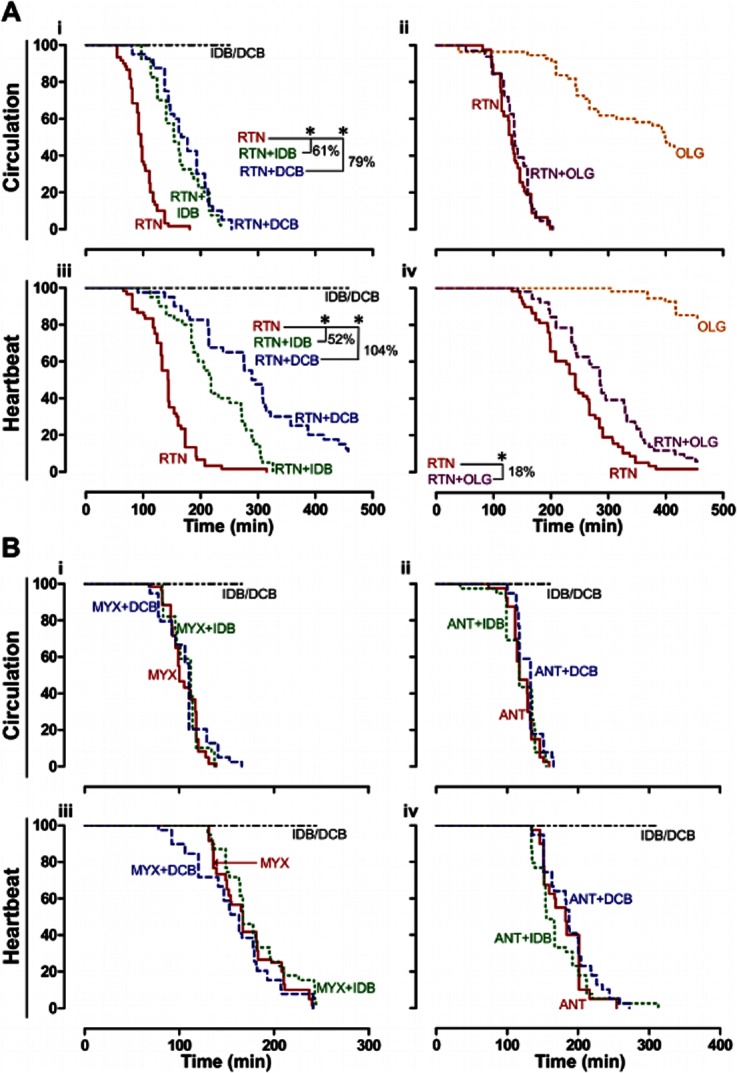
Effect of ubiquinone analogues upon acute mitochondrial inhibition. A,B, Time-dependent changes in proportion of 56 hpf zebrafish with active circulation (i,ii) and heartbeat (iii,iv) following acute exposure to mitochondrial inhibitors (at time = 0 min). Ai,iii, Rotenone (RTN, 1 μM) with/without idebenone (IDB, 3 μM) or decylubiquinone (DCB, 30 μM); Aii,iv, RTN (1 μM) and oligomycin (OLG, 3 μM), alone and combined. Bi-iv, Myxothiazol (MYX, 1 μM) or antimycin (ANT, 0.1 μM), with/without IDB (3 μM) or DCB (30 μM). *P < 0.05 and % delay in circulatory arrest or assystole; Kaplan-Meier with Log Rank. Data are from 40–60 embryos per treatment, from 2–3 independent clutches. IDB or DCB (IDB/DCB) alone maintained 100% circulation or heartbeat throughout the experiment.
Lapachol (10 μM) induced a diverse abnormality profile afflicting ∼80% embryos at 56 hpf, precluding hatching and leading to >50% death at 80 hpf (Figure 4Fi,iii). Lapachol caused decreased pigmentation (melanin), head defects (abnormal eyes, otoliths and craniofacial defects), gastrula arrest, oedema and asystole (Figure 4Fii,iv). Asystole was typically preceded by cardiac oedema and concentration-dependent bradycardia (Figure 5A; with signs of atrioventricular block Figure 5Biii). In contrast, dicoumarol (50 μM) abnormalities were merely decreased melanin (Figure 4G).
Menadione (1 μM) was the single most powerful inducer of hypochromic anaemia among 20 compounds, affecting the majority of embryos at 80 hpf (Figure 4H). We thus investigated hypochromic anaemia, modulating the duration and developmental stage of menadione exposure. Strikingly, a short exposure from 24 to 32 hpf sufficed to evoke over 50% hypochromic anaemia at 80 hpf, whereas longer exposures at earlier (8–24 hpf) or later (48-80 hpf) stages resulted in normally coloured erythrocytes at 80 hpf (Figure 4 K).
Rescue experiments with ubiquinone analogues or precursor during chronic mitochondrial inhibition
Idebenone and decylubiquinone fail to rescue chronic mitochondrial inhibition
Following individual drug titration, we tested whether ubiquinone analogues rescued the effects of chronic mitochondrial inhibition in zebrafish. Embryos were chronically exposed to concentrations inducing the highest percentage of abnormalities at 80 hpf for rotenone (0.1 μM), 3NP (10 mM), myxothiazol (0.03 μM) and atovaquone (1 μM), in the presence or absence of the highest concentration of ubiquinone analogues without detectable chronic toxicity (1 μM idebenone; 3 μM decylubiquinone). Neither idebenone nor decylubiquinone rescued changes in number of normal embryos, hatching, heart rate or oedema induced by chronic mitochondrial inhibition (Figure 6A).
Figure 6.

Effect of ubiquinone analogues and precursor upon chronic mitochondrial inhibition. A,B, Normal embryos (i), hatching (ii), and average heart rate (iii; asystole included) in % of control, as well as oedema in surviving embryos (% live; iii); Readings at 80 hpf, following chronic exposure to the indicated concentrations of drugs isolated vs. combined with idebenone (IDB, 1 μM, A) or decylubiquinone (DCB, 3 μM, A), or with 4-hydroxybenzoate (4HB, 5 μM, B). Data are mean ± SEM on n independent experiments with rotenone (RTN, 0.1 μM, n = 4), 3-nitropropionic acid (3NP, 10 mM, n = 3), myxothiazol (MYX, 0.03 μM, n = 5; 0.1 μM, n = 6), atovaquone (ATV, 1 μM; n = 6, A; n = 6, B), antimycin (ANT, 3 nM, n = 3), and 4-nitrobenzoate (4NB, 10 mM, n = 4). *P < 0.05 for combined vs. isolated drug, unpaired t-test. C, Normal, abnormal and dead embryos upon chronic exposure to 3NP (10 mM), alone or combined with RTN (0.1 μM). Data are mean ± SEM for n = 3 independent experiments, with readings at 32 (i), 56 (ii) and 80 hpf (iii). Note that 3NP toxicity (judged from abnormal and dead embryos vs. normal) does not increase upon combination with rotenone.
Zebrafish has active alternative ubiquinone reduction pathways
Lack of rescuing from chronic complex I or II inhibition with ubiquinone analogues prompted us to test whether combined rotenone and 3NP displayed additive toxicity. Results showed that the combination of rotenone (0.1 μM) with 3NP (10 mM) does not exceed the toxicity of 3NP alone (Figure 6C). Together with development beyond the gastrula stage requiring mitochondrial ATP synthesis (oligomycin effect; Figure 2G), these results suggest that alternative ubiquinone reduction pathways actively feed complex III (Figure 8C), supporting mitochondrial ATP synthesis.
Given the possible contribution of NQO1 in the reduction of idebenone to idebenol and feeding of complex III [(Haefeli et al., 2011); Figure 8D], we appraised the contribution of this enzyme by testing how its inhibitor dicoumarol modulated toxicity of its substrates – menadione and β-lapachone. Results showed that dicoumarol (50 or 500 nM; concentrations devoid of lethality, Figure 4G) did not significantly increase the toxicity of 1 μM menadione [predicted to be detoxified by NQO1; (Kishi et al., 2002)] and did not decrease the toxicity of 3 μM β-lapachone [predicted to be bioactivated by NQO1; (Li et al., 2011)] (Figure 4J, Figure 8D).
The ubiquinone precursor 4HB rescues atovaquone but not myxothiazol or antimycin toxicity
Given the central role of ubiquinone and complex III in energizing the mitochondrial respiratory chain, we tested whether the ubiquinone precursor 4HB (5 μM) could rescue the toxicity of complex III inhibitors (Figure 6B). Although failing to rescue embryos from myxothiazol or antimycin toxicity, 4HB significantly rescued atovaquone toxicity, increasing the proportion of normal embryos (Figure 6Bi; P < 0.05, Δ > 10%) and displaying a trend towards decreased oedema (Figure 6Biii; ATV vs. ATV+4HB). 4HB failed to rescue the effects of the ubiquinone synthesis inhibitor 4NB (Figure 6B). The 2000-fold difference in concentrations required for 4NB toxicity (10 mM; Figure 4D) vs. 4HB efficacy (5 μM; Figure 6B), and the toxicity displayed by ≥5 mM 4HB (Figure 4E) precluded equimolar competition in rescue assays with 4HB and 4NB.
Rescue experiments with ubiquinone analogues upon acute mitochondrial inhibition
Acute complex I inhibition with rotenone induces cardiac insufficiency and asystole via ATP depletion
Following literature data supporting in vitro rescuing by ubiquinone analogues of decreased mitochondrial oxygen consumption or ATP levels in synaptosomes (Telford et al., 2010), cell lines (Haefeli et al., 2011) or isolated mitochondria (James et al., 2005), we aimed to establish an in vivo testing paradigm for ubiquinone analogues in zebrafish. Thus, we monitored circulation and heartbeat in 56 hpf embryos acutely challenged with mitochondrial inhibitors with or without ubiquinone analogues (idebenone or decylubiquinone; Figure 7). Circulatory arrest upon acute mitochondrial inhibition was associated with bradycardia and atrioventricular block (e.g. Figure 5Biii) or supraventricular arrhythmia (e.g. Figure 5Biv), and followed by asystole. Mitochondrial ATPase inhibition (oligomycin) evoked circulatory arrest. When combined, oligomycin did not modify rotenone's circulatory arrest (Figure 7Aii), but delayed rotenone induced asystole by 18% (Figure 7Aiv, P < 0.05). The latter can be explained by preventing ATP consumption upon rotenone induced ATPase reversal.
Idebenone and decylubiquinone delay the onset of cardiac dysfunction evoked by rotenone but not by myxothiazol or antimycin
Idebenone (3 μM) and decylubiquinone (30 μM) delayed rotenone induced cardiac insufficiency, respectively, by 61 and 79% (Figure 7Ai, P < 0.05), and also delayed asystole, respectively by 52 and 104% (Figure 7Aiii, P < 0.05). In contrast, both ubiquinone analogues failed to protect from cardiac dysfunction induced by either myxothiazol (1 μM; Figure 7Bi,iii) or antimycin (100nM, Figure 7Bii,iv).
Discussion and conclusions
Modelling mitochondrial dysfunction in zebrafish
Mitochondrial respiratory chain and inhibitors
Here, we show that zebrafish and mammalian mitochondria display high genetic and functional homology, and characterize the developmental and cardiovascular consequences of mitochondrial dysfunction evoked by respiratory chain inhibitors. Chronic complex I or II inhibition induced developmental abnormalities, whereas complex III inhibition directly converted normal into dead embryos, plausibly by lack of downstream alternatives to sustain mitochondrial ATP synthesis. Interestingly, when combining effective concentrations of rotenone and 3NP, lack of additive effects suggests a negligible residual complex I activity during complex II inhibition and vice versa. Yet, albeit abnormal, embryos undergo organogenesis and survive past 80 hpf. Because this cannot happen without mitochondrial ATP synthesis (oligomycin arrests gastrulation), data suggests alternative mitochondrial dehydrogenases [ETFDH, glycerol-3-phosphate dehydrogenase or dihydroorotate dehydrogenase (DHODH); Figure 8] actively reduce endogenous ubiquinone to support mitochondrial ATP production. This is important for interpreting zebrafish models of complex I or II deficiencies, for example in primary mitochondrial disease or in neurodegenerative disorders which have been associated with deficits in complexes I or II, such as Parkinson and Huntington's diseases respectively. Specifically, alternative feeding of complex III may explain why parkin knockout zebrafish presented 45% reduction of complex I activity without significant changes in swimming behaviour (Flinn et al., 2009).
While the concentrations we have used for mitochondrial inhibitors are within the range commonly used in the literature, it is conceivable that in zebrafish, as with other models, there may be some contribution of off-target effects, such as microtubule depolymerization by rotenone (Choi et al., 2011), and fumarase inhibition by 3NP (Porter and Bright, 1980).
Cytosolic dehydrogenase: NQO1
In contrast with mitochondrial dehydrogenases, cytosolic NQO1 unlikely feeds complex III physiologically, since ubiquinone hydrophobicity precludes significant cytosolic-mitochondria shuttling. Pharmacologically, however, NQO1 may be quite relevant in reducing exogenous ubiquinone analogues (Haefeli et al., 2011; Giorgio et al., 2012). Indeed, exogenous idebenone or decylubiquinone must travel the cytosol, where they may be reduced by NQO1, before feeding electrons to mitochondrial complex III. NQO1 inhibition was reported to decrease melanogenesis in zebrafish and melanoma cell lines (Choi et al., 2010). Here, we tested the NQO1 inhibitors dicoumarol and lapachol and both caused melanin defects in zebrafish embryos. Interestingly, lapachol was more potent than dicoumarol for melanin defects and presented a wider spectrum of abnormalities. Considering the higher NQO1 inhibiting potency of dicoumarol versus lapachol [IC50 = 10nM vs. 150nM, respectively; (Preusch, 1986; Preusch et al., 1991)], we argue that effects may instead be mediated by DHODH inhibition, where lapachol was shown more potent than dicoumarol [IC50 = 61nM vs. 5 μM, respectively; (Knecht et al., 2000; Gonzalez-Aragon et al., 2007)]. Together, these findings suggest that the overall level of NQO1 activity during early zebrafish development is low, in contrast with that of DHODH. Consistently, dicoumarol failed to modulate chronic toxicity of menadione or β-lapachone suggesting they are not being significantly detoxified or bioactivated by NQO1. Still, specific organs like the heart may exhibit higher NQO1 activity, as addressed below.
Cardiovascular abnormalities and ubiquinone
Mitochondrial respiratory chain dysfunction and ubiquinone deficiency disorders are frequently associated with cardiovascular manifestations (Berardo et al., 2011). Consistently, we show that mitochondrial inhibitors induce cardiac oedema associated with bradycardia and arrhythmia in zebrafish. Interestingly, we identified an unusual cardiovascular phenotype for 4NB and atovaquone, consisting in focal bleeding and necrosis, with pronounced cardiac oedema but normal heart rate. Meaningfully, 4NB inhibits ubiquinone biosynthesis (Forsman et al., 2010), atovaquone was suggested to cause feedback inhibition of ubiquinone biosynthesis (Kaneshiro et al., 2000), and the ubiquinone precursor 4HB reduces atovaquone but not myxothiazol or antimycin toxicity. Therefore, data suggests that this unusual cardiovascular phenotype reflects ubiquinone deficiency, and may thus assist future study of related disorders in zebrafish.
In vivo testing of quinone analogues
Quinone analogues as research tools and chemotherapeutics
Quinone analogues are widespread in nature, playing key roles in electron transport chains and in interspecies chemical warfare. Their ability to interfere with biological processes stems from redox properties, and has promoted their use as research tools and as putative anticancer or antiparasitic drugs (O'Brien, 1991).
Menadione is a research tool for inducing oxidative stress, increasing mitochondria superoxide by redox cycling (Johnson-Cadwell et al., 2007). While sometimes used as precursor for vitamin K, menadione may induce haemolysis in patients lacking glucose-6-phosphate dehydrogenase (Roberts-Harewood, 2009). Here, we report that menadione causes hypochromic anaemia in zebrafish when present between 24 and 32 hpf, following primitive erythropoiesis (Chen and Zon, 2009), but not when present for longer subsequent periods. Hence, instead of acute haemolysis, data suggests that menadione impairs zebrafish haematopoiesis, possibly by depolarizing mitochondria via redox-cycling or complex I inhibition (Dong et al., 2009) and disturbing membrane potential-dependent biosynthesis of the iron-sulfur cluster haem (Figure 8D) (Ye and Rouault, 2010). Thus, we suggest that menadione may be a useful research tool for studying erythropoiesis and its mitochondrial dependence in zebrafish.
Lapachol and β-lapachone are putative anticancer lead compounds, with their presence in plant extracts (e.g. Tabebuia impetiginosa) being associated with cytotoxicity against multiple cell lines (Gomez Castellanos et al., 2009). Similar reasoning applies to diospyrin, diosquinone, juglone and plumbagin. Here, we show that the LC50 of these quinone analogues in developing zebrafish is similar or even smaller than reported for cancer cells. Considering the similarities between cellular divisions in a developing embryo and in cancer cells, results suggest caution in interpreting data from zebrafish embryos whilst testing for selective anticancer toxicity.
Atovaquone is used therapeutically against Pl. falciparum malaria and Pn. jiroveci pneumonia. Selective toxicity for parasites versus humans is achieved by structural differences in atovaquone's target – the cytochrome b Qo site in complex III (Kessl et al., 2003). Here, we report a high amino acid homology of zebrafish, mice and humans at the Qo and Qi sites, confirming predicted functional differences by comparing LC50 of atovaquone, myxothiazol and antimycin with that reported for parasites. Together, data supports using in vivo zebrafish assays for comparative toxicity of mitochondria-targeted quinone analogues, namely in antimalaric drug research.
Ubiquinone analogues and treatment of mitochondrial dysfunction
Ubiquinone analogues are among the few specific therapeutic options for mitochondrial diseases (Napolitano et al., 2000). Idebenone improved cardiac function in Friedrich ataxia (Rustin et al., 1999), with higher doses required for improving neurological function (Di Prospero et al., 2007). Idebenone effects have been ascribed to antioxidant properties (Geromel et al., 2002). Further, idebenone can mimic ubiquinone, transferring to complex III the electrons received from mitochondrial dehydrogenases, thus promoting mitochondrial ATP production (Brière et al., 2004). Although ubiquinone analogues have failed to rescue ubiquinone deficient cells (Lopez et al., 2010), there are also evidences that idebenone's ability to rescue ATP levels correlates with NQO1 activity (Haefeli et al., 2011), promoting idebenone reduction and subsequent complex III feeding. Current support for this hypothesis stems from in vitro evidence, using cells or isolated mitochondria and predictive parameters such as ATP levels and oxygen consumption. Here, we explore this hypothesis in vivo, using zebrafish and cardiovascular parameters, testing the ability of ubiquinone analogues to prevent/delay cardiac insufficiency following induced mitochondrial dysfunction.
Idebenone and decylubiquinone significantly delayed cardiac insufficiency and asystole evoked by acute rotenone exposure. However, both drugs were ineffective against chronic rotenone exposure, as ubiquinone analogues clearly cannot sustainably protect from chronic multi-organ toxicity. The mechanisms of cardioprotection by idebenone and decylubiquinone in our zebrafish model are more likely related to increased ATP production efficiency than to antioxidant or reactive oxygen species (ROS) scavenging properties. Indeed, these ubiquinone analogues were without effect against both acute and chronic complex III inhibition, particularly with antimycin that is also an inducer of mitochondrial ROS production (Chandel et al., 2000). Thus, transient cardiac protection from acute rotenone exposure in zebrafish might be explained by higher NQO1 activity in the heart [as in mammals: (Palming et al., 2007; Zhu and Li, 2012)], allowing enhanced local ubiquinone analogue reduction (Haefeli et al., 2011; Giorgio et al., 2012), and consequently more feeding of complex III and ATP production in the heart.
In future studies of mitochondrial dysfunction in zebrafish, it would be valuable to further explore ATP/ADP ratios as well as ROS production in this model organism. Significantly, Mendelsohn et al. (2008) have monitored AMP/ATP by HPLC and total ATP by luciferase assays in zebrafish. Also, Niethammer et al. (2009) have successfully used a genetically encoded H2O2 sensor in zebrafish.
Concluding remarks
In summary, the present work supports zebrafish for studying mitochondrial dysfunction and testing mitochondria-targeted treatments. Our reported developmental and cardiovascular phenotypes should assist further research on mitochondrial diseases in this model organism. The present data, on mitochondrial sequence analysis and atovaquone effects, highlights zebrafish's potential for the in vivo differential toxicity screening of mitochondria-targeted antiparasitic drugs. Further, our in vivo assay identifying a delay of cardiac failure by idebenone and decylubiquinone may assist comparisons of other ubiquinone analogues, currently being developed as mitochondria-targeted drugs. Data on other quinone analogues (especially menadione) should assist their use as research tools. Lastly, our data showing a relatively low dependence of young zebrafish on mitochondrial complex I/II for ATP production should help interpret phenotypes of disease models linked to complex I/II inhibition.
Acknowledgments
This work was supported by Fundação para a Ciência e a Tecnologia (FCT), Strategic Project: PEst-C/EQB/LA006/2011, and by Research Grants (PI: J. M. A. O.) PTDC/NEU-NMC/0237/2012 (FCT), FCOMP-01-0124-FEDER-029649 (COMPETE), and PPII_CARDIAC and PPII_ZEBRA (Universidade do Porto and Santander-Totta). B. P. is grateful to FCT for her PhD grant (SFRH/BD/63852/2009).
Glossary
- 3NP
3-nitropropionic acid
- 4HB
4-hydroxybenzoic acid
- 4NB
4-nitrobenzoic acid
- ATV
atovaquone
- bpm
beats per minute
- COX
cytochrome c oxidase
- DCB
decylubiquinone
- DCM
dicoumarol
- DHODH
dihydroorotate dehydrogenase
- DPR
diospyrin
- DQN
diosquinone
- ETFDH
electron transfer flavoprotein dehydrogenase
- hpf
hours post fertilization
- IDB
idebenone
- JGL
juglone
- LPC
lapachol
- mDNA
mitochondrial DNA
- MND
menadione
- ND1
NADH dehydrogenase subunit 1
- NQO1
NAD(P)H:quinone oxidoreductase
- NTZ
naphthazarin
- PLB
plumbagin
- ROS
reactive oxygen species
- VPA
valproic acid
- βLPC
β-lapachone
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Drug categories and structures.
Appendix S1 References of Table 2.
References
- Artuso L, Romano A, Verri T, Domenichini A, Argenton F, Santorelli FM, et al. Mitochondrial DNA metabolism in early development of zebrafish (Danio rerio. Biochim Biophys Acta. 2012;1817:1002–1011. doi: 10.1016/j.bbabio.2012.03.019. [DOI] [PubMed] [Google Scholar]
- Azzolin L, Basso E, Argenton F, Bernardi P. Mitochondrial Ca2+ transport and permeability transition in zebrafish (Danio rerio. Biochim Biophys Acta. 2010;1797:1775–1779. doi: 10.1016/j.bbabio.2010.07.002. [DOI] [PubMed] [Google Scholar]
- Baden KN, Murray J, Capaldi RA, Guillemin K. Early developmental pathology due to cytochrome c oxidase deficiency is revealed by a new zebrafish model. J Biol Chem. 2007;282:34839–34849. doi: 10.1074/jbc.M703528200. [DOI] [PubMed] [Google Scholar]
- Bentinger M, Tekle M, Dallner G. Coenzyme Q-biosynthesis and functions. Biochem Biophys Res Commun. 2010;396:74–79. doi: 10.1016/j.bbrc.2010.02.147. [DOI] [PubMed] [Google Scholar]
- Berardo A, Musumeci O, Toscano A. Cardiological manifestations of mitochondrial respiratory chain disorders. Acta Myol. 2011;30:9–15. [PMC free article] [PubMed] [Google Scholar]
- Bretaud S, Lee S, Guo S. Sensitivity of zebrafish to environmental toxins implicated in Parkinson's disease. Neurotoxicol Teratol. 2004;26:857–864. doi: 10.1016/j.ntt.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Brière JJ, Schlemmer D, Chretien D, Rustin P. Quinone analogues regulate mitochondrial substrate competitive oxidation. Biochem Biophys Res Commun. 2004;316:1138–1142. doi: 10.1016/j.bbrc.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Broughton RE, Milam JE, Roe BA. The complete sequence of the zebrafish (Danio rerio) mitochondrial genome and evolutionary patterns in vertebrate mitochondrial DNA. Genome Res. 2001;11:1958–1967. doi: 10.1101/gr.156801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- Chen AT, Zon LI. Zebrafish blood stem cells. J Cell Biochem. 2009;108:35–42. doi: 10.1002/jcb.22251. [DOI] [PubMed] [Google Scholar]
- Choi TY, Sohn KC, Kim JH, Kim SM, Kim CH, Hwang JS, et al. Impact of NAD(P)H:quinone oxidoreductase-1 on pigmentation. J Invest Dermatol. 2010;130:784–792. doi: 10.1038/jid.2009.280. [DOI] [PubMed] [Google Scholar]
- Choi W-S, Palmiter RD, Xia Z. Loss of mitochondrial complex I activity potentiates dopamine neuron death induced by microtubule dysfunction in a Parkinson's disease model. J Cell Biol. 2011;192:873–882. doi: 10.1083/jcb.201009132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa MA, Alves AC, Seabra RM, Andrade PB. Naphthoquinones of Diospyros chamaethamnus. Planta Med. 1998;64:391. doi: 10.1055/s-2006-957464. [DOI] [PubMed] [Google Scholar]
- Devenish RJ, Prescott M, Boyle GM, Nagley P. The oligomycin axis of mitochondrial ATP synthase: OSCP and the proton channel. J Bioenerg Biomembr. 2000;32:507–515. doi: 10.1023/a:1005621125812. [DOI] [PubMed] [Google Scholar]
- Di Prospero NA, Baker A, Jeffries N, Fischbeck KH. Neurological effects of high-dose idebenone in patients with Friedreich's ataxia: a randomised, placebo-controlled trial. Lancet Neurol. 2007;6:878–886. doi: 10.1016/S1474-4422(07)70220-X. [DOI] [PubMed] [Google Scholar]
- Dong CK, Patel V, Yang JC, Dvorin JD, Duraisingh MT, Clardy J, et al. Type II NADH dehydrogenase of the respiratory chain of Plasmodium falciparum and its inhibitors. Bioorg Med Chem Lett. 2009;19:972–975. doi: 10.1016/j.bmcl.2008.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley FG, Patel SD, Ragan I, Attardi G. Photolabelling of a mitochondrially encoded subunit of NADH dehydrogenase with [3H]dihydrorotenone. FEBS Lett. 1987;219:108–112. doi: 10.1016/0014-5793(87)81200-0. [DOI] [PubMed] [Google Scholar]
- Finsterer J. Treatment of mitochondrial disorders. Eur J Paediatr Neurol. 2010;14:29–44. doi: 10.1016/j.ejpn.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Flinn L, Mortiboys H, Volkmann K, Koster RW, Ingham PW, Bandmann O. Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio. Brain. 2009;132(Pt 6):1613–1623. doi: 10.1093/brain/awp108. [DOI] [PubMed] [Google Scholar]
- Forsman U, Sjoberg M, Turunen M, Sindelar PJ. 4-Nitrobenzoate inhibits coenzyme Q biosynthesis in mammalian cell cultures. Nat Chem Biol. 2010;6:515–517. doi: 10.1038/nchembio.372. [DOI] [PubMed] [Google Scholar]
- Fry M, Pudney M. Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4′-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80) Biochem Pharmacol. 1992;43:1545–1553. doi: 10.1016/0006-2952(92)90213-3. [DOI] [PubMed] [Google Scholar]
- Geromel V, Darin N, Chretien D, Benit P, DeLonlay P, Rotig A, et al. Coenzyme Q(10) and idebenone in the therapy of respiratory chain diseases: rationale and comparative benefits. Mol Genet Metab. 2002;77:21–30. doi: 10.1016/s1096-7192(02)00145-2. [DOI] [PubMed] [Google Scholar]
- Giorgio V, Petronilli V, Ghelli A, Carelli V, Rugolo M, Lenaz G, et al. The effects of idebenone on mitochondrial bioenergetics. Biochim Biophys Acta. 2012;1817:363–369. doi: 10.1016/j.bbabio.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez Castellanos JR, Prieto JM, Heinrich M. Red Lapacho (Tabebuia impetiginosa)–a global ethnopharmacological commodity? J Ethnopharmacol. 2009;121:1–13. doi: 10.1016/j.jep.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Aragon D, Ariza J, Villalba JM. Dicoumarol impairs mitochondrial electron transport and pyrimidine biosynthesis in human myeloid leukemia HL-60 cells. Biochem Pharmacol. 2007;73:427–439. doi: 10.1016/j.bcp.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Gurvich N, Berman MG, Wittner BS, Gentleman RC, Klein PS, Green JB. Association of valproate-induced teratogenesis with histone deacetylase inhibition in vivo. FASEB J. 2005;19:1166–1168. doi: 10.1096/fj.04-3425fje. [DOI] [PubMed] [Google Scholar]
- Haefeli RH, Erb M, Gemperli AC, Robay D, Courdier Fruh I, Anklin C, et al. NQO1-dependent redox cycling of idebenone: effects on cellular redox potential and energy levels. PLoS ONE. 2011;6:e17963. doi: 10.1371/journal.pone.0017963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höltta-Vuori M, Salo VT, Nyberg L, Brackmann C, Enejder A, Panula P, et al. Zebrafish: gaining popularity in lipid research. Biochem J. 2010;429:235–242. doi: 10.1042/BJ20100293. [DOI] [PubMed] [Google Scholar]
- Huang LS, Sun G, Cobessi D, Wang AC, Shen JT, Tung EY, et al. 3-nitropropionic acid is a suicide inhibitor of mitochondrial respiration that, upon oxidation by complex II, forms a covalent adduct with a catalytic base arginine in the active site of the enzyme. J Biol Chem. 2006;281:5965–5972. doi: 10.1074/jbc.M511270200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes WT, Gray VL, Gutteridge WE, Latter VS, Pudney M. Efficacy of a hydroxynaphthoquinone, 566C80, in experimental Pneumocystis carinii pneumonitis. Antimicrob Agents Chemother. 1990;34:225–228. doi: 10.1128/aac.34.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham PW. The power of the zebrafish for disease analysis. Hum Mol Genet. 2009;18(R1):R107–R112. doi: 10.1093/hmg/ddp091. [DOI] [PubMed] [Google Scholar]
- James AM, Cocheme HM, Smith RA, Murphy MP. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J Biol Chem. 2005;280:21295–21312. doi: 10.1074/jbc.M501527200. [DOI] [PubMed] [Google Scholar]
- Johnson-Cadwell LI, Jekabsons MB, Wang A, Polster BM, Nicholls DG. ‘Mild Uncoupling’ does not decrease mitochondrial superoxide levels in cultured cerebellar granule neurons but decreases spare respiratory capacity and increases toxicity to glutamate and oxidative stress. J Neurochem. 2007;101:1619–1631. doi: 10.1111/j.1471-4159.2007.04516.x. [DOI] [PubMed] [Google Scholar]
- Kaneshiro ES, Sul D, Hazra B. Effects of atovaquone and diospyrin-based drugs on ubiquinone biosynthesis in Pneumocystis carinii organisms. Antimicrob Agents Chemother. 2000;44:14–18. doi: 10.1128/aac.44.1.14-18.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessl JJ, Lange BB, Merbitz-Zahradnik T, Zwicker K, Hill P, Meunier B, et al. Molecular basis for atovaquone binding to the cytochrome bc1 complex. J Biol Chem. 2003;278:31312–31318. doi: 10.1074/jbc.M304042200. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Kishi T, Takahashi T, Mizobuchi S, Mori K, Okamoto T. Effect of dicumarol, a Nad(P)h: quinone acceptor oxidoreductase 1 (DT-diaphorase) inhibitor on ubiquinone redox cycling in cultured rat hepatocytes. Free Radic Res. 2002;36:413–419. doi: 10.1080/10715760290021261. [DOI] [PubMed] [Google Scholar]
- Knecht W, Henseling J, Loffler M. Kinetics of inhibition of human and rat dihydroorotate dehydrogenase by atovaquone, lawsone derivatives, brequinar sodium and polyporic acid. Chem Biol Interact. 2000;124:61–76. doi: 10.1016/s0009-2797(99)00144-1. [DOI] [PubMed] [Google Scholar]
- Krungkrai J. The multiple roles of the mitochondrion of the malarial parasite. Parasitology. 2004;129(Pt 5):511–524. doi: 10.1017/s0031182004005888. [DOI] [PubMed] [Google Scholar]
- Li LS, Bey EA, Dong Y, Meng J, Patra B, Yan J, et al. Modulating endogenous NQO1 levels identifies key regulatory mechanisms of action of β-lapachone for pancreatic cancer therapy. Clin Cancer Res. 2011;17:275–285. doi: 10.1158/1078-0432.CCR-10-1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez LC, Quinzii CM, Area E, Naini A, Rahman S, Schuelke M, et al. Treatment of CoQ10 deficient fibroblasts with ubiquinone, CoQ analogs, and vitamin C: time- and compound-dependent effects. PLoS ONE. 2010;5:e11897. doi: 10.1371/journal.pone.0011897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mather MW, Henry KW, Vaidya AB. Mitochondrial drug targets in apicomplexan parasites. Curr Drug Targets. 2007;8:49–60. doi: 10.2174/138945007779315632. [DOI] [PubMed] [Google Scholar]
- Mendelsohn BA, Kassebaum BL, Gitlin JD. The zebrafish embryo as a dynamic model of anoxia tolerance. Dev Dyn. 2008;237:1780–1788. doi: 10.1002/dvdy.21581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano A, Salvetti S, Vista M, Lombardi V, Siciliano G, Giraldi C. Long-term treatment with idebenone and riboflavin in a patient with MELAS. Neurol Sci. 2000;21(5 Suppl):S981–S982. doi: 10.1007/s100720070015. [DOI] [PubMed] [Google Scholar]
- Niethammer P, Grabher C, Look AT, Mitchison TJ. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. 2009;459:996–999. doi: 10.1038/nature08119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien PJ. Molecular mechanisms of quinone cytotoxicity. Chem Biol Interact. 1991;80:1–41. doi: 10.1016/0009-2797(91)90029-7. [DOI] [PubMed] [Google Scholar]
- Oliveira JM. Nature and cause of mitochondrial dysfunction in Huntington's disease: focusing on huntingtin and the striatum. J Neurochem. 2010;114:1–12. doi: 10.1111/j.1471-4159.2010.06741.x. [DOI] [PubMed] [Google Scholar]
- Palming J, Sjoholm K, Jernas M, Lystig TC, Gummesson A, Romeo S, et al. The expression of NAD(P)H:quinone oxidoreductase 1 is high in human adipose tissue, reduced by weight loss, and correlates with adiposity, insulin sensitivity, and markers of liver dysfunction. J Clin Endocrinol Metab. 2007;92:2346–2352. doi: 10.1210/jc.2006-2476. [DOI] [PubMed] [Google Scholar]
- Pei W, Kratz LE, Bernardini I, Sood R, Yokogawa T, Dorward H, et al. A model of Costeff Syndrome reveals metabolic and protective functions of mitochondrial OPA3. Development. 2010;137:2587–2596. doi: 10.1242/dev.043745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DJT, Bright HJ. 3-Carbanionic substrate analogues bind very tightly to fumarase and aspartase. J Biol Chem. 1980;255:4772–4780. [PubMed] [Google Scholar]
- Preusch PC. Lapachol inhibition of DT-diaphorase (NAD(P)H:quinone dehydrogenase. Biochem Biophys Res Commun. 1986;137:781–787. doi: 10.1016/0006-291x(86)91147-2. [DOI] [PubMed] [Google Scholar]
- Preusch PC, Siegel D, Gibson NW, Ross D. A note on the inhibition of DT-diaphorase by dicoumarol. Free Radic Biol Med. 1991;11:77–80. doi: 10.1016/0891-5849(91)90191-5. [DOI] [PubMed] [Google Scholar]
- Roberts-Harewood M. Inherited haemolytic anaemias. Medicine. 2009;37:143–148. [Google Scholar]
- Rustin P, von Kleist-Retzow JC, Chantrel-Groussard K, Sidi D, Munnich A, Rotig A. Effect of idebenone on cardiomyopathy in Friedreich's ataxia: a preliminary study. Lancet. 1999;354:477–479. doi: 10.1016/S0140-6736(99)01341-0. [DOI] [PubMed] [Google Scholar]
- Soares J, Coimbra AM, Reis-Henriques MA, Monteiro NM, Vieira MN, Oliveira JM, et al. Disruption of zebrafish (Danio rerio) embryonic development after full life-cycle parental exposure to low levels of ethinylestradiol. Aquat Toxicol. 2009;95:330–338. doi: 10.1016/j.aquatox.2009.07.021. [DOI] [PubMed] [Google Scholar]
- Song Y, Selak MA, Watson CT, Coutts C, Scherer PC, Panzer JA, et al. Mechanisms underlying metabolic and neural defects in zebrafish and human multiple acyl-CoA dehydrogenase deficiency (MADD) Plos One. 2009;4:e8329. doi: 10.1371/journal.pone.0008329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stackley KD, Beeson CC, Rahn JJ, Chan SS. Bioenergetic profiling of zebrafish embryonic development. PLoS ONE. 2011;6:e25652. doi: 10.1371/journal.pone.0025652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart JB, Freyer C, Elson JL, Wredenberg A, Cansu Z, Trifunovic A, et al. Strong purifying selection in transmission of mammalian mitochondrial DNA. Plos Biol. 2008;6:e10. doi: 10.1371/journal.pbio.0060010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telford JE, Kilbride SM, Davey GP. Decylubiquinone increases mitochondrial function in synaptosomes. J Biol Chem. 2010;285:8639–8645. doi: 10.1074/jbc.M109.079780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuppen HA, Blakely EL, Turnbull DM, Taylor RW. Mitochondrial DNA mutations and human disease. Biochim Biophys Acta. 2010;1797:113–128. doi: 10.1016/j.bbabio.2009.09.005. [DOI] [PubMed] [Google Scholar]
- Vettori A, Bergamin G, Moro E, Vazza G, Polo G, Tiso N, et al. Developmental defects and neuromuscular alterations due to mitofusin 2 gene (MFN2) silencing in zebrafish: a new model for Charcot-Marie-Tooth type 2A neuropathy. Neuromuscul Disord. 2011;21:58–67. doi: 10.1016/j.nmd.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Ye H, Rouault TA. Erythropoiesis and iron sulfur cluster biogenesis. Adv Hematol. 2010 doi: 10.1155/2010/329394. 2010: Article ID 329394. DOI:10.1155/2010/329394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Li Y. NAD(P)H: quinone oxidoreductase 1 and its potential protective role in cardiovascular diseases and related conditions. Cardiovasc Toxicol. 2012;12:39–45. doi: 10.1007/s12012-011-9136-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.