

Abstract
Background and Purpose
Neurodegenerative diseases present progressive neurological disorder induced by cell death or apoptosis. Catalpol, an iridoid glucoside isolated from the root of Rehmannia glutinosa Libosch, is present in a wide range of plant families. Although catalpol is an effective anti-apoptotic agent in LPS-induced neurodegeneration, the underlying mechanism has not been established. Here we have identified some of the mechanisms involved the prevention by catalpol of apoptosis induced by LPS in an experimental model of neurodegeneration in vitro.
Experimental Approach
Apoptosis was induced by adding LPS (80 ng·mL−1) to pheochromocytoma (PC12) cells, pretreated with catalpol for 12 h. We measured intracellular reactive oxygen species (ROS), apoptosis and intracellular calcium concentration ( [Ca2+]i) by flow cytometry or laser confocal scanning microscopy. We also analysed the protein expression of Bcl-2, Bax and Ca2+-calmodulin-dependent protein kinase II (CaMKII)-dependent apoptosis signal-regulating kinase-1 (ASK-1)/JNK/p38 signalling pathway in PC12 cells by Western blot.
Key Results
Catalpol stimulated expression of Bcl-2 and inhibited the expression of Bax. Catalpol also attenuated the increase in Ca2+ concentration induced by LPS in PC12 cells and down-regulated CaMK phosphorylation. The CaMKII-dependent ASK-1/JNK/p38 signalling cascade was blocked by catalpol. All these changes were accompanied by a decrease of apoptosis induced by LPS in PC12 cells.
Conclusions and Implications
The data presented here provide new mechanistic insights into the links between the CaMKII-dependent ASK-1/JNK/p38 signalling pathway and the protective effect of catalpol on apoptosis induced by LPS in PC12 cells.
Keywords: Catalpol, neurodegenerative diseases, LPS, apoptosis, CaMKII, ASK-1/JNK/p38
Introduction
Neurodegenerative conditions, such as Parkinson's, Alzheimer's and Huntington's diseases, occur as progressive neurological disorders characterized by typical protein assemblies and induced cell death or apoptosis (Bredesen et al., 2006; Rubinsztein, 2006). Despite extensive research, the aetiology of this type of diseases is still unknown, but may depend on multiple factors, including both hereditary and environmental causes. There are many similarities that relate these diseases to one another on a subcellular level. But the knowledge about the cause of cell death or apoptosis in neurodegenerative diseases is not clear. Elucidating these causes can offer hope for therapeutic advances that could ameliorate many diseases at the same time and would be particularly important for pre-symptomatic treatments. The rat pheochromocytoma (PC12) cell line, which exhibits a number of properties and characteristics of sympathetic neurons (Jumblatt and Tischler, 1982) and is one of the most widely used neuronal cell lines for research on the mechanisms of Parkinson's and Alzheimer's diseases. The endotoxin LPS, is known to induce a strong response from normal animal immune systems. Thus, in vitro models induced by LPS, are often used in research on inflammation. Among the many disorders induced by LPS, neurodegenerative diseases, such as Parkinson's disease, are of particular interest here (Li et al., 2010; Burguillos et al., 2011). In our study, PC12 cells exposed to LPS were chosen as models of neurological degeneration to evaluate the anti-apoptotic effect of pharmacologically active ingredients of medicaments.
Catalpol, an iridoid glucoside isolated from the root of Rehmannia glutinosa Libosch, has a wide distribution in many plant families. So far, catalpol exerts a wide variety of biological activities including anti-microbial, anti-tumour, analgesic, purgative, liver protective, sedative and anti-inflammatory and is known to attenuate apoptosis in avariety of cells in vivo and in vitro (Jiang et al., 2008; Zhu et al., 2010).
Catalpol induced neuronal differentiation in PC12 cells through activation of intracellular signal transduction pathways (Yamazaki et al., 1996). Catalpol can also inhibit apoptosis in hydrogen peroxide-induced PC12 cells (Jiang et al., 2004). Although catalpol is an effective anti-apoptotic agent in LPS-induced neurodegeneration (Tian et al., 2006), the underlying mechanism has not been established. Here we have identified some of the mechanisms involved in the prevention by catalpol of apoptosis induced by LPS in an experimental model of neurodegeneration in vitro. We used PC12 cells treated with LPS and assessed apoptosis mediated by Ca2+-mediated signalling pathways, including activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII) and the apoptosis signal-regulating kinase-1 (ASK-1).
Methods
Cell culture and drug treatment
Differentiated PC12 cells were obtained from Shanghai Institutes for Biological Science, Chinese Academy of Sciences, and maintained in DMEM (Gibco BRL, Grand Island, NY, USA) supplemented with 10% heat-inactivated FCS, penicillin (100 U·mL−1), and streptomycin 100 mg·mL−1 (all from Gibco). Cells were cultured at 37°C in a humidified atmosphere containing 5% CO2. Cells exposed to vehicle alone (10% FCS culture medium) were used as the control group. Morphological analysis was performed with inverted microscope linked to a digital CCD camera (Olympus, Tokyo, Japan). Images were taken with an average of 100 cells per field.
MTT tests for determining optimum concentrations of catalpol and LPS
In a previous study (data not shown), catalpol was ineffective in concentrations below 10 nM, but did exert significant protective effects from 0.1–10 μM. In our present experiments, PC12 cells were treated with different concentrations of catalpol for 12 or 24 h. The viability of cells were analysed using MTT. The optical density of metabolized MTT was quantified by a Micro-plate reader (Bio-Rad Laboratories, Hercules, CA, USA) at 490 nm. Cell viabilities were expressed as percentages of cell viability in the control group, set to 100%.
Following these experiments, PC12 cells were pretreated with catalpol (10 μM) for 12 h. Apoptosis was then induced by adding LPS (20–160 ng·mL−1) to the serum-free medium for 12 h. Then, after being washed with DMEM, the cells were divided into two groups: the cells in LPS 12 h + Catalpol group were incubated in DMEM with catalpol (10 μM) for an additional 12 h, and the cells in LPS 12 h group were incubated in DMEM medium for 12 h. The viability of cells in the two groups was measured by the MTT test.
Measurement of intracellular ROS
Intracellular ROS can be detected using 2,7-dichlorofluorescein diacetate (DCFH-DA), which is freely permeable to cell membranes, and hydrolysed by intracellular esterases to non-fluorescent DCFH, which is then rapidly oxidized in the presence of ROS to highly fluorescent dichlorofluorescein (DCF). Briefly, the pretreated cells were incubated with DCFH-DA for 20 min at 37°C. The fluorescence of DCF was detected with flow cytometry with excitation at 488 nm and emission at 530 nm (Chen et al., 2012). Data were processed using the Cell Quest program (BD Biosciences, San Jose, CA, USA). All measurements were done in triplicate.
Release of LDH from PC12 cells
The release of LDH from PC12 cells, as a marker of cell damage, was determined in aliquots of the culture medium. At the end of the incubation, the cells and the supernatant medium were collected. The remaining intracellular LDH was obtained by lysing cells with 0.2% Tween-20 in PBS. The activities of LDH in 100 μL medium or cell lysates was measured using an LDH assay kit according to the manufacturer's instructions. The OD values were measured at 550 nm with a Microplate Reader. The percentage cell lysis was determined as the ratio of LDH in the medium/total LDH per well (medium plus cell lysates).
Assay of apoptosis in PC12 cells
Double staining for Annexin V/FITC and propidium iodide (PI) was performed to evaluate apoptosis in PC12 cells. Briefly, PC12 cells were treated as described above. 1 × 106 cells were suspended in binding buffer and double-stained with Annexin V/FITC and PI for 30 min at room temperature. After that, the fluorescence of each sample was quantitatively analysed by flow cytometry (FACS Caliber, BD Biosciences) with FCS Express Version 3 software. Cell sorting and data processing were carried out on 10 000 cells.
The results were interpreted as follows: cells negative for both PI and Annexin V/FITC staining were considered as live cells, PI-negative, Annexin V/FITC-positive cells were considered to be in early apoptosis, and PI positive, Annexin V/FITC-positive cells were considered to be in late apoptosis or necrosis.
Measurements of [Ca2+]i
Intracellular Ca2+ concentration ([Ca2+]i) was measured with the ratiometric fluorescent dye Fluo-3/AM utilizing a laser confocal scanning microscope (LEICA TCS SP5, Germany). For confocal microscopy imaging in the visible light excitation range, we used the Ca2+-sensitive dye Fluo-3, which is commonly used to image [Ca2+]i in living cells (Meng et al., 2007).
Pluronic F127 (0.1% ) was added to Fluo 3-AM in DMSO (500 μM) to prevent aggregation of Fluo 3-AM in HBSS and help uptake into cells. The DMSO solution of Fluo 3-AM was diluted with HBSS to yield a working solution of 5 μM Fluo 3-AM. PC12 cells were pretreated with catalpol or vehicle, as described earlier, and the supernatant removed. The cells then were incubated with Fluo-3/AM for 30 min in the dark at room temperature. The dye-loaded cells were gently washed three times with Ca2+-free HEPES buffered saline. The cells were kept in the medium for a further 20 min in the dark to allow the hydrolysis of Fluo-3-AM into Ca2+-sensitive free acid form (Fluo-3) by cell esterases. Fluorescence was elicited by excitation with a 488 nm argon laser at an approximate rate of two frames per second. The fluorescence intensities were detected at 528 nm. Fluorescence images were scanned and stored as a time series. LPS (final concentration 1 mg·mL−1) was applied to the cells in the extracellular medium (140 mM NaCl, 4.6 mM KCl, 2 mM CaCl2,10 mM glucose) after the scanning began. Emitted fluorescence was collected by the 20 × objective. The data obtained from the first and second scans (at 1 and 5 s after addition of LPS) were averaged and taken as the basal value of [Ca2+]i, and the maxima of fluorescence intensities in each sample of cells was considered as the peak data of [Ca2+]i. Ten to 15 individual cells in each scan were randomly selected in each group for determination of the average fluorescence intensities.
Analysis of mRNA expression of Bcl-2 and Bax in PC12 cells
Monolayers of adherent PC12 cells were prepared as described above. Total RNA from these cells was extracted using Trizol (Invitrogen). The RNA was quantified by OD at 260 and 280 nm with a Gene Quant Pro spectrophotometer (Beijing Technologies, Beijing, China). SYBR Green I PCR was performed in duplicate for each sample using the ABI 7500 PCR Sequence Detector (Applied Biosystems Inc., Foster City, CA, USA) and the SYBR Exscript™ RT-PCR Kit (TaKaRa Bio Inc.). The following primers were used as bcl-2: (i) forward primer, 5′- CGGGAGAACAGGGTATGA -3′; and (ii) reverse primer, 5′- CAGGCTGGAAGGAGAAGAT -3′), fragment length was 149 bp; Bax: (i) forward primer, 5′- TGGGCTGGACACTGGACT -3′; and (ii) reverse primer, 5′- GTGAGTGAGACGGTGAGGA -3′, fragment length was 148 bp; β-actin was amplified: (i) forward primer, 5′- GTAAAGACCTCTATGCCAACA -3′; and (ii) reverse primer, 5′- GGACTCATCGTACTCCTGCT -3′, fragment length was 227 bp; and used as a standard for each PCR analysis. These primers were designed and synthesized by TaKaRa Bio Inc. The conditions for the PCR were as follows: (i) 50 cycles of denaturation at 94°C for 30 s; (ii) annealing at 60°C for 30 s; (iii) and extension at 72°C for 30 s. The fold-change in gene expression was determined by the ΔΔCT method.
Analysis of protein expression of Bcl-2 and Bax in mitochondrial and cytosolic fractions by Western blot
Mitochondria were isolated using a QProteome Mitochondria Isolation Kit (Qiagen, Valencia, CA, USA). PC12 cells were washed and lysed. The supernatant was centrifuged (1000× g) for 10 min to separate the mitochondrial fractions (the pellets) from the cytosolic fractions (the supernatant). The cytosolic fractions were concentrated using acetone precipitation on ice for 1 h, followed by centrifugation (10 000× g) for 10 min and resuspension of the pellets in 1% SDS. The mitochondrial fractions were resuspended and sheared by passing 10 times through a 26 gauge needle. These lysates were then centrifuged (1000× g) for 10 min and the supernatant (mitochondrial fractions) was centrifuged again at 6000× g for 10 min. The pellets were resuspended in 1% SDS. After assay of protein concentration, the fractions were analysed by SDS-PAGE and immunoblotting.
Analysis of protein expression of CaMKII-dependent ASK-1/JNK/p38 signalling cascade in PC12 cells by Western blot
We measured the level of CaMKII and the ASK-1/JNK/p38 signalling cascade by Western blot analysis, with or without calcimycin (A-23187; 100 nM). At the same time, KN93 (a CaMKII inhibitor; 10 μM) was used as a positive control. PC12 cells were seeded out in 25 cm2 culture flasks (Nalge Nunc, Rochester, NY, USA) at a concentration of 5 × 105·cells mL−1. Western blots were performed following standard protocols. Total protein lysates were extracted with lysis buffer (125 mM of Tris-HCl, pH 6.8), 4% SDS, phosphatase inhibitors and protease inhibitor mixture. Protein concentration was measured by a quantification kit (Boster Biological Technology, Wuhan, China). Equal amounts of protein were separated by gel electrophoresis and electrophoretically transferred to PVDF membranes. After blocking with 5% non-fat dry milk, membranes were incubated with the following primary antibodies overnight at 4°C followed by HRP-conjugated secondary antibody (Santa Cruz, CA, USA) and developed with ECL reagent (Beyotime Institute of Biotechnology, Shanghai, China). Proteins were quantified with the Scan Pro software as a proportion of the signal of the housekeeping protein band (β-actin).
Data analyses
Data are presented as mean ± SD and differences among the groups were evaluated by anova for multiple groups using Prism GraphPad Software (San Diego, CA, USA). P-values below 0.05 were considered statistically significant.
Materials
Catalpol was purchased from the Chinese Institute for Drug and Biological Product Control (Beijing, China) and then was dissolved in DMSO. DMEM, penicillin, streptomycin and fetal calf serum (FCS) were purchased from Gibco BRL (Gaithersburg, MD, USA). LPS (Escherichia coli 055:B5) were obtained from Sigma-Aldrich, St. Louis, MO, USA. 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) and DMSO, Fluo-3/AM and Pluronic F127 were obtained from Sigma-Aldrich. Annexin V fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis detection kit and LDH were purchased by Nanjing Key-Gen Biotech Co., Ltd. (Nanjing, China). Trizol was obtained from Invitrogen (Carlsbad, CA, USA). The reverse transcriptase PCR (RT-PCR) kit (AMV Ver.3.0) and Genefinder were purchased from TaKaRa Bio Inc. (Dalian, China). Reactive oxygen species (ROS) assays and the ECL reagent kit were produced by Beyotime Institute of Biotechnology (Shanghai, China). Rabbit anti-rat CaMKII, phospho-CaMKII (Thr286), apoptosis signal-regulating kinase-1 (ASK-1), pASK-1 (Ser83), JNK, pJNK (Thr183/Tyr185), p38, p-p38 (Thr180/Tyr182) antibody and HRP-conjugated goat anti-rabbit IgG were supplied from Santa Cruz Biotechnology (Santa Cruz, CA, USA). KN93 and calcimycin (A-23187) were purchased from Sigma-Aldrich. All other reagents were of the highest purity commercially available.
Results
Determination of optimum concentration of Catalpol and LPS
The effects of catalpol on the viability of PC12 cells were time and concentration-dependent. We tested two exposure times – 12 and 24h- and a range of concentrations (10nM–100mM) and found that, for either incubation time, the maximum effect was exerted by 10 μM catalpol (Figure 1A). This effect (increased proliferation) was also significant at 1 and 100 μM catalpol after a 24h incubation but, after 12h incubation, only 10 μM was effective (Figure 1B). We therefore chose to use a 12h exposure to 10 μM catalpol as our standard pre-treatment.
Figure 1.
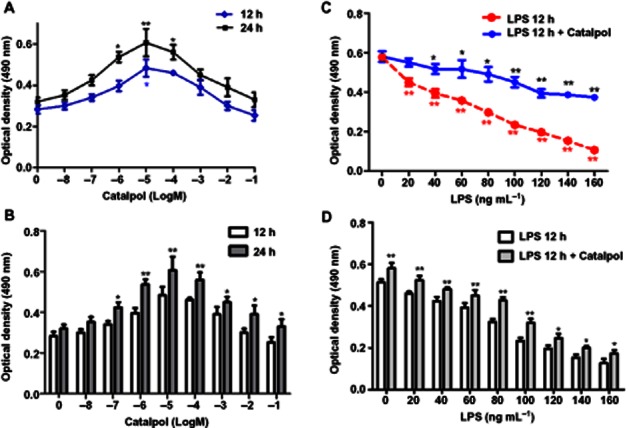
Viability of PC12 cells pretreated with catalpol and/or LPS. (A) PC12 cells were pretreated with different concentrations of catalpol for 12 or 24 h. The viability of cells was measured by the MTT assay. *P < 0.05, **P < 0.01 versus DMEM control. (B) PC12 cells were treated and the viability of cells was measured as in (A). Effects of two exposures (12 or 24h) to catalpol are shown. *P < 0.05, **P < 0.01, significantly different from 12h values. (C) PC12 cells were pretreated with catalpol (10 μM) for 12 h. Then apoptosis was induced by adding LPS (20–160 ng·mL−1) to the serum-free medium for 12 h. After washing with DMEM, the cells in LPS 12 h + Catalpol group were incubated in DMEM with catalpol (10 μM) for an additional 12 h. The cells in LPS 12 h group were incubated in DMEM medium for the additional 12 h. Cell viability of cells was measured by MTT assay. *P < 0.05, **P < 0.01 versus DMEM control. (D) PC12 cells were treated and the viability of cells assayed as in (C). The effects of adding catalpol for the additional 12 h is shown. Data are presented as means ± SD, n = 5. *P < 0.05, **P < 0.01, significant effect of catalpol.
The effect of LPS was assessed in PC12 cells given the standard pretreatment with catalpol (10 μM, 12h). As shown in Figure 1C, adding LPS (20–160 ng·mL−1) for 12h induced a concentration-dependent apoptosis, shown by the loss of viability. However when catalpol (10μM) was combined with LPS for 12h, the apoptosis was decreased, at every concentration of LPS (Figure 1D).
As the effect of catalpol appeared to be greatest at 80ng mL-1, LPS we chose this concentration as our standard stimulus in subsequent experiments.
Morphology of PC12 cells
Morphology of PC12 cells after catalpol treatment in the presence of LPS and the controls are shown in Figure 2A. After incubation with DMEM, differentiated PC12 cells displayed typical neuronal phenotype, which resembled that of sympathetic neurons. LPS-treated cells showed heterogeneity in their shape and were mostly detached from the culture plate surface. Cells pretreated with catalpol showed resistance to the injury induced by LPS, displaying almost uniform and typical neuronal shape, being mostly attached to the plate surface and maintaining visible networks.
Figure 2.
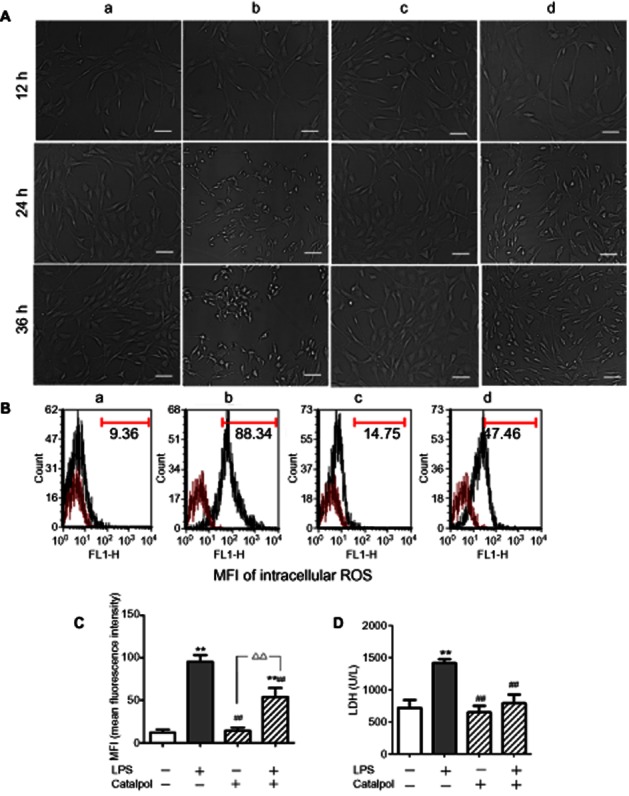
(A) Morphology of PC12 cells at different times (magnification: ×200, bar: 50 μm). (B) Intracellular ROS levels in PC12 cells pretreated with catalpol and LPS. Mean fluorescence intensity (MFI) was measured with DCFH-DA by flow cytometry. (C) MFI of intracellular ROS levels in PC12 cells pretreated with catalpol /LPS. (D) Viability assay in PC12 cells pretreated with catalpol and LPS. (E) Analysis of LDH leakage from PC12 cells pretreated with catalpol and LPS. A, DMEM control; B, LPS (80 ng·mL−1); C, catalpol (10 μM); D, LPS (80 ng·mL−1) + catalpol (10 μM). Data are presented as means ± SD, n = 5 **P < 0.01 versus DMEM control; ##P < 0.01 versus LPS, ▵▵P < 0.01, significantly different as shown.
Effect of catalpol on the generation of LPS-induced ROS in PC12 cells
The intracellular ROS level was determined by flow cytometry using DCFH-DA. As shown in Figure 2B, C, when PC12 cells were exposed to LPS, the mean fluorescence intensity of intracellular ROS levels increased significantly compared with DMEM control (P < 0.01). The increase was partially prevented by pre-incubation with catalpol, compared with the LPS only group (P < 0.01). The results suggested that catalpol could prevent the generation of LPS-induced ROS in PC12 cells.
Effect of catalpol on LDH leakage
To further investigate the protective effect of catalpol on LPS-exposed PC12 cells, release of LDH was assayed. As shown in Figure 2D, incubation of the cells with LPS caused a significant increase of LDH leakage into the cell medium, compared with that from control cells. In contrast, pre-incubation of the cells with catalpol considerably attenuated the leakage of LDH induced by LPS, compared with that from cells exposed to LPS only.
Effect of catalpol on protecting PC12 cells against LPS-induced apoptosis
Apoptosis in PC12 cells exposed to LPS was assayed by flow cytometry (Figure 3). The results showed that incubation of PC12 cells with LPS induced significantly increased levels of apoptosis, compared with the control group. After pretreatment with catalpol, apoptosis in PC12 cells induced by LPS was decreased significantly. These results suggested that catalpol was protecting the cells against LPS-induced apoptosis.
Figure 3.

Effect of catalpol on LPS-induced apoptosis in PC12 cells measured by flow cytometry. (A) The proportion (%) of cell number in each quadrant is shown. Lower left quadrant (absence of both markers) indicates viable cells; upper left quadrant [propidium iodide (PI) positive] indicated cellular necrosis. The total apoptotic cells (early- and late-stage apoptosis) were represented by the right side of the panel (Annexin V staining alone or together with PI). (B) Mean values of percent apoptotic PC12 cells pretreated with catalpol and LPS. A, DMEM control; B, LPS (80 ng·mL−1); C, catalpol (10 μM); D, LPS (80 ng·mL−1) + catalpol (10 μM). Data are presented as means ± SD, n = 5 **P < 0.01 versus DMEM control; ##P < 0.01 versus LPS, ▵▵P < 0.01, significantly different as shown.
Effect of catalpol on the intracellular calcium concentration ([Ca2+]i)
As shown in Figure 4, incubation with LPS increased both the basal and peak values of [Ca2+]i in PC12 cells, compared with control. However, in cells pretreated with catalpol, these changes in [Ca2+]i induced by LPS were reduced and the time of the peak delayed.
Figure 4.
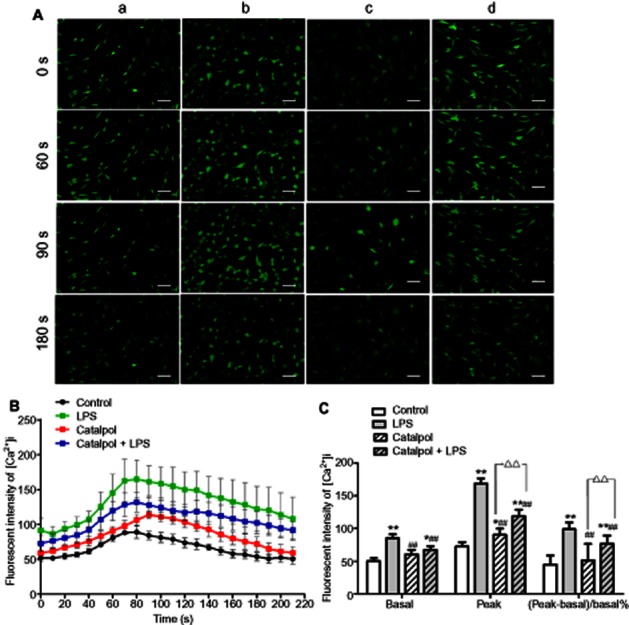
Effects of catalpol on intracellular calcium concentration ([Ca2+]i) in PC12 cells as measured by the Ca2+ indicator Fluo-3 by laser confocal scanning microscopy. (A) Images of [Ca2+]i (magnification: ×200, Bar: 50 μm), A, DMEM control; B, LPS (80 ng·mL−1); C, catalpol (10 μM); D, LPS (80 ng·mL−1) + catalpol (10 μM); (B) The time-course of changes in mean fluorescence intensity following addition of LPS. Mean values from 10–15 cells in each scan are shown, after the different treatments. (C) Basal and peak fluorescence intensity of [Ca2+]i in each group. Data are presented as means ± SD, n = 10. *P < 0.05, **P < 0.01 versus DMEM control; ##P < 0.01 versus LPS, ▵▵P < 0.01, significantly different as shown.
Effect of catalpol on the expression of Bcl-2 and Bax mRNA in PC12 cells
To determine whether the inhibitory effects of catalpol on apoptosis in PC12 cells were related to changes in Bcl-2 and Bax, the transcription levels of Bcl-2 and Bax mRNA were assessed using real-time RT-PCR (Figure 5). We found that LPS significantly decreased expression of Bcl-2 mRNA (Figure 5A), while significantly increasing expression of Bax mRNA (Figure 5B), compared with the control group. Pre-treatment with catalpol alone did not change either Bcl-2 or Bax expression but such pre-treatment did reverse the changes induced by LPS. Furthermore, as indicated in Figure 5C, the Bax/Bcl-2 ratios were increased by incubation with LPS, while treatment with catalpol before LPS decreased the Bax/Bcl-2 ratio.
Figure 5.
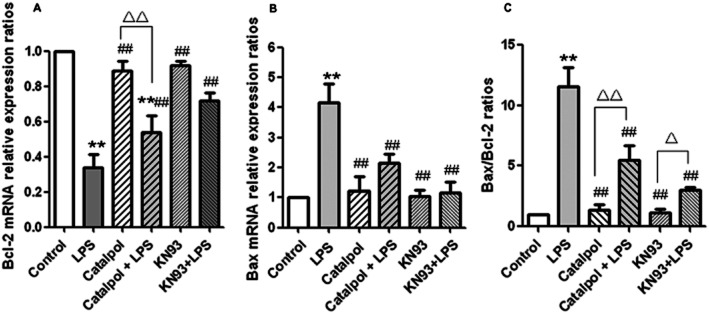
Effect of catalpol on the expression of mRNA for Bcl-2 and Bax in PC12 cells. The expression of Bcl-2 and Bax were estimated by quantitative real-time RT-PCR. Results are shown as relative expression ratio of Bcl-2 mRNA (A), Bax mRNA (B) and Bax/Bcl-2 ratio (C) in PC12 cells with respect to control group (ratio =1) and normalized by β-actin reference gene expression. Data are presented as means ± SD, n = 3. **P < 0.01 versus DMEM control; ##P < 0.01 versus LPS, ▵▵P < 0.01, significantly different as shown.
In the same series of experiments, we used KN93, a specific inhibitor of CaMKII, as a positive control to check whether these effects on expression of Bcl-2 and Bax were dependent on activation of CaMKII. In the presence of KN93, the changes induced by LPS in the expression of Bcl-2 and Bax mRNA and in their ratio was blocked, suggesting that the expression of Bcl-2 and Bax in the cells treated with LPS was dependent on CaMKII activation. As the changes in Bcl-2 and Bax after catalpol were similar to those after KN93, it is likely that the cytoprotective effects of catalpol were also dependent on CaMKII inhibition.
Effect of catalpol on expression of Bcl-2 and Bax proteins in mitochondrial and cytosolic fractions of PC12 cell
The expression of the pro-apoptotic factor Bax and that of the anti-apoptotic factor Bcl-2 were further analysed by Western blot analysis (Figure 6). Bcl-2 is predominantly localized to the mitochondria in non-myocytic cell types, whereas Bax is found primarily in the cytosol (Kim et al., 2009). In our cultures of PC12 cells, Bcl-2 was found in the mitochondrial fraction along with the mitochondrial marker, cytochrome oxidase and only a small fraction of Bcl-2 was detected in the cytosolic fraction. Bax was detected in both the mitochondrial and cytosolic fractions, with a greater proportion in the cytosol (Figure 6). As indicated in Figure 6A, in the mitochondrial fraction of PC12 cells after treatment with LPS, the expression of Bcl-2 protein was decreased whereas the expression of Bax was increased, compared with the control group. However, treatment with catalpol reversed these effects of LPS, increasing Bcl-2 expression and inhibiting Bax expression. Calculation of the Bax/Bcl-2 ratio in these cultures showed that incubation with LPS increased this ratio, compared with the control group, while catalpol decreased the raised Bax/Bcl-2 ratio after LPS.
Figure 6.
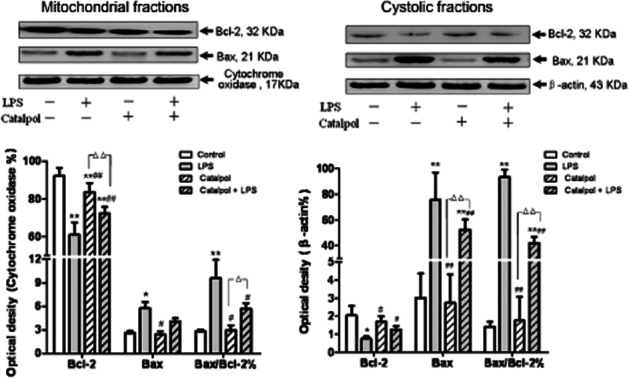
Effect of catalpol on levels of Bcl-2 and Bax proteins in mitochondrial and cytosolic fractions of PC12 cells. Electropherogram of Western blotting: lane 1, DMEM control; lane 2, LPS (80 ng·mL−1); lane 3, catalpol (10 μM); lane 4, catalpol (10 μM) + LPS (80 ng·mL−1). Summary data on expression levels of Bcl-2, Bax and ratio of Bax/Bcl-2 are presented as means ± SD, n = 3. *P < 0.05, **P < 0.01 versus DMEM control; #P < 0.05, ##P < 0.01 versus LPS; ▵▵P < 0.01, significantly different as shown.
In the cytosolic fraction of PC12 cells incubated with LPS (Figure 6B), the expression of Bcl-2 protein was decreased, while the expression of Bax increased significantly compared with the control group (P < 0.01). Pre-treatment with catalpol reversed these LPS-induced changes and those in the Bax/Bcl-2 ratios.
Effect of catalpol on the CaMKII signal transduction pathway in PC12 cells
CaMKII is a ubiquitous mediator of Ca2+-linked signalling that phosphorylates a wide range of substrates to coordinate and regulate Ca2+-mediated apoptosis (Salas et al., 2010) and activation of this pathway requires phosphorylation of CaMKII itself. To assess the involvement of CaMKII in the anti-apoptotic effect of catalpol after LPS, we measured levels of CaMKII and pCaMKII by Western blot. At the same time, KN93, a specific inhibitor of CaMKII, was used as a positive control and . We found that in PC12 cells exposed to LPS, the levels of pCaMKII increased significantly compared with control (Figure 7A, B), and this increase was abolished by pretreatment with catalpol (10 μM). This abolition by catalpol was reversed by adding the Ca2+ ionophore calcimycin As shown in Figure 7C, the percentage of apoptotic cells in catalpol-pretreated PC12 cells was decreased and calcimycin reversed this effect of catalpol. These results show a correlation between decreased levels of phosphorylation of CaMKII and anti-apoptotic effects, both due to catalpol.
Figure 7.

Effect of catalpol on levels of pCaMKII and CaMKIIin PC12 cells to mediate anti-apoptosis. (A) Electropherogram of Western blotting: lane 1, DMEM control; lane 2, LPS (80 ng·mL−1); lane 3, 10−5 M catalpol; lane 4, 10−5 M catalpol + LPS (80 ng·mL−1); lane 5, 1 mM calcimycin; lane 6, 10−5 M catalpol + LPS (80 ng·mL−1) + 1 mM calcimycin; lane7, KN93; lane8, LPS (80 ng·mL−1) + KN93. (B) Mean values of the pCaMKII / CaMKII ratio (as %) in PC12 cells. (C) Mean values of apoptotic cells (% total cells) measured by flow cytometry. The apoptotic cells include the cells in early- and late-stage apoptosis, which were represented in the right quadrants in the dot plot (Annexin V staining alone or together with PI) as described in Figure 3A. Data are presented as means ± SD, n = 3. *P < 0.05, **P < 0.01 versus DMEM control; #P < 0.05, ##P < 0.01 versus LPS; ▵P < 0.05, ▵▵P < 0.01, significantly different as shown.
Catalpol regulates anti-apoptosis through a CaMKII-dependent ASK-1/JNK/p38 signalling cascade in PC12 cells
ASK-1 is a key MAP kinase kinase kinase (MAPKKK) that activates both the JNK/SAPK and the p38 signalling cascades by directly phosphorylating and, therefore, activating MKK4 and 7 (SAPK pathway) and MKK3 and 6 (p38 pathway) (Arpad et al., 2009). Over-expression of ASK-1 is known to induce apoptotic cell death (Tobiume et al., 2001). We therefore tested the effect of catalpol on the Ca2+-induced ASK-1/JNK/p38 signalling cascade in PC12 cells exposed to LPS. We measured the levels of ASK-1, JNK, and p38 and their phosphorylated forms by Western blot analysis in the presence of bradykinin (to stimulate CaMK kinase). We found that in PC12 cells exposed to LPS, the levels of pASK-1, pJNK and p-p38 were significantly increased, compared with the control group (Figure 8). This increase was abolished by pretreatment of the cells with catalpol (10μM) and the effect of catalpol was itself reversed by treatment with calcimycin. These results showed that catalpol prevented CaMKII-dependent activation of the ASK-1/JNK/p38 signalling cascade.
Figure 8.

Catalpol regulates anti-apoptosis through CaMKII-dependent ASK-1/JNK/p38 signalling cascade in PC12 cells. (A) Electropherogram of Western blotting: lane 1, DMEM control; lane 2, LPS (80 ng·mL−1); lane 3, catalpol (10 μM); lane 4, catalpol (10 μM) + LPS (80 ng·mL−1); lane 5, 1 mM calcimycin; lane 6, catalpol (10 μM) + LPS (80 ng·mL−1) + 1 mM calcimycin; lane 7, KN93; lane 8, LPS (80 ng·mL−1) + KN93. (B–D) Mean values of ratios of pASK/ASK (B), p-p38/p38 (C), and pJNK/JNK (D) in PC12 cells. Data are presented as means ± SD, n = 3. *P < 0.05, **P < 0.01 versus DMEM control; #P < 0.05, ##P < 0.01 versus LPS, ▵P < 0.05, ▵▵P < 0.01, significantly different as shown.
Discussion and conclusions
Apoptosis is a highly regulated process that controls normal development and homeostasis of multicellular organisms. The ratio of anti-apoptotic (such as Bcl-2) versus pro-apoptotic proteins (such as Bax) has been shown to dictate the ultimate sensitivity or resistance of cells to various apoptotic stimuli. The balance of Bax and Bcl-2 an important determinant of cell death or survival (Liu et al., 2011; Martinou and Youle, 2011). Bax translocates from the cytosol to the outer mitochondrial membrane where it can form Bax: Bcl-2 heterodimers and tetramers, leading to pore formation and cytochrome C release, and resulting in caspase activation (Roy et al., 2009; Zhang et al., 2011). Many studies have shown that the Bax/Bcl-2 ratio plays a key role in determining whether cells could undergo apoptosis (Raisova et al., 2001; Reagan-Shaw et al., 2008). In our present study, real-time RT-PCR and Western blotting showed that catalpol stimulated the expression of Bcl-2 protein and significantly decreased the Bax/Bcl-2 ratio. At the same time, catalpol decreased the translocation of Bax from the cytosol to the outer mitochondrial membrane and ultimately mediated anti-apoptosis.
Apoptosis can be triggered by increased [Ca2+]i and CaMK plays a crucial role in apoptosis in neurons by activating transcription factors (see Wayman et al., 2008). The CaMKII transcriptional pathway regulates the expression of both Bcl-2 and Bax through phosphorylation (Vanderheyden et al., 2009). Exposure to LPS results in Ca2+ influx into neurons, which led to the activation of Ca2+-mediated signal transduction, including activation of CaMK and PKC isoenzymes (Fährmann et al., 2002).
Our results indicated that LPS activated CaMKII by increasing [Ca2+]i and that the phosphorylated CaMKII could reinforce Ca2+-mediated apoptosis. We found that treatment with catalpol inhibited this phosphorylation of CaMKII and, in turn, attenuated Ca2+-mediated apoptosis. At the same time, inhibition by catalpol was blocked by calcimycin, a potent Ca2+ ionophore. These results showed that the cytoprotective effects of catalpol in PC12 cells treated with LPS could be due to inhibition of the phosphorylation of CaMKII and activation of anti-apoptotic mechanisms. Our results also suggest that LPS-induced apoptosis activated several protein kinases in PC12 cells, including CaMKII and ASK-1, through a Ca2+-dependent mechanism. It has been suggested that phosphorylation of CaMK could control its ability to regulate transcription in apoptosis cells (Nutt et al., 2005). Our results show that catalpol contributed to the down-regulation of CaMK phosphorylation in PC12 cells.
CaMKII activity is likely to be sustained through autophosphorylation and possibly oxidation of the auto-inhibitory domain, but inhibition of CaMKII phosphatase may also sustain activation. Activated CaMKII then could induce apoptosis through at least three pathways, all of which are necessary for cell death: (i) activation of JNK, which induces the activation of Fas (Timmins et al., 2009); (ii) stimulation of Ca2+ uptake by the mitochondria, leading to outer mitochondrial membrane permeabilization and release of apoptogens, as well as loss of the mitochondrial membrane potential (O'Connell and Stenson-Cox, 2007); and (iii) activation of STAT1 (Tabas, 2009), which has previously been shown to promote apoptosis in diverse cell types by a number of transcriptional and perhaps non-transcriptional mechanisms.
Apoptosis occurs through an orchestrated sequence of intracellular signalling cascades. In particular, the MAPK signalling pathways have been known as highly conserved cascades for regulation of cell death and survival (Wada and Penninger, 2004). There are at least six independent MAPK signalling units in mammalian systems. Three of them play a critical role in apoptotic cell death; (i) the ERK pathway; (ii) JNK (also known as stress-activated protein kinase, SAPK) pathway; and (iii) the p38 pathway (Yang et al., 2000). Another important input into the MAPK system is ASK-1, also known as MAP3K5) which is a key MAPKKK that activates both the JNK/SAPK and the p38 signalling cascades by directly phosphorylating its substrates in response to an array of stresses such as oxidative stress, endoplasmic reticulum stress and calcium influx (Tobiume et al., 2001). ASK-1 activity is involved in cancer, diabetes, cardiovascular and neurodegenerative diseases and over-expression of ASK-1-induced apoptotic cell death (Homma et al., 2009; Limón-Pacheco and Gonsebatt, 2009). However, whether and how Ca2+ signals regulate p38 remains to be elucidated. In this study, we demonstrated that Ca2+ signals regulated the ASK-1–p38 MAPK cascade and that CaMKII served as an activator of the ASK-1–p38 axis in Ca2+-mediated signal transduction. In our work, using cultured cells, MTT assay, real-time PCR and Western blotting, we found that catalpol inhibited the apoptosis of PC12 cells induced by LPS. Moreover a possible mechanism was through up-regulation of the CaMK mRNA and a decrease of [Ca2+]i through the CaMKII-dependent ASK-1/JNK/p38 signalling cascade.
Rat PC12 cells differentiated by nerve growth factor (NGF) have been extensively used to study the differentiation and apoptosis of neurons. These PC12-derived cells are a useful model system for primary neurons because, upon exposure to NGF, they undergo partial growth arrest, cease to divide, extend processes and exhibit specific changes in protein expression, developing a phenotype resembling mature sympathetic neurons (Sombers et al., 2002; Martinez et al., 2010; Tomas et al., 2012; Zhang et al., 2012). Thus, we used differentiated PC12 cells, as a model of neurons, to study the protective effect of catalpol on apoptosis. However, cultures of cells from the embryonic subependymal zone (SEZ), hippocampal gyrus or dentate gyrus are closer models of Alzheimer's or Parkinson's diseases (Masliah et al., 2012; Nagahara et al., 2009). So, we plan to investigate the effects of catapol in other models, such as the cultures from the embryonic SEZ, in the next step of our research in order to corroborate and extend the conclusions of this study.
In this study, we investigated the intracellular signalling pathways of Ca2+-induced apoptosis, which were initiated by phosphorylation of CaMKII, and followed by the serial activation of other protein kinases, such as ASK-1, JNK and p38. Using a model of inflammation in PC12 cells, we found that activation of CaMKII was critically involved in the response to LPS. Also, the JNK/p38 signalling cascade was involved in the regulation of phosphorylation of CaMKII. Inhibition of apoptosis in PC12 cells treated with LPS by catalpol was effected through suppressing phosphorylation of CaMKII, which couples its activation to further downstream signalling processes. This is the first demonstration that the anti-apoptotic activity of catalpol involved CaMKII-dependent ASK-1/JNK/p38 signalling pathways during LPS-induced apoptosis in PC12 cells, as a model of neurons.
The data presented here provide new mechanistic insights into the links between the CaMKII-dependent ASK-1/JNK/p38 signalling cascade and the protective effect of catalpol against apoptosis induced by LPS in PC12 cells. In summary, catalpol protected PC12 cells against LPS-induced apoptosis and this action suggests it might be a potential neuroprotective agent.
Acknowledgments
The study was supported by the Science Foundation of Liaoning University of Traditional Chinese Medicine.
Glossary
- ASK-1
apoptosis signal-regulating kinase-1
- CaMKII
Ca2+-calmodulin-dependent protein kinase II
- DCF
dichlorofluorescein
- DCFH-DA
2,7-dichlorofluorescein diacetate
- FCS
fetal calf serum
- FITC
fluorescein isothiocyanate
- MTT
3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl-tetrazolium bromide
- NGF
nerve growth factor
- PC12 cell
pheochromocytoma cell
- PI
propidium iodide
- ROS
reactive oxygen pecies
- RT-PCR
reverse transcriptase PCR
Authors' contributions
WC contributed to the general design, performed the experiments, and contributed to the statistical data analysis and the writing of the manuscript. XL contributed to the writing and review of the manuscript. LZ and JW performed the statistical analysis. LJ contributed to the analysis of the studies. JW and DH participated in its design and coordination. LR contributed to the overall experimental design, data interpretation and critical manuscript review. All authors read and approved the final manuscript.
Conflict of interest
The authors declare that they have no competing interests.
References
- Arpad B, Szabolcs B, Andras S, Eva P, Zita B, Balazs S, et al. Potentiation of paclitaxel-induced apoptosis by galectin-13 overexpression via activation of Ask-1-p38-MAP kinase and JNK/SAPK pathways and suppression of Akt and ERK1/2 activation in U-937 human macrophage cells. Eur J Cell Biol. 2009;88:753–763. doi: 10.1016/j.ejcb.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burguillos MA, Hajji N, Englund E, Persson A, Cenci AM, Machado A, et al. Apoptosis-inducing factor mediates dopaminergic cell death in response to LPS-induced inflammatory stimulus: evidence in Parkinson's disease patients. Neurobiol Dis. 2011;41:177–188. doi: 10.1016/j.nbd.2010.09.005. [DOI] [PubMed] [Google Scholar]
- Chen WN, Liu JL, Meng JJ, Lu CL, Li XM, Wang EH, et al. Macrophage polarization induced by neuropeptide-methionine enkephalin (MENK) promotes tumoricidal responses. Cancer Immunol Immunother. 2012;61:1755–1768. doi: 10.1007/s00262-012-1240-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fährmann M, Kaufhold M, Rieg T, Seidler U. Different actions of protein kinase C isoforms α and ε on gastric acid secretion. Br J Pharmacol. 2002;136:938–946. doi: 10.1038/sj.bjp.0704790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma K, Katagiri K, Nishitoh H, Ichijo H. Targeting ASK1 in ER stress-related neurodegenerative diseases. Expert Opinion on Therapeutic Targets. 2009;13:653–664. doi: 10.1517/14728220902980249. [DOI] [PubMed] [Google Scholar]
- Jiang B, Du J, Liu JH, Bao YM, An LJ. Catalpol attenuates the neurotoxicity induced by beta-amyloid (1-42) in cortical neuron-glia cultures. Brain Res. 2008;1188:139–147. doi: 10.1016/j.brainres.2007.07.105. [DOI] [PubMed] [Google Scholar]
- Jiang B, Liu JH, Bao YM, An LJ. Catalpol inhibits apoptosis in hydrogen peroxide-induced PC12 cells by preventing cytochrome c release and inactivating of caspase cascade. Toxicon. 2004;43:53–59. doi: 10.1016/j.toxicon.2003.10.017. [DOI] [PubMed] [Google Scholar]
- Jumblatt JE, Tischler AS. Regulation of muscarinic ligand binding sites by nerve growth factor in PC12 phaeochromocytoma cells. Nature. 1982;297:152–154. doi: 10.1038/297152a0. [DOI] [PubMed] [Google Scholar]
- Kim H, Tu HC, Ren DC, Takeuchi O, Jeffers JR, Zambetti GP, et al. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell. 2009;36:487–499. doi: 10.1016/j.molcel.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Patrick MF, Hong JS. Neuroinflammation is a key player in Parkinson's disease and a prime target for therapy. J Neural Transm. 2010;117:971–979. doi: 10.1007/s00702-010-0428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limón-Pacheco J, Gonsebatt ME. The role of antioxidants and antioxidant-related enzymes in protective responses to environmentally induced oxidative stress. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 2009;674:137–147. doi: 10.1016/j.mrgentox.2008.09.015. [DOI] [PubMed] [Google Scholar]
- Liu N, Zheng YJ, Zhu Y, Xiong SD, Chu YW. Selective Impairment of CD4+ CD25+ Foxp3+ regulatory T cells by paclitaxel is explained by Bcl-2/Bax mediated apoptosis. Int Immunopharmacol. 2011;11:212–219. doi: 10.1016/j.intimp.2010.11.021. [DOI] [PubMed] [Google Scholar]
- Martinez JA, Zhang ZQ, Svetlov SI, Hayes RL, Wang KK, Larner SF. Calpain and caspase processing of caspase-12 contribute to the ER stress-induced cell death pathway in differentiated PC12 cells. Apoptosis. 2010;15:1480–1493. doi: 10.1007/s10495-010-0526-4. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21:92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Hansen LA. Alzheimer disease: AD pathology-emerging subtypes or age-of-onset spectrum? Nature Reviews Neurology. 2012;8:11–12. doi: 10.1038/nrneurol.2011.205. [DOI] [PubMed] [Google Scholar]
- Meng HW, Li C, Feng L, Cheng BH, Wu FX, Wang XH, et al. Effects of ginkgolide B on 6-OHDA-induced apoptosis and calcium over load in cultured PC12. Int J Dev Neurosci. 2007;25:509–514. doi: 10.1016/j.ijdevneu.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nature Medicine. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt LK, Margolis SS, Jensen M, Herman CE, Dunphy WG, Rathmell JC, et al. Metabolic regulation of oocyte cell death through the CaMKII-mediated phosphorylation of caspase-2. Cell. 2005;123:89–103. doi: 10.1016/j.cell.2005.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell AR, Stenson-Cox C. A more serine way to die: defining the characteristics of serine protease-mediated cell death cascades. Biochim Biophys Acta. 2007;1773:1491–1499. doi: 10.1016/j.bbamcr.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Raisova M, Hossini AM, Eberle J, Riebeling C, Wieder T, Sturm I, et al. The Bax/Bcl-2 ratio determines the susceptibility of human melanoma cells to CD95/Fas-mediated apoptosis. J Invest Dermatol. 2001;117:333–340. doi: 10.1046/j.0022-202x.2001.01409.x. [DOI] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahsan H, Mukhtar H, Ahmad N. Combination of vitamin E and selenium causes an induction of apoptosis of human prostate cancer cells by enhancing Bax/Bcl-2 ratio. Prostate. 2008;68:1624–1634. doi: 10.1002/pros.20824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy SS, Madesh M, Davies E, Antonsson B, Danial N, Hajnóczky G. Bad targets the permeability transition pore independent of Bax or Bak to switch between Ca2+-dependent cell survival and death. Mol Cell. 2009;33:377–388. doi: 10.1016/j.molcel.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- Salas MA, Valverde CA, Sánchez G, Said M, Rodriguez JS, Portiansky EL, et al. The signaling pathway of CaMKII-mediated apoptosis and necrosis in the ischemia/reperfusion injury. J Mol Cell Cardiol. 2010;48:1298–1306. doi: 10.1016/j.yjmcc.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sombers LA, Colliver TL, Ewing AG. Differentiated PC12 cells: a better model system for the study of the VMAT's effects on neuronal communication. Ann N Y Acad Sci. 2002;971:86–88. doi: 10.1111/j.1749-6632.2002.tb04439.x. [DOI] [PubMed] [Google Scholar]
- Tabas I. Macrophage apoptosis in atherosclerosis: consequences on plaque progression and the role of endoplasmic reticulum stress. Antioxid Redox Signal. 2009;11:2333–2339. doi: 10.1089/ars.2009.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian YY, An LJ, Jiang L, Duan YL, Chen J, Jiang B. Catalpol protects dopaminergic neurons from LPS-induced neurotoxicity in mesencephalic neuron-glia cultures. Life Sciences. 2006;80:193–199. doi: 10.1016/j.lfs.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Timmins JM, Ozcan L, Seimon TA, Gang L, Malagelada C, Backs J, et al. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Invest. 2009;119:2925–2941. doi: 10.1172/JCI38857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001;2:222–228. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomas M, Marin MP, Martinez-Alonso E, Esteban-Pretel G, Diaz-Ruiz A, Vazquez -Martinez R, et al. Alcohol induces Golgi fragmentation in differentiated PC12 cells by deregulating Rab1-dependent ER-to-Golgi transport. Histochem Cell Biol. 2012;138:489–501. doi: 10.1007/s00418-012-0970-z. [DOI] [PubMed] [Google Scholar]
- Vanderheyden V, Devogelaere B, Missiaen L, Smedthd HD, Bultynck G, Parys JB. Regulation of inositol 1,4,5-trisphosphate-induced Ca2+ release by reversible phosphorylation and dephosphorylation. Biochim Biophys Acta. 2009;1793:959–970. doi: 10.1016/j.bbamcr.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- Wayman GA, Lee YS, Tokumitsu H, Silva A, Soderling TR. Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron. 2008;59:914–931. doi: 10.1016/j.neuron.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki M, Chiba K, Mohri T. Neuritogenic effect of natural iridoid compounds on PC12h cells and its possible relation to signaling protein kinases. Biological & Pharmaceutical Bulletin. 1996;19:791–795. doi: 10.1248/bpb.19.791. [DOI] [PubMed] [Google Scholar]
- Yang GH, Jarvisc BB, Chung YJ, Pestka JJ. Apoptosis induction by the satratoxins and other trichothecene mycotoxins: relationship to ERK, p38 MAPK, and SAPK/JNK activation. Toxicol Appl Pharmacol. 2000;164:149–160. doi: 10.1006/taap.1999.8888. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Huang WD, Lv XY, Yang YM. Puerarin protects differentiated PC12 cells from H2O2-induced apoptosis through the PI3K/Akt signalling pathway. Cell Biol Int. 2012;36:419–426. doi: 10.1042/CBI20100900. [DOI] [PubMed] [Google Scholar]
- Zhang ZC, Song T, Zhang TT, Gao J, Wu GY, An LJ, et al. A novel BH3 mimetic S1 potently induces Bax/Bak-dependent apoptosis by targeting both Bcl-2 and Mcl-1. Int J Cancer. 2011;128:1724–1735. doi: 10.1002/ijc.25484. [DOI] [PubMed] [Google Scholar]
- Zhu HF, Wan D, Luo Y, Zhou JL, Chen L, Xu XY. Catalpol increases brain angiogenesis and up-regulates VEGF and EPO in the rat after permanent middle cerebral artery occlusion. Int J Biol Sci. 2010;6:443–453. doi: 10.7150/ijbs.6.443. [DOI] [PMC free article] [PubMed] [Google Scholar]