

Abstract
Background and Purpose
Synaptic vesicle protein 2A (SV2A) is the specific binding site of the anti-epileptic drug levetiracetam (LEV) and its higher affinity analogue UCB30889. Moreover, the protein has been well validated as a target for anticonvulsant therapy. Here, we report the identification of UCB1244283 acting as a SV2A positive allosteric modulator of UCB30889.
Experimental Approach
UCB1244283 was characterized in vitro using radioligand binding assays with [3H]UCB30889 on recombinant SV2A expressed in HEK cells and on rat cortex. In vivo, the compound was tested in sound-sensitive mice.
Key Results
Saturation binding experiments in the presence of UCB1244283 demonstrated a fivefold increase in the affinity of [3H]UCB30889 for human recombinant SV2A, combined with a twofold increase of the total number of binding sites. Similar results were obtained on rat cortex. In competition binding experiments, UCB1244283 potentiated the affinity of UCB30889 while the affinity of LEV remained unchanged. UCB1244283 significantly slowed down both the association and dissociation kinetics of [3H]UCB30889. Following i.c.v. administration in sound-sensitive mice, UCB1244283 showed a clear protective effect against both tonic and clonic convulsions.
Conclusions and Implications
These results indicate that UCB1244283 can modulate the conformation of SV2A, thereby inducing a higher affinity state for UCB30889. Our results also suggest that the conformation of SV2A per se might be an important determinant of its functioning, especially during epileptic seizures. Therefore, agents that act on the conformation of SV2A might hold great potential in the search for new SV2A-based anticonvulsant therapies.
Keywords: SV2A, levetiracetam, epilepsy, positive allosteric modulation, anticonvulsant
Introduction
The synaptic vesicle protein 2A (SV2) proteins belong to the Major Facilitator Superfamily (MFS) of transporters and are integral 12-transmembrane glycoproteins, specifically localized to secretory vesicles in both neurons and endocrine cells (Bajjalieh et al., 1992; Feany et al., 1992). The SV2s exist in three isoforms (A, B and C) of which SV2A is the most abundant and also the most studied one.
SV2A -/- knockout mice are characterized by a lethal seizure phenotype without any obvious abnormalities in synaptic vesicle biogenesis, supporting an important role of the protein in synaptic transmission (Crowder et al., 1999). Although, the exact function of SV2A in this process is not known, current evidence points towards an involvement of the protein in calcium-dependent exocytosis (Xu and Bajjalieh, 2001; Nowack et al., 2010; Wan et al., 2010; Yao et al., 2010), but also neurotransmitter loading/retention in synaptic vesicles (Budzinski et al., 2009; Vautrin, 2009), synaptic vesicle priming (Chang and Sudhof, 2009) and transport of vesicle constituents (Bajjalieh et al., 1994) have been suggested. The latter suggestion is based on the homology of SV2A with other MFS transporters like LacY. Transport of substrates via these transporters is believed to be mediated by a conformational switch between alternate states open to different sides of the membrane (Holyoake and Sansom, 2007; Kaback et al., 2007). Also for SV2A, the presence of two alternate conformations has been reported using protein tomography (Lynch et al., 2008). However, no endogenous SV2A transport substrate has been reported to date.
SV2A is the binding site of the anti-epileptic drug levetiracetam (LEV) (Lynch et al., 2004). The principal anticonvulsant action of LEV does not involve the classical anti-seizure mechanisms, for example GABAergic facilitation or inhibition of Na+ currents. Instead, LEV seems to have a unique mechanism of action based on SV2A binding as indicated by several lines of evidence accumulated in the recent years (reviewed in Kaminski et al., 2012). First, a strong positive correlation between SV2A binding affinity and anticonvulsant potency has been demonstrated in several epilepsy models (Lynch et al., 2004; Kaminski et al., 2008; Gillard et al., 2011). Second, seizure protection by LEV and other SV2A ligands strongly correlates with the degree of SV2A occupancy in vivo (Gillard et al., 2011). Finally, the anticonvulsant efficacy of LEV is reduced in SV2A-deficient animals (Kaminski et al., 2009). One of the functional consequences of LEV binding to the SV2A protein in brain slices was found to be a reduction in exocytosis (Yang et al., 2007). However, the underlying mechanistic effect of ligand binding is not fully elucidated. One hypothesis would be that ligand binding to SV2A would stabilize a certain conformation of the protein resulting in a potentiation or optimization of its function thereby providing seizure protection.
In a screening effort to search for new SV2A ligands, we identified a positive allosteric modulator (PAM), UCB1244283, which strongly potentiated the affinity of UCB30889, a higher affinity analogue of LEV, for SV2A. Subsequently, UCB1244283 was used to investigate the implications of allosteric modulation on SV2A-ligand binding. Finally, we made an initial assessment of the potential of SV2A positive allosteric modulation as a possible anticonvulsant therapy using the audiogenic mouse model.
Methods
Radioligands, drugs and chemicals
LEV (2S-(2-oxo-1-pyrrolidinyl)butanamide), UCB30889 ((2S)-2-[4-(3-azidophenyl)-2-oxopyrrolidin-1-yl]butanamide) and UCB1244283 ((4-(3,5-dimethylphenyl)-N-(2-methoxyphenyl)-3-methylbutanamide) were synthesized at UCB (Braine-l'Alleud, Belgium). The chemical structures of UCB1244283, LEV and UCB30889 are depicted in Figure 1. [3H]UCB30889 (47 Ci·mmol−1) was custom labelled by Amersham Biosciences (Roosendaal, The Netherlands). HEK cells (Flp-In™-293) and zeocin were purchased from Life Technologies (Merelbeke, Belgium). PBS, DMEM, L-glutamine, trypsin and FBS were purchased from Lonza (Verviers, Belgium). Complete protease inhibitor cocktail was purchased from Roche (Vilvoorde, Belgium) and DNAse (Deoxyribonuclease I, Type II from Bovine Pancreas) from Sigma-Aldrich (Bornem, Belgium). GF/B filters and GF/C filters were from Brandel (Alpha Biotech Ltd, London, UK) and Pall (Zaventem, Belgium) respectively. All other reagents were of analytical grade and obtained from conventional commercial sources.
Figure 1.
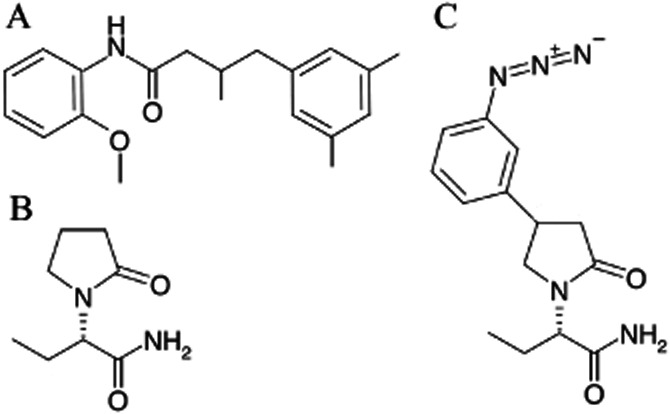
Chemical structures of (A) UCB1244283, (B) levetiracetam and (C) UCB30889.
Membrane preparations from rat cortex
Membrane proteins from rat brain were prepared as described previously (Gillard et al., 2003). Briefly, Sprague Dawley male rats (200–300 g) were killed by decapitation. Brains were quickly removed and the cerebral tissues were dissected on ice. All subsequent operations were performed at 4°C. The tissues were homogenized (10% w/v) in 20 mM Tris-HCl buffer (pH 7.4) containing 250 mM of sucrose (buffer A). The homogenates were spun at 30 000 g for 15 min and the pellets were resuspended in the same buffer. After incubation at 37°C for 15 min, the membranes were washed three times using the same centrifugation protocol. The final pellets were resuspended in buffer A at a protein concentration of 10 to 15 mg·mL−1 and stored in liquid nitrogen. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Preparation of membrane proteins from HEK cells
Human SV2A was cloned from a fetal brain cDNA library as described in (Lynch et al., 2004) and stably expressed in HEK cells. Cells were subcultured in DMEM containing 200 mM of L-glutamine and 100 μg·mL−1 zeocin, supplemented with 10% FBS. The cells were grown in a humidified atmosphere of 5% CO2 at 37°C. Confluent cells were harvested by trypsinization and pelleted by centrifugation at 1500 g for 10 min at 4°C. The pellet was washed once with ice-cold PBS using the same centrifugation protocol. The resulting pellet was homogenized in a buffer containing 15 mM Tris-HCl, 1 mM EGTA, 0.3 mM EDTA and 2 mM MgCl2 (pH 7.5) supplemented with complete protease inhibitor cocktail Roche. The homogenate was freeze-thawed twice and equilibrated at 25°C followed by a 10 min DNAse (10 U·mL−1) treatment. Subsequently, the solution was centrifuged for 25 min at 40 000 g and 4°C. Finally, the pellet was resuspended in buffer A at a protein concentration of 5 to 10 mg·mL−1 and stored in liquid nitrogen.
Binding experiments with [3H]UCB30889
Experiments were performed essentially as described before (Gillard et al., 2003). For all assays, membrane proteins (5–100 μg per assay) were incubated for 120 min at 4°C or 60 min at 25°C or 37°C in 0.2 mL of a 50 mM Tris-HCl buffer (pH 7.4) containing 2 mM MgCl2.
Competition binding experiments
Increasing concentrations of unlabelled competing drugs with or without 10 μM UCB1244283, were added in the presence of 5.25 nM of [3H]UCB30889. At the end of the incubation period, the membrane-bound radioligand was recovered by rapid filtration through GF/B glass fibre filter plates pre-soaked in 0.1% polyethyleneimine (PEI). Plates were washed rapidly with 0.8 mL of ice-cold Tris buffer (50 mM, pH 7.4); the total washing procedure did not exceed 10 s. To determine the dose-response profile of UCB1244283, increasing concentrations of this compound were incubated in the presence of 5.25 nM of [3H]UCB30889 and an identical experimental procedure was followed as indicated above.
Kinetic experiments
Specific [3H]UCB30889 binding in association experiments was measured at the indicated times after addition of the membrane proteins at 25°C. 10 μM of UCB1244283 was added 60 min before, 30 min after or simultaneously with the initiation of the association reaction. Dissociation was induced by addition of 100 μM of unlabelled UCB30889 to the association reaction mixture at the indicated times. Samples were filtered on GF/C glass fibre filters pre-soaked in 0.1% PEI and washed with 5 mL of ice-cold Tris buffer (50 mM, pH 7.4). The total filtration time per sample did not exceed 2 s. In the association experiments, kobs is the observed rate constant and equals kon*L + koff (with kon the association rate constant, koff the dissociation rate constant and L the concentration of the radioligand). It should be noted that at the low radioligand concentration used in these experiments, it was not possible to accurately calculate the kon values given the fact that at low radioligand concentrations, the kobs value is mainly driven by dissociation (koff).
Saturation binding studies
Membrane proteins were incubated with concentrations of [3H]UCB30889 ranging from 1 to 360 nM. Samples were filtered on GF/B filters using a Brandel harvester. The total filtration time did not exceed 10 s.
In all experiments, non-specific binding was defined as the residual binding observed in the presence of 1 mM of LEV. Radioactivity was determined by liquid scintillation.
In vivo seizure testing
Female mice (24–29 g) prone for sound-induced seizures were used. The mice derived from the DBA/2 strain originally selected by Dr. Lehmann from the Laboratory of Acoustic Physiology (Paris) and purchased from Charles River (Italy). They were maintained on a 12/12-h light/dark cycle with lights on at 06:00 h and had free access to food and drinking water. The temperature in the husbandry was maintained at 20–21°C with 40% humidity. All mice were selected for sound-induced seizure response before surgery and kept in the husbandry for 2 weeks prior to i.c.v. cannula implantation. All tests were performed according to the Helsinki declaration and complied with the guidelines of the European Community Council directive 86/609/EEC. A local ethical committee approved the experimental protocols.
The mice were anaesthetized with a mixture of medetomidine hydrochloride (1 mg·kg−1 i.p., Domitor, Pfizer) and ketamine (1000 mg·kg−1 i.p., Imalgene, Rhône-Mérieux) and surgically implanted in the right lateral ventricle (−1 mm anteroposterior, −0.9 mm lateral, 2.3 depth) with i.c.v. injection guide cannula (Plastics One). The pedestal of the guide cannula was held in place with dental acrylic cement (Powder Grip Cement, Caulk Dentistry, USA) applied to the exposed skull surface. A recovery period of 3 weeks, in individual cages, was allowed before compound injection and seizure testing.
UCB1244283 was solubilized in DMSO and diluted to 10% in a solution of 5% Cremophor/40% HPBCD (in-house formulation) in water to give a final concentration of 1.55 mg·mL−1 at pH 5.8.
The testing for sound-seizures was performed in a sound-attenuated wooden chamber (150 × 50 × 63 cm), containing five Makrolon cages (267 × 207 × 140 mm). The light intensity of this chamber was approximately 1500 lux. A loudspeaker (AD0141T4, Philips, Anderlecht, Belgium), through which the acoustic stimulus is delivered, was positioned above each cage. A white noise generator (custom made, UCB) provided a 90 dB sound, at frequencies between 10 and 20 kHz, to five cages simultaneously for a duration of 30 s. The sound level was verified before testing, using a sound level meter (Realistic, Radio Shack, Fort Worth, TX, USA), and adjusted if necessary. The sound-induced seizure response was verified in all the implanted animal the day before compound testing.
On the day of compound testing, each mouse was gently taken by the neck and an injection needle was inserted into the i.c.v. cannula and connected to a polyethylene tubing (Plactics One, C313C, Roanoke, VA, USA) for slow infusion of the compound using a 250 μL-Hamilton syringe. The compound solutions were administered in 10 μL volume and the injection needle was kept in the cannula for 30 s after finishing the injection to avoid fluid reflux. Each animal was placed in the acoustic chamber 5–15 min after infusion and after 30 s habituation the sound stimulus (90 dB, 10–20 kHz) was delivered for 30 s. During this interval, the mouse was observed for the presence of the three phases of the seizure activity: wild running, clonic convulsions and tonic hindlimb extension.
Data analysis
Data analysis was performed by computerized nonlinear curve fitting methods (Graphpad Prism 5 software, San Diego, CA, USA), according to equations describing several binding models (Molinoff et al., 1981). IC50 values were corrected to Ki by applying the Cheng and Prusoff equation (Cheng and Prusoff, 1973). Comparison between two groups was done using an unpaired Student's t-test. Comparisons between different curve fits were done using an extra-sum-of-squares F-test and the decision level was set at 0.05. The curve fits on the graphs represent the predicted values of the best fit. In vivo experiments were analysed using the one tailed Fisher's exact test (Graphpad Prism 5 software).
Results
UCB1244283 is a SV2A positive allosteric modulator of [3H]UCB30889
Competition binding experiments using UCB1244283, showed a strong potentiation of the binding of [3H]UCB30889 both on rat cortex and on recombinant human and rat SV2A expressed in HEK cells (Figure 2, Supporting Information Figure S1). The experiments were performed at 4, 25 and 37°C and the corresponding pEC50 values for UCB1244283 ranged from 4.8 to 6.1 and are given in Table 1. The maximal effect of UCB1244283 on the binding of [3H]UCB30889 ranged from a 500 to 900% increase compared to the control condition and the potency of UCB1244283 was approximately 10- to 20-fold lower on rat cortex compared to recombinant SV2A at 4°C. The exact pEC50 and Emax values could not be calculated for rat cortex at 25°C and 37°C since the plateau was not reached at these temperatures in the concentration range tested. For both recombinant SV2A and rat cortex, a slight decrease in potency of UCB1244283 with increasing temperature was observed. The data were fit using a sigmoidal dose-response model with variable slope. The Hill slopes were not significantly different from unity.
Figure 2.
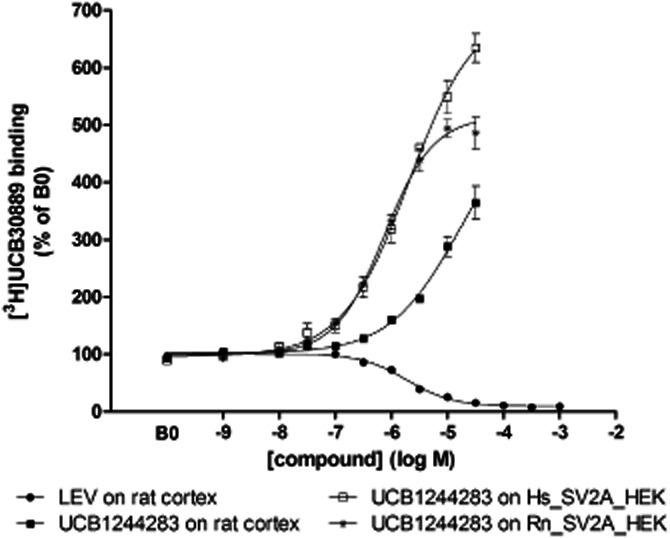
Effect of UCB1244283 and LEV on [3H]UCB30889 binding at 4°C on rat cortex and human (Hs) and rat (Rn) recombinant SV2A. Increasing concentrations of LEV or UCB1244283 were incubated with 5.25 nM of [3H]UCB30889 for 120 min at 4°C as described in materials and methods. Data were normalized to the binding of [3H]UCB30889 in the absence of any compound (B0), n = 3 to 5.
Table 1.
pEC50 and Emax values of UCB1244283 for sites labelled with [3H]UCB30889 on rat cortex and human and rat recombinant SV2A expressed in HEK cells at different temperatures
| T (°C) | Rat cortex | Human recombinant SV2A | Rat recombinant SV2A | |
|---|---|---|---|---|
| pEC50 | 4 | 4.75 ± 0.45 | 5.72 ± 0.03 | 6.05 ± 0.19 |
| 25 | 5.52 ± 0.11 | 5.64 ± 0.08 | ||
| 37 | 5.47 ± 0.16 | 5.43 ± 0.19 | ||
| Emax (% of B0) | 4 | 571 ± 181 | 706 ± 47 | 522 ± 34 |
| 25 | 735 ± 70 | 824 ± 141 | ||
| 37 | 880 ± 118 | 835 ± 45 |
Data are presented as mean ± SD (n = 3) and were obtained by analysis of competition curves as presented in Figure 2, using a non-linear regression model with variable slope as described in materials and methods. Hill slopes were not significantly different from unity.
UCB1244283 alters the binding properties of UCB30889
Saturation binding curves of [3H]UCB30889 both on rat cortex and on human recombinant SV2A, at 4 and 37°C, were compatible with the labelling of a homogenous population of binding sites (Figure 3). The Kd and Bmax values for rat cortex (88 ± 8 nM and 14.6 ± 3.8 pmol·mg−1 protein, n = 3) and human recombinant SV2A (64 ± 5 nM and 66.6 ± 17.1 pmol·mg−1 protein, n = 3) at 4°C, were in line with previous reports (Gillard et al., 2003; 2006). Addition of 10 μM of UCB1244283 to the saturation binding experiments resulted in an approximate doubling of the number of binding sites and a strong increase of the affinity. The latter effect was most pronounced on human recombinant SV2A (Table 2) which would be expected at a concentration of 10 μM of UCB1244283, given the observed lower potency of this compound on rat cortex compared to human recombinant SV2A (Table 1). The saturation profile however, remained compatible with a homogenous population of binding sites.
Figure 3.
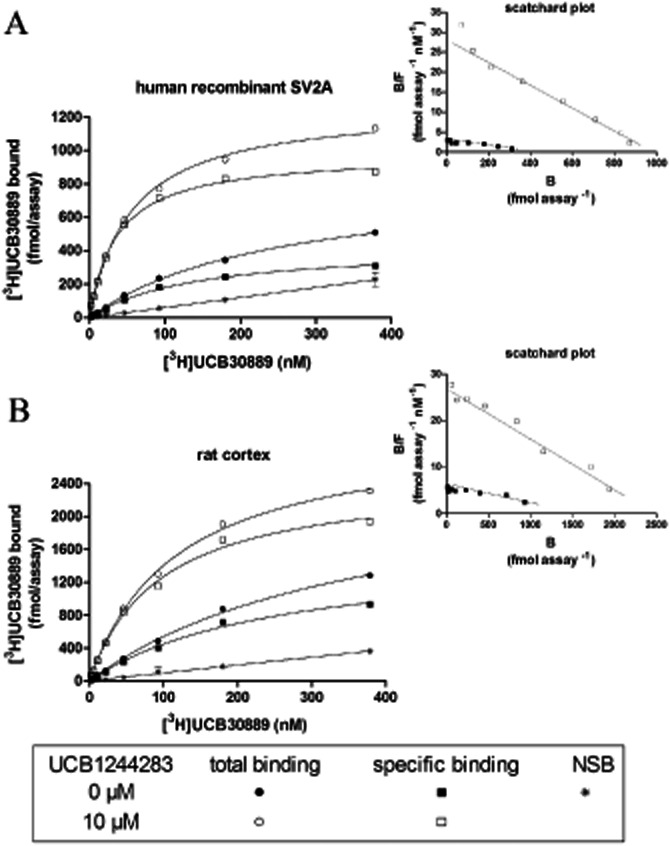
Effect of UCB1244283 on the binding characteristics of [3H]UCB30889. Saturation binding curves of [3H]UCB30889 (A) on human recombinant SV2A expressed in HEK cells and (B) on rat cortex. Membranes were incubated with increasing concentrations of [3H]UCB30889 for 60 min at 37°C with (open symbols) or without (filled symbols) 10 μM of UCB1244283. Non-specific binding (NSB) was determined as the residual binding observed in the presence of 1 mM levetiracetam. The insets are the Scatschard plots from the transformed data. Specific binding = total binding – NSB. Results are representative of three independent experiments.
Table 2.
Effect of UCB1244283 on the Kd and Bmax values of [3H]UCB30889 on rat cortex and human recombinant SV2A expressed in HEK cells at 4°C and 37°C
| Rat cortex | Human recombinant SV2A | |||
|---|---|---|---|---|
| + UCB1244283 | + UCB1244283 | |||
| Kd (nM) | ||||
| 4°C | 88 ± 8 | 37 ± 9** | 64 ± 5 | 13 ± 5*** |
| 37°C | 247 ± 10 | 83 ± 11*** | 153 ± 23 | 33 ± 8*** |
| Bmax (pmol·mg−1) | ||||
| 4°C | 14.6 ± 3.8 | 22.0 ± 7.3 | 66.6 ± 17.1 | 118.2 ± 20.4* |
| 37°C | 14.9 ± 1.7 | 25.1 ± 0.9*** | 69.1 ± 13.8 | 130.2 ± 14.5** |
Data are represented as mean ± SD (n = 3, *P < 0.05; **P < 0.01; ***P < 0.001, unpaired Student's t-test between condition with and without UCB1244283).
The effect of UCB1244283 on the affinity of UCB30889 was also evident from competition curves on recombinant human SV2A and on rat cortex labelled with [3H]UCB30889 (Figure 4). The corresponding pKi values are presented in Table 3. UCB1244283 induced a three- to 10-fold increase in the affinity of UCB30889 depending on the temperature and the protein source used, with the strongest effects again observed on human recombinant SV2A. On the other hand, the affinity of LEV trended to be reduced in the presence of UCB1244283 but this trend only reached significance at 4°C on human recombinant SV2A and at 37°C on rat cortex (unpaired Student's t-test). Nevertheless, the physiological relevance of a 0.1 log reduction in the affinity of LEV in the presence of UCB1244283 is questionable.
Figure 4.
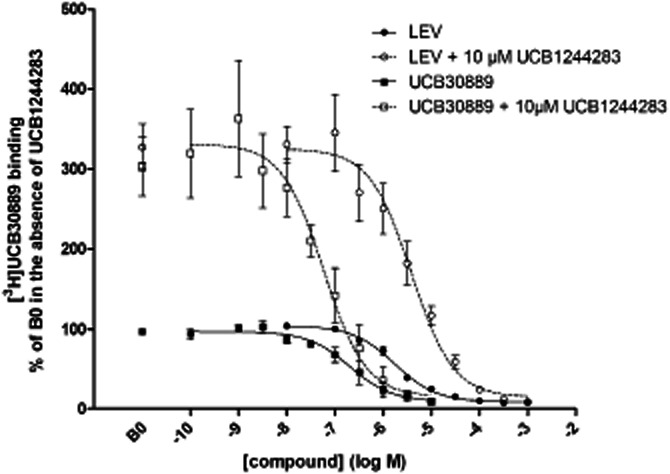
Effect of UCB1244283 on the competition profile of UCB30889 and levetiracetam against [3H]UCB30889 on rat cortex. A concentration range of both compounds was incubated with 2.6 nM of [3H]UCB30889 in the presence (open symbols) or absence (filled symbols) of 10 μM of UCB1244283 during 120 min at 4°C. B0 is the binding of [3H]UCB30889 in the absence of any competing compound. Data represent the mean ± SD of three to six independent experiments. Analysis was performed using a nonlinear regression model with variable slope as described in materials and methods.
Table 3.
Effect of UCB1244283 on the affinities of UCB30889 and LEV for sites labelled with [3H]UCB30889 on rat cortex and human recombinant SV2A expressed in HEK cells at 4°C and 37°C
| Rat cortex pKi ± SD | Human recombinant SV2A pKi ± SD | |||
|---|---|---|---|---|
| + UCB1244283 | + UCB1244283 | |||
| 4°C | ||||
| LEV | 5.75 ± 0.14 | 5.48 ± 0.10 | 5.47 ± 0.04 | 5.38 ± 0.03* |
| UCB30889 | 7.03 ± 0.06 | 7.36 ± 0.08*** | 6.85 ± 0.43 | 7.79 ± 0.05** |
| 37°C | ||||
| LEV | 5.32 ± 0.03 | 5.19 ± 0.04** | 5.16 ± 0.07 | 5.23 ± 0.03 |
| UCB30889 | 6.46 ± 0.16 | 7.07 ± 0.08*** | 6.37 ± 0.48 | 7.31 ± 0.05* |
Data are presented as mean ± SD (n = 3 to 6) and were obtained by analysis of competition curves as presented in Figure 4, using a non-linear regression model with variable slope as described in materials and methods. Hill slopes were not significantly different from unity. *P < 0.05, **P < 0.01, ***P < 0.001 (unpaired Student's t-test between condition with and without UCB1244283).
UCB1244283 modifies the binding kinetics of [3H]UCB30889
Detailed binding kinetic studies were only performed at 25°C since kinetics at 4 and 37°C were too slow and too fast respectively to allow accurate analysis (Supporting Information Figure S2). At 25°C, both the association and the dissociation kinetics on rat cortex and on recombinant human SV2A were best described by a biphasic model (Figure 5, Table 4 and Supporting Information Table S1). The kinetic constants were similar between rat cortex and human recombinant SV2A. However, the slow component of dissociation was markedly slower on rat cortex than on human recombinant SV2A (t1/2_rat cortex 3.5 ± 0.6 min vs. t1/2_recombinant SV2A 2.0 ± 0.2 min, n = 3, P < 0.05). These observations are in line with previous reports at 4°C (Gillard et al., 2006). Given the high degree of similarity in the kinetic profile between rat cortex and human recombinant SV2A, only the detailed curve analysis graphs on rat cortex are displayed in the main document, while the analysis results for human recombinant SV2A can be found in Supporting Information Table S1.
Figure 5.

Effect of UCB1244283 on the binding kinetics of [3H]UCB30889 on rat cortex at 25°C. UCB1244283 was added to the reaction mixture at different time points as indicated in the figure legend and the concentration of [3H]UCB30889 was 5.25 nM. The experiments were performed as described in materials and methods. Non-specific binding (NSB) was determined as the residual binding observed in the presence of 100 μM UCB30889. Arrows indicate the onset of dissociation by addition of 100 μM unlabeled UCB30889. (B) Close-up of the association curves presented in (A). All curves were shifted to the origin in order to allow better visual comparison of the curves. Data were fit according to a biphasic association model as described in materials and methods. (C) Close-up of the dissociation curves as presented in (A). All curves were again shifted to the origin and data were fit with a two phase (continuous line) and three phase (dotted line) dissociation model as described. Data are representative of at least three independent experiments.
Table 4.
Kinetic constants of [3H]UCB30889 binding on rat cortex at 25°C
| No UCB1244283 | 10 μM UCB1244283 | |
|---|---|---|
| Association | • (n = 3) | ○ □ ▪ (n = 2 + 2 + 2) |
| kobs fast (min−1) | 3.53 ± 0.27 | 1.40 ± 0.19*** |
| % fast | 49 ± 2 | 63 ± 6 |
| kobs mid (min−1) | / | / |
| % mid | / | / |
| kobs slow (min−1) | 0.32 ± 0.05 | 0.19 ± 0.03** |
| t1/2 fast (min) | 0.20 ± 0.02 | 0.50 ± 0.07*** |
| t1/2 mid (min) | / | / |
| t1/2 slow (min) | 2.18 ± 0.35 | 3.72 ± 0.69** |
| Dissociation | • (n = 3) | ○ (n = 3) |
|---|---|---|
| koff fast (min−1) | 2.71 ± 0.38 | 1.24 ± 0.10 |
| % fast | 53 ± 6 | 44 ± 5 |
| koff mid (min−1) | / | 0.24 ± 0.02 |
| % mid | / | 35 ± 3 |
| koff slow (min−1) | 0.20 ± 0.03 | 0.05 ± 0.003 |
| t1/2 fast (min) | 0.26 ± 0.06 | 0.56 ± 0.04 |
| t1/2 mid (min) | / | 2.85 ± 0.26 |
| t1/2 slow (min) | 3.50 ± 0.55 | 12.92 ± 0.59 |
The binding kinetic constants of [H]UCB30889 were calculated from kinetic experiments as depicted in Figure 5 by non-linear regression according to a model with two or three association or dissociation components (indicated as fast, mid and slow). Data represent mean ± SD and the n-values are indicated in the column headers. The symbols in the column headers correspond to the ones in Figure 5 (UCB1244283 added: ▪ simultaneously with the radioligand, □ 60 min before the radioligand, ○ 30 min after the radioligand). koff is the dissociation kinetic constant and kobs the observed rate constant = konL + koff with kon the association kinetic constant and L the concentration of radioligand (5.25 nM in these experiments). t1/2 is the half-time of association or dissociation. Stars indicate the significance level of the difference between the corresponding parameters in the presence and absence of UCB1244283 (biphasic models, **P < 0.01, ***P < 0.001, unpaired Student's t-test).
In order to study the effect of UCB1244283 on the association kinetics of [3H]UCB30889, three experimental approaches were followed. In the first one (Figure 5, ○), UCB1244283 was added to the [3H]UCB30889 association reaction after 30 min, when the equilibrium was reached. This resulted in the initiation of a second biphasic association event with twofold lower kobs values. In addition, the new equilibrium value of bound [3H]UCB30889 was approximately sixfold higher than that in the absence of UCB1244283 (Figure 5B). This magnitude of increase corresponded well with the increase of the bound fraction of [3H]UCB30889 in the saturation isotherms in the presence of UCB1244283 at a radioligand concentration of 5.25 nM (Figure 3). In the second (Figure 5, □) and third (Figure 5, ▪) approach, UCB1244283 was pre-incubated during 60 min with the protein in the reaction mixture before addition of [3H]UCB30889 or added simultaneously with the radioligand respectively. Both approaches resulted in an identical binding profile that could be fit with the same kinetic constants and fractions as obtained from the curve fits of the first approach. In conclusion, [3H]UCB30889 showed a biphasic association profile and both components were slowed down in the presence of 10 μM of UCB1244283.
The effect of UCB1244283 on the dissociation kinetics of [3H]UCB30889 at equilibrium was assessed by adding 100 μM of unlabelled UCB30889 in the presence of 10 μM of UCB1244283 (Figure 5A and C). UCB1244283, clearly slowed down the dissociation kinetics of [3H]UCB30889 and the data were best described by a triphasic model while in the absence of UCB1244283 the dissociation profile was biphasic as indicated earlier [biphasic vs. triphasic model, P < 0.001, F-test (α = 0.05; 40, 2)] (Figure 5C). The fact that UCB1244283 clearly converted the dissociation kinetics from a two to a (at least) triphasic model, supports the induction of a conformational change. Similar results were obtained from the kinetic experiments on human recombinant SV2A. However, the reduction of the kobs values in the presence of UCB1244283 was even more pronounced on human recombinant SV2A compared to rat cortex. The koff values were very similar on rat cortex and on human recombinant SV2A, except for the slowest phase which was significantly slower on recombinant human SV2A (t1/2_recombinant SV2A 16.3 ± 0.6 min vs. t1/2_rat cortex 12.9 ± 0.6 min, P < 0.001, n = 3). Finally, the proportions of the different fractions were different between rat cortex and human recombinant SV2A with the largest proportion of all binding sites corresponding to the fastest fraction on rat cortex (koff fast, ± 44%) while on human recombinant SV2A the largest proportion corresponded to the middle fraction (koff mid, ± 56%).
UCB1244283 has anticonvulsant properties in sound-sensitive mice
Given the fact that UCB1244283 is a positive allosteric modulator of the SV2A protein which is the target of the anticonvulsant LEV (Lynch et al., 2004), this compound was tested for potential protective effects in the audiogenic mouse model. The experiments were conducted as described in materials and methods and the results are given in Table 5. Due to the poor blood-brain-barrier passage of the compound, it had to be administered i.c.v. (data not shown). A clear protective effect against both tonic and clonic convulsions was evident at the highest dose tested, 15 min after i.c.v. administration (P < 0.01).
Table 5.
Effect of UCB1244283 against clonic and tonic convulsions in sound-sensitive mice at the indicated time after administration
| UCB1244283 (μg) | Time (min) | Protected/total (number of animals) |
|---|---|---|
| 7.75 | 5 | 0/3 |
| 7.75 | 15 | 1/6 |
| 15.5 | 15 | 5/6** |
P < 0.01, one-tailed Fisher's exact test.
Discussion and conclusions
In this study, we report for the first time that the SV2A protein contains multiple interacting binding sites and we identified UCB1244283 acting as a SV2A PAM of UCB30889.
The SV2A PAM was discovered in a screening effort to identify high affinity SV2A ligands based on competitive profiling against [3H]UCB30889. This radioligand was also used to study the effects of positive allosteric modulation of SV2A on the binding characteristics of SV2A ligands in more detail since it has been demonstrated to be better suited for this purpose than labelled LEV itself, given its higher affinity for the LEV binding site (LBS) (Gillard et al., 2003).
Initial dose-response curves showed that the potency of UCB1244283 was higher on human and rat recombinant protein compared to rat cortex with EC50 values for recombinant protein ranging from 1 to 4 μM and EC50 values for rat cortex above 8 μM. Therefore, the results support a higher potency of the PAM on recombinant protein compared to native tissue irrespective of the species source of SV2A. In this respect, it should be acknowledged that the potency of a PAM increases with increasing occupancy of the receptor by the orthosteric ligand (Christopoulos and Kenakin, 2002). However, in the experimental conditions of this study with a concentration of radioligand well below the Kd, the occupancy by [3H]UCB30889 was very low, both on rat cortex and on recombinant SV2A. Therefore, the fact that the affinity of [3H]UCB30889 was higher on human recombinant SV2A compared to rat cortex, is unlikely to have a large impact on the difference in potency between the recombinant protein and the native tissue. Other possible contributing factors to the observed difference include differences in interacting proteins and differences in protein conformation between the recombinant and the native expression systems. Also, a decrease in potency with increasing temperature was observed. This is in line with the previously reported temperature dependency of ligand binding to SV2A (Noyer et al., 1995; Gillard et al., 2011) and the observations made in this study (Table 2).
Due to the poor solubility of UCB1244283, the applied concentration in the follow-up binding assays was limited to 10 μM. This concentration is slightly above the EC50 value on recombinant protein. Therefore, the experimental observations are only likely to display a partial effect of the PAM, being highest on recombinant protein compared to rat cortex. However, since a general characteristic of allosteric modulators is a saturation of their effect with increasing concentrations (Christopoulos and Kenakin, 2002), UCB1244283 is likely to display more than half of its effect at a concentration of 10 μM, at least on recombinant SV2A. At a concentration of 10 μM, UCB1244283 caused a doubling of the Bmax value and a significant increase in the affinity of [3H]UCB30889 for the SV2A protein in saturation binding experiments. The doubling of the SV2A binding sites may be due to the modulation of the SV2A protein from a low to a high affinity state for [3H]UCB30889 in the presence of UCB1244283, thereby unveiling previously very low affinity sites. The fact that these low affinity sites could not be detected in the absence of UCB1244283 can be explained by the sensitivity limit of the saturation binding assays we performed, which did not allow the detection of binding sites with an affinity greater than 20 μM (calculated from theoretical extrapolation of the data). It should also be noted that a fraction of the increase in the Bmax value in the presence of UCB1244283 might be the consequence of the slowing down of the dissociation rate of the radioligand by UCB1244283 thereby decreasing the potential loss of specific binding due to a fast dissociation rate. However, assuming a scenario with a washing time of 10 s at 25°C on rat cortex, this slowing effect of UCB1244283 on the dissociation speed would only account for a 10% difference in the Bmax values in the presence and absence of UCB1244283 which is way below the observed increase in Bmax of more than 150%. In reality, this effect is predicted to be even lower given the fact that the washing procedure is performed at 4°C. The affinity of [3H]UCB30889 for recombinant human SV2A was increased by fivefold in the presence of UCB1244283 while the results were still compatible with the labelling of a homogenous population of binding sites. The observed increase in affinity cannot solely be explained by the potentiation of very low affinity sites since these sites would only account for 50% of the total population of binding sites (given the doubling of the Bmax value) and as a consequence only contribute for 50% to the Kd value. Therefore, the modulation of SV2A to a higher affinity state for [3H]UCB30889 would not only imply an affinity increase of previously very low affinity sites but also the potentiation of the affinity of (part of) the original sites detected in the absence of UCB1244283. This is supported by the model of De Lean et al. (De Lean et al., 1982) in which the affinity difference between two potential subpopulations in a homogenous population of binding sites cannot be more than approximately threefold, otherwise two sites would be seen. One explanation for the observed potentiation in affinity is that UCB1244283 induces an overall conformational change of the protein, leading to the unveiling of previously very low affinity states and a conformational change in the SV2A protein, resulting in an increased affinity for [3H]UCB30889. This idea is strongly supported by the effect of UCB1244283 on the binding kinetics of [3H]UCB30889: upon addition of 10 μM of UCB1244283, both association and dissociation kinetics slowed down significantly and the corresponding kinetic models shifted from biphasic to triphasic. The effect of UCB1244283 on the binding kinetics was quite instantaneous which is in line with observations from other allosteric modulators (Christopoulos and Kenakin, 2002). The observations from the kinetic studies support the above predictions from the saturation isotherms: the PAM not only induces an additional kinetic component, which could be translated into an additional conformation, but also seems to change the characteristics of the original components. Nevertheless, it should be noted again that at a concentration of 10 μM, UCB1244283 is unlikely to reach its full potential. Therefore, the data are most likely to represent a situation in which only part of the SV2A protein present is affected by the PAM while the other part remains in its original state. The lower potency of UCB1244283 on rat cortex compared to recombinant protein probably underlies the more modest magnitude of the PAM effect seen on rat cortex. Moreover, the existence of at least two different conformations has already been reported for SV2A (Lynch et al., 2008). Our data do not support a scenario in which the effect of UCB1244283 is mediated although binding to an endogenous interaction partner of SV2A for two main reasons. First, HEK cells are non-neuronal cells that do not possess synaptic vesicles (the residence place of SV2A) and only possess very low endogenous SV2A levels. Overexpression of SV2A in HEK cells results in high protein levels (as evidenced by Western blotting) and therefore, it is not very likely that endogenous interaction partners will be able to greatly affect the total pool of overexpressed SV2A, which is many times bigger than the endogenous pool. Second, the effect of UCB1244283 was observed on both rat cortex and recombinant SV2A expressed in HEK cells, having very different protein backgrounds.
The results from the kinetic experiments are also consistent with an increase in affinity caused by UCB1244283. A strong decrease in the koff values was evident which, at least in part, drives the Kd (= koff kon−1It should be noted that it was not possible to determine the effect of UCB1244283 on the kon values in our experimental settings because the kobs values were mainly driven by the koff values at the low concentration of radioligand used. Performing the association experiments at higher radioligand concentrations resulted in a decrease of the specific binding window and an increase in the reaction rate, thereby strongly reducing the accuracy of the measurement of the kobs values (data not shown). Therefore kinetic Kd values could not be calculated based on the kinetic parameters. The triphasic kinetic model was the model, with least complexity, which best fitted the dissociation data in the presence of UCB1244283. However, this does not mean that the true conformational complexity of the protein in the presence of the PAM is limited to three conformations. Therefore, based on the current data, it is not possible to make more accurate predictions on the different conformations. In general, the kinetic constants were very similar on rat cortex and on human recombinant protein, except for the slow phase of dissociation. Significant differences were also observed between the proportions of the different fractions on rat cortex and on human recombinant protein. All these differences might be explained by a higher potency of UCB1244283 on human recombinant SV2A compared to rat cortex as evidenced by the dose-response curves and the saturation isotherms.
The SV2A protein has been demonstrated to be the binding site of LEV (Lynch et al., 2004) and to mediate its anticonvulsant activity in vivo (Kaminski et al., 2009). In order to understand the physiological relevance of a conformational effect on SV2A in this context, UCB1244283 was tested in the audiogenic-mouse model which has been shown to be highly suited to study SV2A related anticonvulsant effects (Kaminski et al., 2008). A clear, protective effect of the compound against both tonic and clonic convulsions was observed after i.c.v. administration. UCB1244283 was also tested for selectivity on 65 other potential targets including GPCRs, ion channels, enzymes and transporters in an in-house screen and showed great selectivity for SV2A (data not shown). These results support the idea that a conformational change of SV2A per se may be sufficient to protect against convulsions in the audiogenic mouse model. As a consequence, it can be hypothesized that the SV2A PAM promotes/stabilizes the protein in a conformation that enables the protein to fulfil a protective role during seizures which might be reinforced by SV2A-ligand binding to the LBS. In this scenario, also LEV, which is insensitive to the conformational change induced by UCB1244283, would be more effective since the proportion of SV2A in the more ‘effective’ conformation would be increased by the SV2A PAM. However, additional experiments are needed to determine the real value of SV2A positive allosteric modulators as potential antiepileptic drugs. Key questions are whether the effect of the PAM is additive or synergistic to the effect provided by a SV2A-anticonvulsant like LEV and whether a PAM only increases the potency or also the efficacy of these SV2A ligands. The latter would imply that a SV2A PAM would hold the capacity to overcome a certain level of resistance to SV2A-anticonvulsants and that the cause of resistance to this kind of drugs would lay – in part – in the conformation of the protein. Indeed, it would be important to know what determines/affects the conformational state of SV2A in its biological context. Possible candidates are: interactions with other proteins like synaptotagmin (Schivell et al., 1996; 2005; Pyle et al., 2000; Wan et al., 2010) or other small substances like nucleotides (Yao and Bajjalieh, 2008), peptides, sugars, lipids etc., post-translational modifications [phosphorylation (Gross et al., 1995), glycosylation (Dong et al., 2008; Chang and Sudhof, 2009), etc.] or dimerization which has been reported for several other members of the MFS (Geertsma et al., 2005; Tanabe et al., 2009). The fact that the SV2A PAM effect on UCB30889 was stronger on a recombinant SV2A expression system compared to a native one might be related to differences in one or more of these candidate mechanisms in a recombinant versus a native expression system. Finally, since SV2A is an integral transmembrane protein of synaptic vesicles, the conformational status of the protein might be dependent on the specific step of the synaptic vesicle cycle during neuronal activity. This idea is supported by the observation that considerable incubation period under spontaneous neuronal activity or certain loading conditions are required in order for LEV to exert an effect on neurotransmission in brain slices (Yang et al., 2007).
In conclusion, we show that the SV2A protein contains multiple interacting binding sites that are linked via the conformation of the protein. The sensitivity to the conformational state of the protein does not seem to be a shared characteristic of SV2A ligands given the fact that UCB1244283 only acts as a SV2A PAM of UCB30889 while LEV is insensitive to this effect. However, since UCB1244283 on its own is protective in the sound-sensitive mouse model, manipulation of the conformation of the protein might be a novel and additional way of exploiting SV2A as a target for anticonvulsants.
Acknowledgments
The authors thank C. Chaussée for skilful technical assistance and also the technical staff of the Cellular and Screening Sciences team for their general support. We thank F. Lebon for the many inspiring discussions.
V.D. is the recipient of a grant from the Walloon Region (Belgium) – DGO6 (Convention 6536).
Glossary
- LBS
levetiracetam binding site
- LEV
levetiracetam; SV2A, synaptic vesicle protein 2A
Conflicts of interest
All authors are employees of UCB Pharma.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Effect of UCB1244283 and LEV on [3H]UCB30889 binding at different temperatures on rat cortex and human and rat recombinant SV2A expressed in HEK cells. Increasing concentrations of LEV or UCB1244283 were incubated with 5.25 nM of [3H]UCB30889 for 60 min at 37°C or 25°C as described in materials and methods, n = 3 to 5.
Figure S2 Effect of UCB1244283 on the binding kinetics of [3H]UCB30889 on human recombinant SV2A expressed in HEK cells at (A) 4°C and (B) 37°C. Experiments were performed as described in materials and methods. Non-specific binding (NSB) was determined as the residual binding observed in the presence of 100 μM UCB30889. Arrows indicate the onset of dissociation. Data are representative of one to three experiments.
Table S1 Kinetic constants of [3H]UCB30889 binding on human recombinant SV2A expressed in HEK cells at 25°C.
References
- Bajjalieh SM, Peterson K, Shinghal R, Scheller RH. SV2, a brain synaptic vesicle protein homologous to bacterial transporters. Science (New York, NY) 1992;257:1271–1273. doi: 10.1126/science.1519064. [DOI] [PubMed] [Google Scholar]
- Bajjalieh SM, Frantz GD, Weimann JM, McConnell SK, Scheller RH. Differential expression of synaptic vesicle protein 2 (SV2) isoforms. J Neurosci. 1994;14:5223–5235. doi: 10.1523/JNEUROSCI.14-09-05223.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budzinski KL, Allen RW, Fujimoto BS, Kensel-Hammes P, Belnap DM, Bajjalieh SM, et al. Large structural change in isolated synaptic vesicles upon loading with neurotransmitter. Biophys J. 2009;97:2577–2584. doi: 10.1016/j.bpj.2009.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang WP, Sudhof TC. SV2 renders primed synaptic vesicles competent for Ca2+ -induced exocytosis. J Neurosci. 2009;29:883–897. doi: 10.1523/JNEUROSCI.4521-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- Crowder KM, Gunther JM, Jones TA, Hale BD, Zhang HZ, Peterson MR, et al. Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A) Proc Natl Acad Sci U S A. 1999;96:15268–15273. doi: 10.1073/pnas.96.26.15268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lean A, Hancock AA, Lefkowitz RJ. Validation and statistical analysis of a computer modeling method for quantitative analysis of radioligand binding data for mixtures of pharmacological receptor subtypes. Mol Pharmacol. 1982;21:5–16. [PubMed] [Google Scholar]
- Dong M, Liu H, Tepp WH, Johnson EA, Janz R, Chapman ER. Glycosylated SV2A and SV2B mediate the entry of botulinum neurotoxin E into neurons. Mol Biol Cell. 2008;19:5226–5237. doi: 10.1091/mbc.E08-07-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feany MB, Lee S, Edwards RH, Buckley KM. The synaptic vesicle protein SV2 is a novel type of transmembrane transporter. Cell. 1992;70:861–867. doi: 10.1016/0092-8674(92)90319-8. [DOI] [PubMed] [Google Scholar]
- Geertsma ER, Duurkens RH, Poolman B. Functional interactions between the subunits of the lactose transporter from Streptococcus thermophilus. J Mol Biol. 2005;350:102–111. doi: 10.1016/j.jmb.2005.04.047. [DOI] [PubMed] [Google Scholar]
- Gillard M, Fuks B, Michel P, Vertongen P, Massingham R, Chatelain P. Binding characteristics of [3H]ucb 30889 to levetiracetam binding sites in rat brain. Eur J Pharmacol. 2003;478:1–9. doi: 10.1016/j.ejphar.2003.08.032. [DOI] [PubMed] [Google Scholar]
- Gillard M, Chatelain P, Fuks B. Binding characteristics of levetiracetam to synaptic vesicle protein 2A (SV2A) in human brain and in CHO cells expressing the human recombinant protein. Eur J Pharmacol. 2006;536:102–108. doi: 10.1016/j.ejphar.2006.02.022. [DOI] [PubMed] [Google Scholar]
- Gillard M, Fuks B, Leclercq K, Matagne A. Binding characteristics of brivaracetam, a selective, high affinity SV2A ligand in rat, mouse and human brain: relationship to anti-convulsant properties. Eur J Pharmacol. 2011;664:36–44. doi: 10.1016/j.ejphar.2011.04.064. [DOI] [PubMed] [Google Scholar]
- Gross SD, Hoffman DP, Fisette PL, Baas P, Anderson RA. A phosphatidylinositol 4,5-bisphosphate-sensitive casein kinase I alpha associates with synaptic vesicles and phosphorylates a subset of vesicle proteins. J Cell Biol. 1995;130:711–724. doi: 10.1083/jcb.130.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holyoake J, Sansom MS. Conformational change in an MFS protein: MD simulations of LacY. Structure. 2007;15:873–884. doi: 10.1016/j.str.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Kaback HR, Dunten R, Frillingos S, Venkatesan P, Kwaw I, Zhang W, et al. Site-directed alkylation and the alternating access model for LacY. Proc Natl Acad Sci U S A. 2007;104:491–494. doi: 10.1073/pnas.0609968104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski RM, Matagne A, Leclercq K, Gillard M, Michel P, Kenda B, et al. SV2A protein is a broad-spectrum anticonvulsant target: functional correlation between protein binding and seizure protection in models of both partial and generalized epilepsy. Neuropharmacology. 2008;54:715–720. doi: 10.1016/j.neuropharm.2007.11.021. [DOI] [PubMed] [Google Scholar]
- Kaminski RM, Gillard M, Leclercq K, Hanon E, Lorent G, Dassesse D, et al. Proepileptic phenotype of SV2A-deficient mice is associated with reduced anticonvulsant efficacy of levetiracetam. Epilepsia. 2009;50:1729–1740. doi: 10.1111/j.1528-1167.2009.02089.x. [DOI] [PubMed] [Google Scholar]
- Kaminski RM, Gillard M, Klitgaard H. Targeting SV2A for discovery of antiepileptic drugs. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper's Basic Mechanisms of the Epilepsies [Internet], 4th edn. Bethesda, MD: National Center for Biotechnology Information; 2012. [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch BA, Lambeng N, Nocka K, Kensel-Hammes P, Bajjalieh SM, Matagne A, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci U S A. 2004;101:9861–9866. doi: 10.1073/pnas.0308208101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch BA, Matagne A, Brannstrom A, von Euler A, Jansson M, Hauzenberger E, et al. Visualization of SV2A conformations in situ by the use of Protein Tomography. Biochem Biophys Res Commun. 2008;375:491–495. doi: 10.1016/j.bbrc.2008.07.145. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinoff PB, Wolfe BB, Weiland GA. Quantitative analysis of drug-receptor interactions: II. Determination of the properties of receptor subtypes. Life Sci. 1981;29:427–443. doi: 10.1016/0024-3205(81)90208-3. [DOI] [PubMed] [Google Scholar]
- Nowack A, Yao J, Custer KL, Bajjalieh SM. SV2 regulates neurotransmitter release via multiple mechanisms. Am J Physiol. 2010;299:C960–C967. doi: 10.1152/ajpcell.00259.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyer M, Gillard M, Matagne A, Henichart JP, Wulfert E. The novel antiepileptic drug levetiracetam (ucb L059) appears to act via a specific binding site in CNS membranes. Eur J Pharmacol. 1995;286:137–146. doi: 10.1016/0014-2999(95)00436-o. [DOI] [PubMed] [Google Scholar]
- Pyle RA, Schivell AE, Hidaka H, Bajjalieh SM. Phosphorylation of synaptic vesicle protein 2 modulates binding to synaptotagmin. J Biol Chem. 2000;275:17195–17200. doi: 10.1074/jbc.M000674200. [DOI] [PubMed] [Google Scholar]
- Schivell AE, Batchelor RH, Bajjalieh SM. Isoform-specific, calcium-regulated interaction of the synaptic vesicle proteins SV2 and synaptotagmin. J Biol Chem. 1996;271:27770–27775. doi: 10.1074/jbc.271.44.27770. [DOI] [PubMed] [Google Scholar]
- Schivell AE, Mochida S, Kensel-Hammes P, Custer KL, Bajjalieh SM. SV2A and SV2C contain a unique synaptotagmin-binding site. Mol Cell Neurosci. 2005;29:56–64. doi: 10.1016/j.mcn.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Tanabe M, Szakonyi G, Brown KA, Henderson PJ, Nield J, Byrne B. The multidrug resistance efflux complex, EmrAB from Escherichia coli forms a dimer in vitro. Biochem Biophys Res Commun. 2009;380:338–342. doi: 10.1016/j.bbrc.2009.01.081. [DOI] [PubMed] [Google Scholar]
- Vautrin J. SV2 frustrating exocytosis at the semi-diffusor synapse. Synapse (New York, NY) 2009;63:319–338. doi: 10.1002/syn.20610. [DOI] [PubMed] [Google Scholar]
- Wan QF, Zhou ZY, Thakur P, Vila A, Sherry DM, Janz R, et al. SV2 acts via presynaptic calcium to regulate neurotransmitter release. Neuron. 2010;66:884–895. doi: 10.1016/j.neuron.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Bajjalieh SM. SV2 modulates the size of the readily releasable pool of secretory vesicles. Nat Cell Biol. 2001;3:691–698. doi: 10.1038/35087000. [DOI] [PubMed] [Google Scholar]
- Yang XF, Weisenfeld A, Rothman SM. Prolonged exposure to levetiracetam reveals a presynaptic effect on neurotransmission. Epilepsia. 2007;48:1861–1869. doi: 10.1111/j.1528-1167.2006.01132.x. [DOI] [PubMed] [Google Scholar]
- Yao J, Bajjalieh SM. Synaptic vesicle protein 2 binds adenine nucleotides. J Biol Chem. 2008;283:20628–20634. doi: 10.1074/jbc.M800738200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Nowack A, Kensel-Hammes P, Gardner RG, Bajjalieh SM. Cotrafficking of SV2 and synaptotagmin at the synapse. J Neurosci. 2010;30:5569–5578. doi: 10.1523/JNEUROSCI.4781-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.