

Abstract
Background and Purpose
Oestrogens can interact directly with membrane receptors and channels and can activate vascular BKCa channels. We hypothesized that novel oestrogen derivatives could relax smooth muscle by an extracllular effect on the α and β1 subunits of the BKCa channel, rather than at an intracellular site.
Experimental Approach
We studied the effects of novel oestrogens on the tension of pre-contracted isolated rat aortic rings, and on the electrophysiological properties of HEK 293 cells expressing the hSloα or hSloα+β1 subunits. Two of the derivatives incorporated a quaternary ammonium side-chain making them membrane impermeable.
Key Results
Oestrone, oestrone oxime and Quat DME-oestradiol relaxed pre-contracted rat aorta, but only Quat DME-oestradiol-induced relaxation was iberiotoxin sensitive. However, only potassium currents recorded in HEK 293 cells over-expressing both hSloα and hSloβ1 were activated by oestrone, oestrone oxime and Quat DME-oestradiol.
Conclusion and Implications
The novel oestrogens were able to relax smooth muscle, but through different mechanisms. In particular, oestrone oxime required the presence of the endothelium to exert much of its effect, whilst Quat DME-oestradiol depended both on NO and BKCa channel activation. The activation of BKCa currents in HEK 293 cells expressing hSloα+β1 by Quat DME-oestradiol is consistent with an extracellular binding site between the two subunits. The binding site resides between the extracellular N terminal of the α subunit and the extracellular loop between TM1 and 2 of the β1 subunit. Membrane-impermeant Quat DME-oestradiol lacks an exchangeable hydrogen on the A ring obviating antioxidant activity.
Keywords: oestrogens, Ca++-activated K+ channel, β1 subunit
Introduction
The large conductance calcium-activated potassium channel, also called BKCa, maxi-K or KCa1.1, (Alexander et al., 2009) responds to both changes in membrane potential and increases in intracellular-free calcium; consequently, the BKCa channel is able to regulate membrane potential in a variety of smooth muscle cells (del Corsso et al., 2006; Layne et al., 2008; Werner et al., 2008). This channel acts as a negative feedback mechanism because it hyperpolarizes and relaxes smooth muscle following calcium entry (McManus et al., 1995; Brenner et al., 2000). In smooth muscle, the BKCa channel is composed of α pore-forming subunits and β1 regulatory subunits. A reduction of BKCa channel expression and function is associated with ageing arteries (Nishimaru et al., 2004), and a reduction of β1 regulatory subunit expression is associated with hypertension (Brenner et al., 2000; Pluger et al., 2000; Pluznick et al., 2003). The β1 regulatory subunit confers increased calcium sensitivity on the BKCa channel, shifting the current voltage relationship to potentials that are more negative and slows the activation of the BKCa current. It was the discovery of these regulatory subunits (β1-β4) that explained the variation of BKCa channel behaviour in differing tissues. The β1 regulatory subunit is, therefore, a potential target for pharmacological intervention, as it co-assembles with the α subunit in arteriole smooth muscle and is responsible for the observed behaviour.
Oestrogens such as 17β-oestradiol are known to relax vascular smooth muscle. A number of mechanisms have been proposed for this effect, including increasing NO release by endothelial cells (Broughton et al., 2010), as well as direct effects on smooth muscle, such as inhibition of calcium channels (Cairrao et al., 2012), elevating cyclic GMP (Rosenfeld et al., 2009), activating oestrogen receptors (Han et al., 2006), activating nNOS (Royal et al., 2011) and finally activating BK channels (White et al., 2002).
We have shown that 17β-oestradiol is able to activate BKCa channels when BKCa channels are inserted into planar lipid bilayers. This activation is stereospecific, 17α-oestradiol being inactive, and requires the presence of the β1 subunit. We have also shown that in order to activate the channel in planar lipid bilayers (De Wet et al., 2006; de Wet et al., 2009), at least two β1 subunits are required to be associating with a functional channel. This work is consistent with those of other investigators who demonstrated that 17β-oestradiol requires the β1 subunit to activate the BKCa channel (Valverde et al., 1999a,b). Deletion of the N terminal of the S0 domain removes the ability of 17β-oestradiol to activate the channel and shift the G–V relationship to negative potentials, but does not remove the ability of the β1 subunit to slow the activation of the channel (Morrow et al., 2006). What these studies imply is that β1 subunit interaction with the α subunit is subtle, with more than one point of interaction between the subunits. These studies also imply that the binding site for 17β-oestradiol is extracellular, as the N-terminal amino acids of BKCa are extracellular (Liu et al., 2010).
Other oestrogens and xenoestrogens are able to regulate the behaviour of a variety of channels; both steroidal oestrogens and non-steroidal antioestrogens have been reported to activate BKCa channels (Dick and Sanders, 2001; Dick et al., 2001; Dick, 2002). However, non-steroidal antioestrogens, such as tamoxifen and toremifene, have been shown to modulate a variety of ion channels (Allen et al., 1998; 1999; 2000; Sahebgharani et al., 2001). We have synthesized membrane impermeable derivatives of tamoxifen, such as ethyl bromide tamoxifen, to investigate the binding site of these compounds and have demonstrated that, for some channels, the binding site is extracellular, while for others, it lies deep within the membrane or on an intracellular component of the channel (Sahebgharani et al., 2001).
The present study has investigated modified steroidal oestrogens and their actions on the BKCa channel. We have synthesized oestrogens, which incorporate some of the features of tamoxifen and its membrane impermeable analogue, ethyl bromide tamoxifen, in order to generate novel BKCa channel activators. If the binding site for oestrogens is on the extracellular domains of the channel, then both steroidal oestrogens and the membrane impermeable analogues will activate the channel; if the binding site for these hormones is between the α and β1 subunits, then the ligand should only activate if the BKCa channel is co-expressed with the β1 subunit.
Methods
Cell culture
HEK 293 cells, stably expressing hSloα-V5-His6, were cultured in MEM supplemented with FBS 10% and 5 μg·mL−1 blasticidin to maintain selection. HEK 293 cells, stably co-expressing hSloα-V5-His6 and hSloβ1-myc-His6, were cultured in MEM supplemented with FBS 10%, 5 μg·mL−1 blasticidin and 1 mg·mL−1 G418 (Invitrogen, Paisley, UK) for selection (De Wet et al., 2006).
Prior to patch clamp recordings, 5 × 104 cells were seeded on to a 35 mm tissue culture dish and incubated for another 48 h at 37°C in 5% CO2.
Whole cell patch-clamp recordings
Macroscopic currents were recorded from whole cells by use of standard patch clamp techniques using a PC501A patch clamp amplifier (Warner Instruments, Hamden, CT, USA). Patch electrodes were fabricated from borosilicate glass tubing and, when filled with the pipette solution detailed later, had resistances of 2–5 MΩ. Cells were viewed through an inverted microscope and continuously superfused with an extracellular solution containing in mM: 136 NaCl, 2.6 CaCl2, 2.4 KCl, 1.2 MgCl2, 15 HEPES, 10 glucose, titrated with 3 M NaOH to pH 7.4. Perfusion of the experimental chamber was at a rate of approximately 1 mL per minute, the volume of fluid within the chamber being held constant by continuous aspiration and the temperature of the perfusion fluid was maintained at 21°C. Patch electrodes were filled with a solution containing in mM: 110 KCl, 3.0 MgCl2, 40 HEPES, 3 EGTA, titrated with 3 M KOH to pH 7.4. Atomic absorption spectroscopy revealed total Ca++ to be 7.04 μM, which is an equivalent free concentration of approximately 0.2 nM at room temperature and pH 7.35. CaCl2 was added to this solution to adjust the free Ca++ concentration, where necessary, and the free concentration calculated using Maxchelator software (Pacific Grove, CA, USA).
Voltage-gated BKCa currents were recorded from cells clamped at a holding potential of −40 mV. Outward BKCa currents were evoked by stepwise changes in membrane potential lasting for 50 ms to a range of potentials (−30 to +60 mV). Raw output currents were filtered with a low band pass filter at 5 kHz, and digitized at 25 kHz, using a CED 1401 (Cambridge, UK) interface connected to a PC computer running winWCP (v4.08) patch clamp software (Strathclyde Electrophysiological Software, University of Strathclyde, Glasgow, UK).
For some of the analysis BKCa conductance was determined from whole cell current according to:
Where GK is the conductance and IK is the K channel current and Vm and VK are the membrane potential at which the current was measured and the current reversal potential (−97 mV) respectively. The computed conductance was normalized to allow better comparison among cells of different sizes.
Preparation of isolated rat thoracic aorta rings
Adolescent male Sprague-Dawley rats, supplied by Charles River (London, UK; weight approximately 180–240 g), were killed by cervical dislocation. Thoracic aortae were removed and cleared of periadvential fat and then dissected into four rings (3–4 mm width). To obviate the effects of NO release, the endothelial layer was removed mechanically in half of the rings by gently rubbing the intimal surface of the artery lumen with a matchstick. All rings were then mounted in organ baths (AD Instruments, DMT, Oxford, UK) which were filled with warmed (37°C) and gas-equilibrated (95% O2, 5% CO2) Krebs' solution containing (in mmol·L−1) CaCl2 1.6, MgSO4 1.17, EDTA 0.026, NaCl 130, NaHCO3 14.9, KCl 4.7, KH2PO4 1.18 and glucose 11. Isometric tension was measured with isometric transducers, digitized using a MacLab A/D converter (AD Instruments), stored, and displayed on a MacIntosh computer. A pre-load tension of 7.5 mN was applied, and the rings were equilibrated for 60 min, changing the Krebs' solution fully every 15 min. Relaxation is expressed as a percentage of the steady-state tension (100%). Phenylephrine (1 nM–100 μM) was used to induce vasoconstriction of the aortic rings, and dose–response curves were generated. Rings were pre-contracted with a sub-maximal concentration of phenylephrine (1 μM). Following pre-contraction, cumulative concentrations (0.3, 1.0, 3.0, 10.0 and 30.0 μM) of oestrone or its derivatives were added at 8 min intervals. The pre-contraction in denuded and intact rings was not significantly different. The artery endothelium viability and integrity were confirmed by dilatory response of the ring to acetylcholine (1 nM–100 μM). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010).
Statistical analysis
Data were analyzed using GraphPad Prism version 5.03. Data are presented as the mean +/– the SEM unless otherwise stated.
Concentration response relationships for rubbed and unrubbed aortic rings were compared using two-way anova. Where possible, sigmoidal curve fitting (Y = 100/(1 + 10((LogEC50-X)*Hill Slope)) was performed using GraphPad Prism software version 5.03 for Windows (GraphPad Software, La Jolla, CA, USA).
The effects of oestrogen derivatives on normalized peak current over time were compared with control recordings using a Kruskal–Wallis test followed by a Dunns multiple comparison test.
The conductance voltage data (G–V data) were fitted with the Boltzmann function using GraphPad Prism 5.03. The rate at which BKCa currents developed were determined by fitting a single exponential to the rising phase of the BKCa current using winWCP (v4.08) acquisition software.
Materials
Oestrogen derivatives were synthesized in the laboratories of the University of Brighton, the starting material was oestrone (Figure 1). DME-oestrone and DME-oestradiol contained the tamoxifen side chain whilst Quat DME-oestrone and Quat DME-oestradiol contained the ethyl bromide tamoxifen (EBT) side chain (Allen et al., 2000; Sahebgharani et al., 2001). TLC, elemental analysis, 1H, 13C NMR and IR spectroscopy confirmed structures and purity.
Figure 1.
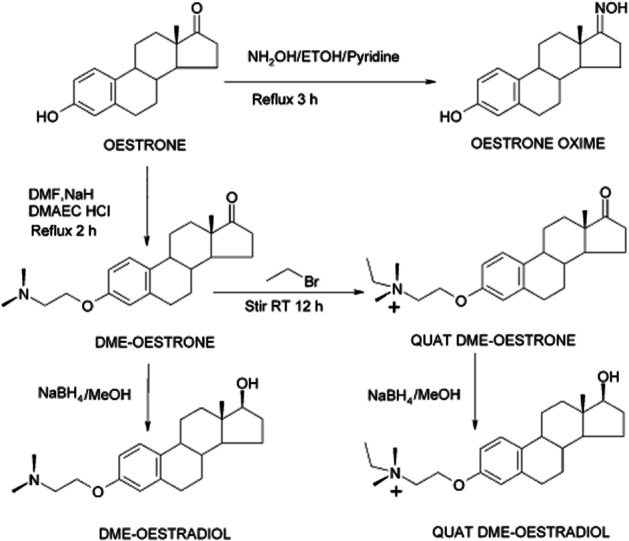
The synthesis of oestrogen derivatives from the parent compound oestrone. The derivatives contain the tamoxifen side chain. Structure and purity were confirmed by NMR, elemental analysis and IR spectroscopy.
Results
The effect of novel steroidal oestrogens on isolated rat thoracic aorta rings
The novel oestrogens were tested for their ability to relax pre-contracted aorta. Aortic rings, with or without intact endothelium, contracted when exposed to phenylephrine (1 nM–100 μM). Removing the endothelium made the aorta more sensitive to phenylephrine, shifting the pEC50 from 6.7 ± 0.15 to 7.71 ± 0.56. Most of the oestrone derivatives could produce a relaxant response in aortic rings pre-contracted with 1 μM phenylephrine (Figure 2). Oestrone oxime appeared to be the most potent analogue of oestrone, but this potency was dependent on the presence of an intact endothelium. The pEC50 was 5.42 ± 0.06 in aortic rings with an intact endothelium and was 4.53 ± 0.07 in aortic rings with the endothelium removed. Quat DME-oestrone appeared to lack relaxant activity, but Quat DME-oestradiol, DME-oestradiol and DME-oestrone were able to relax aorta in an endothelium independent manner. In our hands, only the relaxation by Quat DME-oestradiol showed reversibility with 100 nM iberiotoxin (IBTX) (Figure 3A), implying that the other analogues can induce relaxation independent of BKCa channels. The effects of oestrogens on arterial smooth muscle are known to involve both endothelium-dependent and endothelium-independent mechanisms, and it has been proposed that 17β-oestradiol can directly relax arteries by inducing nNOS activity within arterial smooth muscle. While reversibility of response by IBTX implies BKCa channel involvement, we wanted to be certain that any responses observed were direct rather than indirect, involving gaseous signalling molecules, such as NO. We therefore investigated the relaxant effects of Quat DME-oestradiol in aortic rings with endothelium removed and in the presence of a NOS inhibitor. These experiments demonstrated that 200 μM L-NAME partially reduced the relaxant effects of Quat DME-oestradiol, implying that Quat DME-oestradiol relaxations are at least in part due to nNOS activation (Figure 3B, C, D).
Figure 2.

The effect of novel steroidal oestrogens on isolated rat thoracic aorta rings. Cumulative concentration response curves were constructed in rubbed (•) and unrubbed (□) aortic rings. Ligands tested were (A) 17β-oestradiol (n = 12), (B) DME-oestradiol (n = 6), (C) Quat DME-oestradiol (n = 7), (D) Oestrone (n = 11), (E) DME-oestrone (n = 6), (F) Quat DME-oestrone (n = 6), (G) Oestrone oxime (n = 7). Rubbed versus unrubbed rings were compared by two-way anova (*P < 0.05).
Figure 3.
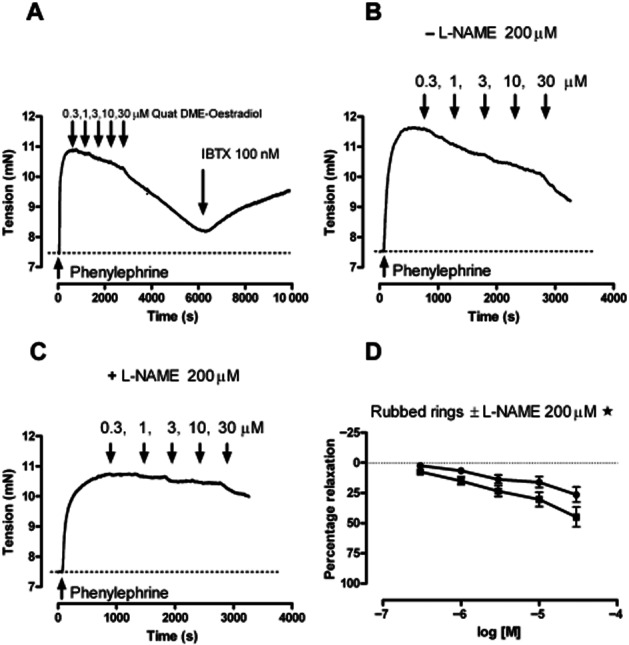
The effects of Quat DME-oestradiol on rat aortic rings. (A) An example of cumulative additions of Quat DME-oestradiol, as indicated, on pre-contracted aortic rings followed by 100 nM of IBTX. (B, C) An example of cumulative addition of Quat DME-oestradiol in the absence of L-NAME (B) and the presence of LNAME (C). L-NAME (•) reduced the response to Quat DME-oestradiol (▪), and this is illustrated in (D). Relaxations in the presence and absence of L-NAME were compared by two-way anova (*P < 0.05, n = 8).
In summary, oestrone, DME-oestrone, oestradiol, DME-oestradiol and Quat DME-oestradiol caused relaxation of the aortic rings that were endothelium-independent. The exception was the oxime derivative of oestrone, which was more potent in unrubbed rings; this analogue, therefore, is likely to require endothelium to generate relaxing factors, such as NO. In contrast, Quat DME-oestrone was ineffective. IBTX reversed the relaxant effects of Quat DME-oestradiol, but the other derivatives were insensitive to IBTX, implying that these compounds primarily target non-BKCa-dependent relaxation mechanisms. This meant that Quat DME-oestradiol was the derivative most likely to directly activate BKCa channels. However, additional experiments with the nNOS inhibitor suggest an indirect as well as a direct mechanism. To investigate the direct effects of our oestrogen derivatives on BKCa channels, we turned from a smooth muscle preparation to HEK 293 cells over-expressing BKCa channel subunits.
HEK 293 cells overexpressing the BKCa α subunit
HEK 293 cells over-expressing the α subunit of the BKCa channel were capable of generating robust BKCa currents (Figure 4Ai; Aii). This current was evoked by stepping the voltage to positive values, the current activated rapidly, did not appear to inactivate and was characteristically noisy.
Figure 4.

The change in evoked BKCa currents with time in HEK 293 cells expressing the α subunit (hSloα) of the BKCa channel. HEK 293 cells were voltage clamped at −40 mV and currents evoked by changing the voltage to a range of test potentials (−40 mV to 60 mV). (Ai) illustrates evoked currents in a HEK 293 cell at the beginning of the recording period; (Aii) illustrates evoked currents after 350 s of recording. (B) The blocking properties of IBTX on BKCa channels formed from α subunits alone. Currents were evoked at +60 mV from a holding potential of −40 mV (n = 5). (C) The activation time constant for currents evoked by changing the potential to +60 mV; the activation of the BKCa current could be fitted to a single exponential (n = 5). The free intracellular calcium ion concentration in panels Ai, Aii, B and C was 0.2 nM. (D) The effects of free intracellular Ca++ on the hSloα G–V relationship. Conductance was calculated from current and voltage, normalized to the maximum, and plotted error bars represent SEM. Solid curves represent fits to the Boltzmann function. V(1/2) for 0.2 nM, 2.4 μM and 55 μM free calcium were 130.9 ± 1.8 mV, 107 ± 0.7 mV and 68.3 ± 1.6 mV, respectively.
Functional expression of BKCa channels in HEK 293 cells was also confirmed by estimating the single channel conductance underlying the evoked current. This was done by fluctuation analysis (Chung and Pulford, 1993), which is a statistical analysis comparing the mean and variance of the current and enables the estimation of the underlying unitary current. Using this method, the underlying single-channel current was estimated to be 35.6 ± 8.5 pA (n = 4), which corresponds to a single channel conductance of 223 pS, close to values reported previously (Lippiat et al., 2000). BKCa currents are sensitive to internal free calcium ion concentration, which proved to be true in our HEK 293 cells expressing the α subunits of the BKCa channel. Raising the free intracellular concentration of Ca++ ions from approximately 0.2 nM to 55 μM shifted the V0.5 for the voltage conductance curve approximately 60 mV in the hyperpolarizing direction (Figure 4D). In our hands, the BKCa currents tended to run up over time, but we saw no appreciable change in the activation rates of the BKCa currents (Figures 4C, 6A). Finally, peak BKCa currents were sensitive to inhibition by 100 nM IBTX, the time course of which could be fitted to a single exponential with a rate of block equal to (2.5 ± 0.48) × 105·M−1·s−1 (Figure 4B).
Figure 6.

The effect of oestrone derivatives on BKCa currents evoked in HEK 293 cells over-expressing α subunits of the BKCa channel. The normalized peak currents over time curves were compared with control and found not to be significantly different apart from Oestrone oxime and Quat DME-oestradiol, which demonstrated a small but significant inhibition. The currents were evoked by stepping the membrane potential from −40 mV to +60 mV. (*P < 0.5 compared with control data, Kruskal–Wallis test followed by a Dunns multiple comparison test).
HEK 293 cells over-expressing the BKCa α and β1 subunit
HEK 293 cells over expressing the α and β1 subunits of the BKCa channel were also capable of generating robust BKCa currents (Figure 5). This current was similarly evoked by stepping the voltage to positive values, did not appear to inactivate and was characteristically noisy. The β1 subunit slowed the macroscopic current kinetics at the subnanomolar [Ca++] employed during these experiments (Figure 5Ai, Aii, C), which is similar to that reported with mSlo (Cox and Aldrich, 2000). Raising the free intracellular concentration of Ca++ ions from approximately 0.2 nM to 55 μM shifted the V0.5 for the voltage conductance curve approximately 100 mV (Figure 5D), and at all concentrations of calcium the α and β1 expressing HEK 293 cells displayed a V0.5 more negative than their α alone expressing counterparts. In these experiments, the BKCa currents ran down with time (Figure 7A), which was different from that observed with HEK 293 cells only expressing the α subunits. Finally, peak BKCa currents were sensitive to 100 nM IBTX. Currents were inhibited by the toxin, the time course of which could be fitted to a single exponential with a rate of block equal to (2.18 ± 0.65) × 104·M−1·s−1. This is appreciably slower than the block in HEK 293 cells only expressing α subunits, but in line with that reported by other investigators (Garcia-Valdes et al., 2001).
Figure 5.

The change in evoked BKCa currents with time in HEK 293 cells expressing the α and β1 subunit (hSloα + β1) of the BKCa channel. HEK 293 cells were voltage clamped at −40 mV and currents evoked by changing the voltage to a range of test potentials (−40 to 60 mV). (Ai) illustrates evoked currents in a HEK 293 cell at the beginning of the recording period; (Aii) illustrates evoked currents after 350 s of recording. (B) The blocking properties of IBTX on BKCa channels formed from α + β1 subunits. Currents were evoked at +60 mV from a holding potential of −40 mV. (n = 6). (C) The activation time constant for currents evoked by changing the potential to +60 mV, the activation of the BKCa current could be fitted to a single exponential (n = 5). The β1 subunit has slowed macroscopic current kinetics at the subnanomolar [Ca++] employed during these experiments. The free intracellular calcium ion concentration in panels Ai, Aii, B and C was 0.2 nM. (D) The effects of free [Ca++] on the hSloα+β1 G-V relationship. Conductance was calculated from current and voltage, normalized to the maximum, and plotted error bars represent SEM. Solid curves represent fits to the Boltzmann function. V(1/2) for 0.2 nM, 2.4 μM and 55 μM free calcium were 108.4 ± 4 mV, 62.6 ± 1.3 mV and 5.1 ± 3.1 mV respectively.
Figure 7.

The effect of oestrone derivatives on BKCa currents evoked in HEK 293 cells over expressing α and β1 subunits of the BKCa channel. The normalized peak currents over time curves were compared to control and found not to be significantly different, apart from oestrone, Oestrone oxime and Quat DME-oestradiol, which demonstrated a significant enhancement in peak current. The currents were evoked by stepping the membrane potential from −40 mV to +60 mV. (*P < 0.5 compared with control data, Kruskal–Wallis test followed by a Dunns multiple comparison test).
The effect of oestrogen derivatives on BKCa currents in HEK 293 cells expressing BKCa α subunits
BKCa channels were studied in whole cell recordings from HEK 293 cells expressing the α subunit. None of the ligands tested increased the whole cell currents (Figure 6). These currents were tested with very low intracellular calcium (≍0.2 nM free [Ca++]), as oestrogens have been reported to be better activators of BKCa channels when the free [Ca++] is low and the open probability for the channel is small. This finding was expected as we, and others, have reported previously that the β1 subunit is required for BKCa activation (Valverde et al., 1999a; De Wet et al., 2006).
The effect of oestrogen derivatives on BKCa currents in HEK 293 cells expressing both BKCa α and β1 subunits
Whole cell recordings from cells expressing both α and β1 subunits responded differently. Three compounds (oestrone, oestrone oxime and Quat DME-oestradiol), seem to activate the BKCa current, the rest being largely inactive (Figures 7 & 8). Interestingly, oestrone oxime and Quat DME-oestradiol were able to activate BKCa currents, even though, in HEK 293 cells only expressing the α subunit, these compounds were weak inhibitors (Figure 6).
Figure 8.

The effect of Quat DME-oestradiol (A), oestrone oxime (D) and oestrone (G) on superimposed evoked currents from HEK 293 cells over-expressing BKCa α and β1 subunits. Cells were held at −40 mV and currents evoked by changing the potential to a range of voltages up to +60 mV. The top trace represents evoked currents before the application of oestrogen (5 μM), the trace immediately below represents evoked currents after application oestrogen. The pipette solution was nominally Ca++ free. The conductance voltage relationship for currents evoked in A, D, and G are shown in B, E and H respectively. (▪) represents the control G–V relationship, while (•) represents the G–V relationship in the presence of 5 μM ligand. The activation time constant for the evoked currents at +60 mV, was fitted to a single exponential and did not change during the application of Quat DME-oestradiol (C) Oestrone oxime (F) and oestrone (I).
Further experiments showed that while these ligands could activate BKCa currents, they were unable to change activation rates (Figure 8).
Discussion and conclusions
The acute effects of 17β-oestradiol have been studied on vascular smooth muscle (Ruehlmann et al., 1998; Nakaya et al., 2007; Asano et al., 2010; Zhang et al., 2010), and a vasorelaxant effect over a large range of concentrations (10 pM–1 mM) (Tep-areenan et al., 2003) was demonstrated.
We studied the acute effects of novel oestrogen derivatives as vasorelaxants in order to obtain a better understanding of their mode of action and selectivity for BKCa channels. We found that most of our oestrogen derivatives can relax pre-contracted aorta with or without an intact endothelium. Oestrone oxime had a particularly potent endothelium-dependent effect, and further studies will be required in order to determine the mechanism of action of this derivative. Only Quat DME-oestradiol caused a relaxation that was sensitive to 100 nM IBTX, suggesting that most of these compounds have other relaxant effects. These properties were not unexpected, as it has been shown that oestradiol can inhibit L-type calcium channels in vascular smooth muscle (Nakajima et al., 1995; Ruehlmann et al., 1998; Cairrao et al., 2012) and modulate endothelial NOS (Haynes et al., 2000; Broughton et al., 2010; Batenburg et al., 2012) and second messengers, such as cGMP (White et al., 1995; 2002), all of which generate a complicated picture and explain the lack of a correlation between the ability of oestrogens to relax aortic rings and activate BKCa channels. Recently, it has been suggested that oestrogens can regulate nNOS within arterial smooth muscle (Han et al., 2007; Royal et al., 2011) and in support of this the relaxant effects of Quat DME-oestradiol were reduced by L-NAME, a non-specific NOS inhibitor. Because Quat DME-oestradiol is membrane impermeant, these data suggest targeting of membrane receptors that modulate nNOS within aortic smooth muscle. Since BKCa channels can be activated by NO (Ahern et al., 1999) and regulated by second messengers, such cGMP (Zhou et al., 2001), it cannot be assumed that relaxant reversal by IBTX is proof of a direct action of these ligands on the BKCa channel. Consequently, we turned to the simpler HEK 293 cell expression system to study the direct action of these compounds on the BKCa channel, as these cells have little, if any, NOS activity (Schmidt et al., 2001; Fang and Silverman, 2009), and do not express ERα, ERβ (Leung et al., 2006; Chantzi et al., 2011) or GPR30/GPER receptors (Filardo et al., 2007).
Previously, we have shown that 17β-oestradiol can activate BKCa channels in planar lipid bilayers (De Wet et al., 2006; de Wet et al., 2009). This activation requires the presence of the β1 subunit and indeed, Bayesian analysis demonstrates that each channel requires at least two β1 subunits for this to occur. While these experiments provided information about the stoichiometry of α and β1 subunits required for oestrogen activation, they did not provide information about the binding site. We postulate that the putative binding site for oestrogens would be at the interface between the β1 subunit and the α subunit. In support of this, Liu et al. (2010) have suggested that the S0 domain of the α subunit is in close proximity to the TM2 of the β1 subunit, and TM1 of the β1 subunit is associated with S1 and S2 domains of the α subunit (Liu et al., 2010). Furthermore, Morrow et al. (2006) have demonstrated that deletion of the extracellular N terminal of the murine α subunit did not prevent the α and β1 subunits interacting. The β1 subunit could still slow the activation and deactivation of BKCa channel gating, but N terminal deletion did effect BKCa modulation by 17β-oestradiol (Morrow et al., 2006).
If our hypothesis is correct, and the binding site for oestrogens resides between the extracellular N terminal of the α subunit and the extracellular loop between TM1 and 2 of the β1 subunit (Figure 9), then membrane-impermeable, as well as membrane-permeable oestrogens will be able to activate BKCa channels. In our hands, Quat DME-oestradiol is, indeed, able to activate channels in HEK cells expressing α and β1 subunits, but does not alter the macroscopic gating kinetics. These data agree with the investigation by Morrow et al., who postulated that the β1 subunit ability to confer oestrogen sensitivity is separate from its ability to alter the macroscopic gating kinetics of BKCa currents. Further, steroidal bile acids, such as lithocholate, are also postulated to bind to β1 subunits prior to the activation of BKCa channels (Bukiya et al., 2008a,b).
Figure 9.
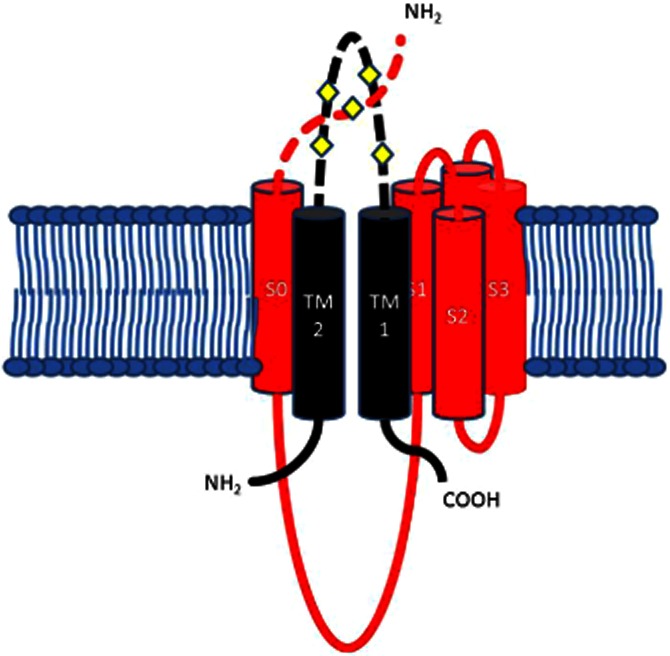
The potential extracellular binding site between the extracellular loop of the β1 subunit and the Nterminal of S0 domain of the α subunit. β1 TM1 and TM2 are black, with TM2 next to S0. Residues 16–20 of the extracellular N-terminal segment preceding S0 have been modelled as a random coil, crossing over the S3–S4 loop. This diagram represents the potential interface and binding site between one of the β1 subunits and one of the four α subunits making up a BKCa channel. The diamonds represent the extracellular cysteines.
Oestrogens are effective activators of the BKCa channel complex when the free intracellular calcium concentration is low (Valverde et al., 1999a; De Wet et al., 2006). At the intracellular concentrations of free calcium employed in these experiments (<0.2 nM), the BKCa complex of α and β1 subunits is thought to be functionally unligated by Ca++ (Meera et al., 1996; Nimigean and Magleby, 2000), and the channel is reported to be purely voltage activated at calcium ion concentrations less than 0.1 μM. Consequently, it appears that the BKCa channel is sensitive to oestrogen activation even in the absence of free intracellular calcium. This has important implications, as BKCa activators based on oestrogens would otherwise require high levels of intracellular Ca++ to activate the channel.
The cysteines (C430 and C911) within the BKCa α subunit seem to show redox sensitivity, and this is responsible for observed run down in isolated patches (Zhang et al., 2006). In addition, hydrogen peroxide virtually eliminates physiological activation of the channel by targeting a cysteine residue near the Ca++ bowl of the BKCa α subunit (Tang et al., 2004). Also, antioxidants such as Tempol (Xu et al., 2005; 2006) are known to activate BKCa channels composed of α subunits. Finally, BKCa β subunits have a cysteine-rich extracellular loop connecting the two transmembrane segments (Figure 9), while the extracellular loops of the β3 subunits are involved in a redox-sensitive gating (Zeng et al., 2003). Consequently, it is likely that these extracellular loops are a redox-sensitive region for all four β subunits and can confer additional redox sensitivity to the BKCa channel.
Oestrogens can act as antioxidants by capturing a ⋅OH hydroxyradical to produce a phenoxyl radical. Further scavenging of a OH produces a quinol, which can be converted back to the parent oestrogen by NADPH-dependent enzymes (Prokai et al., 2005; Prokai-Tatrai et al., 2008). Consequently, it is possible that some, if not all, of the effects of oestrogen could be due to an antioxidant effect in the region of the β1 and α subunit interface. These antioxidant properties could account for the effects of phenolic oestrogens, such as oestrone and 17β-oestradiol, but cannot account for the effects of oestrogens substituted in the three position, such as Quat DME-oestradiol, as these compounds cannot form quinols and act as antioxidants. It appears unlikely, therefore, that the BKCa effects of oestrogens are due to oestrogenic antioxidant activity.
Our studies, to date, reveal that the 17β position on D ring of steroidal oestrogens is important. Previous work has shown that 17β-oestradiol, which possesses an axial secondary alcohol in the 17β position, is a BKCa activator (Valverde et al., 1999a; De Wet et al., 2006; Morrow et al., 2006), whereas the stereoisomer 17α-oestradiol, which possesses an axial secondary alcohol pointing in the opposite direction, is inactive (De Wet et al., 2006). Oestrone has an equatorial or planar carbonyl group and has weak BKCa-activating properties. Thus, it appears that as oxygen moves from an axial 17α position through a planar configuration to an axial 17β position, activity increases. Consistent with this, oestrone oxime, which possesses a planar oxime group, is the weakest activator of BKCa currents. This compound produced marginal activation only noticeable at +60 mV, a potential that cells hardly ever experience. Paradoxically, this fits with the aortic ring data, which demonstrated that oestrone oxime was the most potent relaxant, but that this relaxation was endothelium dependent and was not reversed by IBTX; in short, the relaxation is not a BKCa effect.
In summary, we have synthesized novel oestrogen derivatives, some of which are membrane impermeable. Most of these compounds relax aortic smooth muscle through an endothelium-independent mechanism or induction of NOS in smooth muscle. A number of these derivatives directly activate BKCa currents in HEK 293 cells expressing both α and β1 subunits, the putative smooth muscle channel complex, and this contributes to muscle relaxation. The putative binding site is between the N terminal of the α subunit and the extracellular loop of the β1 subunit. An antioxidant mode of action seems unlikely.
Acknowledgments
This project was supported by a University of Brighton Studentship.
Glossary
- BKCa
large conductance calcium-activated potassium channel
- DMAEC HCl
2-dimethylamine ethyl chloride
- DMF
dimethylformamide
- hSloα
the alpha subunit of the human BKCa channel
- hSloβ1
the beta1 subunit of the human BKCa channel
- IBTX
iberiotoxin
- IKCa
intermediate-conductance calcium-activated potassium channels
- SKCa
small-conductance calcium-activated potassium channels
Conflict of interest
The authors declare no conflicts of interest.
References
- Ahern GP, Hsu SF, Jackson MB. Direct actions of nitric oxide on rat neurohypophysial K+ channels. J Physiol. 1999;520(Pt 1):165–176. doi: 10.1111/j.1469-7793.1999.00165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158:S1–S239. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MC, Newland C, Valverde MA, Hardy SP. Inhibition of ligand-gated cation-selective channels by tamoxifen. Eur J Pharmacol. 1998;354:261–269. doi: 10.1016/s0014-2999(98)00454-3. [DOI] [PubMed] [Google Scholar]
- Allen MC, Gale PA, Newland C, Hunter AC, Lloyd AW, Hardy SP. Inhibition of ligand-gated cation channels by the quaternary ethylbromide derivative of tamoxifen. J Physiol (Cambridge) 1999;515P:16P. [Google Scholar]
- Allen MC, Gale PA, Hunter AC, Lloyd A, Hardy SP. Membrane impermeant antioestrogens discriminate between ligand- and voltage-gated cation channels in NG108-15 cells. Biochim Biophys Acta. 2000;1509:229–236. doi: 10.1016/s0005-2736(00)00297-2. [DOI] [PubMed] [Google Scholar]
- Asano S, Tune JD, Dick GM. Bisphenol A activates Maxi-K (K(Ca)1.1) channels in coronary smooth muscle. Br J Pharmacol. 2010;160:160–170. doi: 10.1111/j.1476-5381.2010.00687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batenburg WW, Jansen PM, van den Bogaerdt AJ, Danser AH. Angiotensin II-aldosterone interaction in human coronary microarteries involves GPR30, EGFR, and endothelial NO synthase. Cardiovasc Res. 2012;94:136–143. doi: 10.1093/cvr/cvs016. [DOI] [PubMed] [Google Scholar]
- Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, et al. Vasoregulation by the beta 1 subunit of the calcium-activated potassium channel. Nature. 2000;407:870–876. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- Broughton BRS, Miller AA, Sobey CG. Endothelium-dependent relaxation by G protein-coupled receptor 30 agonists in rat carotid arteries. Am J Physiol Heart Circ Physiol. 2010;298:H1055–H1061. doi: 10.1152/ajpheart.00878.2009. [DOI] [PubMed] [Google Scholar]
- Bukiya AN, McMillan J, Parrill AL, Dopico AM. Structural determinants of monohydroxylated bile acids to activate beta 1 subunit-containing BK channels. J Lipid Res. 2008a;49:2441–2451. doi: 10.1194/jlr.M800286-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukiya AN, Vaithianathan T, Toro L, Dopico AM. The second transmembrane domain of the large conductance, voltage- and calcium-gated potassium channel beta(1) subunit is a lithocholate sensor. FEBS Lett. 2008b;582:673–678. doi: 10.1016/j.febslet.2008.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairrao E, Alvarez E, Carvas JM, Santos-Silva AJ, Verde I. Non-genomic vasorelaxant effects of 17beta-estradiol and progesterone in rat aorta are mediated by L-type Ca(2+) current inhibition. Acta Pharmacol Sin. 2012;33:615–624. doi: 10.1038/aps.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantzi NI, Meligova AK, Dhimolea E, Petrou CC, Mitsiou DJ, Magafa V, et al. Insights into ectopic estrogen receptor expression, nucleocytoplasmic distribution and interaction with chromatin obtained with new antibodies to estrogen receptors alpha and beta. Steroids. 2011;76:974–985. doi: 10.1016/j.steroids.2011.05.010. [DOI] [PubMed] [Google Scholar]
- Chung SH, Pulford G. Fluctuation analysis of patch-clamp or whole-cell recordings containing many single channels. J Neurosci Methods. 1993;50:369–384. doi: 10.1016/0165-0270(93)90043-q. [DOI] [PubMed] [Google Scholar]
- del Corsso C, Ostrovskaya O, McAllister CE, Murray K, Hatton WJ, Gurney AM, et al. Effects of aging on Ca2+ signaling in murine mesenteric arterial smooth muscle cells. Mech Ageing Dev. 2006;127:315–323. doi: 10.1016/j.mad.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Cox DH, Aldrich RW. Role of the beta 1 subunit in large-conductance Ca2+-activated K+ channel gating energetics – Mechanisms of enhanced Ca2+ sensitivity. J Gen Physiol. 2000;116:411–432. doi: 10.1085/jgp.116.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wet H, Allen M, Holmes C, Stobbart M, Lippiat JD, Callaghan R. Modulation of the BK channel by estrogens: examination at single channel level. Mol Membr Biol. 2006;23:420–429. doi: 10.1080/09687860600802803. [DOI] [PubMed] [Google Scholar]
- Dick GM. The pure anti-oestrogen ICI 182,780 (Faslodex (TM)) activates large conductance Ca2+-activated K+ channels in smooth muscle. Br J Pharmacol. 2002;136:961–964. doi: 10.1038/sj.bjp.0704807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick GM, Sanders KM. (Xeno)estrogen sensitivity of smooth muscle BK channels conferred by the regulatory beta1 subunit: a study of beta1 knockout mice. J Biol Chem. 2001;276:44835–44840. doi: 10.1074/jbc.M106851200. [DOI] [PubMed] [Google Scholar]
- Dick GM, Rossow CF, Smirnov S, Horowitz B, Sanders KM. Tamoxifen activates smooth muscle BK channels through the regulatory beta 1 subunit. J Biol Chem. 2001;276:34594–34599. doi: 10.1074/jbc.M104689200. [DOI] [PubMed] [Google Scholar]
- Fang J, Silverman RB. A cellular model for screening neuronal nitric oxide synthase inhibitors. Anal Biochem. 2009;390:74–78. doi: 10.1016/j.ab.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filardo E, Quinn J, Pang Y, Graeber C, Shaw S, Dong J, et al. Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology. 2007;148:3236–3245. doi: 10.1210/en.2006-1605. [DOI] [PubMed] [Google Scholar]
- Garcia-Valdes J, Zamudio FZ, Toro L, Possan LD. Slotoxin, alpha KTx1.11, a new scorpion peptide blocker of MaxiK channels that differentiates between alpha and alpha+beta (beta 1 or beta 4) complexes. FEBS Lett. 2001;505:369–373. doi: 10.1016/s0014-5793(01)02791-0. [DOI] [PubMed] [Google Scholar]
- Han G, Yu X, Lu L, Li S, Ma H, Zhu S, et al. Estrogen receptor alpha mediates acute potassium channel stimulation in human coronary artery smooth muscle cells. J Pharmacol Exp Ther. 2006;316:1025–1030. doi: 10.1124/jpet.105.093542. [DOI] [PubMed] [Google Scholar]
- Han GC, Ma HD, Chintala R, Miyake K, Fulton DJR, Barman SA, et al. Nongenomic, endothelium-independent effects of estrogen on human coronary smooth muscle are mediated by type I (neuronal) NOS and PI3-kinase-Akt signaling. Am J Physiol Heart Circ Physiol. 2007;293:H314–H321. doi: 10.1152/ajpheart.01342.2006. [DOI] [PubMed] [Google Scholar]
- Haynes MP, Sinha D, Russell KS, Collinge M, Fulton D, Morales-Ruiz M, et al. Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ Res. 2000;87:677–682. doi: 10.1161/01.res.87.8.677. [DOI] [PubMed] [Google Scholar]
- Layne JJ, Werner ME, Hill-Eubanks DC, Nelson MT. NFATc3 regulates BK channel function in murine urinary bladder smooth muscle. Am J Physiol Cell Physiol. 2008;295:C611–C623. doi: 10.1152/ajpcell.00435.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung YK, Mak P, Hassan S, Ho SM. Estrogen receptor (ER)-beta isoforms: a key to understanding ER-beta signaling. Proc Natl Acad Sci U S A. 2006;103:13162–13167. doi: 10.1073/pnas.0605676103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippiat JD, Standen NB, Davies NW. A residue in the intracellular vestibule of the pore is critical for gating and permeation in Ca2+-activated K+ (BKCa) channels. J Physiol (Lond) 2000;529:131–138. doi: 10.1111/j.1469-7793.2000.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GX, Niu XW, Wu RS, Chudasama N, Yao YN, Jin X, et al. Location of modulatory beta subunits in BK potassium channels. J Gen Physiol. 2010;135:449–459. doi: 10.1085/jgp.201010417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus OB, Helms LMH, Pallanck L, Ganetzky B, Swanson R, Leonard RJ. Functional-role of the beta-subunit of high-conductance calcium-activated potassium channels. Neuron. 1995;14:645–650. doi: 10.1016/0896-6273(95)90321-6. [DOI] [PubMed] [Google Scholar]
- Meera P, Wallner M, Jiang Z, Toro L. A calcium switch for the functional coupling between a (hslo) and beta subunits (K-v,K-Ca beta) of maxi K channels. FEBS Lett. 1996;382:84–88. doi: 10.1016/0014-5793(96)00151-2. [DOI] [PubMed] [Google Scholar]
- Morrow JP, Zakharov SI, Liu G, Yang L, Sok AJ, Marx SO. Defining the BK channel domains required for beta1-subunit modulation. Proc Natl Acad Sci U S A. 2006;103:5096–5101. doi: 10.1073/pnas.0600907103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima T, Kitazawa T, Hamada E, Hazama H, Omata M, Kurachi Y. 17beta-Estradiol inhibits the voltage-dependent L-type Ca2+ currents in aortic smooth muscle cells. Eur J Pharmacol. 1995;294:625–635. doi: 10.1016/0014-2999(95)00602-8. [DOI] [PubMed] [Google Scholar]
- Nakaya Y, Mawatari K, Takahashi A, Harada N, Hata A, Yasui S. The phytoestrogen ginsensoside Re activates potassium channels of vascular smooth muscle cells through PI3K/Akt and nitric oxide pathways. J Med Invest. 2007;54:381–384. doi: 10.2152/jmi.54.381. [DOI] [PubMed] [Google Scholar]
- Nimigean CM, Magleby KL. Functional coupling of the beta(1) subunit to the large conductance Ca(2+)-activated K(+) channel in the absence of Ca(2+). Increased Ca(2+) sensitivity from a Ca(2+)-independent mechanism. J Gen Physiol. 2000;115:719–736. doi: 10.1085/jgp.115.6.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimaru K, Eghbali M, Rong L, Marijic J, Stefani E, Toro L. Functional and molecular evidence of MaxiK channel beta 1 subunit decrease with coronary artery ageing in the rat. J Physiol (Lond) 2004;559:849–862. doi: 10.1113/jphysiol.2004.068676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluger S, Faulhaber J, Furstenau M, Lohn M, Waldschutz R, Gollasch M, et al. Mice with disrupted BK channel beta 1 subunit gene feature abnormal Ca2+ spark/STOC coupling and elevated blood pressure. Circ Res. 2000;87:E53–E60. doi: 10.1161/01.res.87.11.e53. [DOI] [PubMed] [Google Scholar]
- Pluznick JL, Wei PL, Carmines PK, Sansom SC. Renal fluid and electrolyte handling in BKCa-beta 1(-/-) mice. Am J Physiol Renal Physiol. 2003;284:F1274–F1279. doi: 10.1152/ajprenal.00010.2003. [DOI] [PubMed] [Google Scholar]
- Prokai L, Prokai-Tatrai K, Perjesi P, Simpkins JW. Mechanistic insights into the direct antioxidant effects of estrogens. Drug Dev Res. 2005;66:118–125. [Google Scholar]
- Prokai-Tatrai K, Perjesi P, Rivera-Portalatin NM, Simpkins JW, Prokai L. Mechanistic investigations on the antioxidant action of a neuroprotective estrogen derivative. Steroids. 2008;73:280–288. doi: 10.1016/j.steroids.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld CR, Liu XT, DeSpain K. Pregnancy modifies the large conductance Ca2+-activated K+ channel and cGMP-dependent signaling pathway in uterine vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2009;296:H1878–H1887. doi: 10.1152/ajpheart.01185.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royal CR, Ma H, Walker R, White RE. Estrogen signaling in microvascular arteries: parturition reduces vasodilation by reducing 17beta-estradiol and nNOS. Steroids. 2011;76:991–997. doi: 10.1016/j.steroids.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruehlmann DO, Steinert JR, Valverde MA, Jacob R, Mann GE. Environmental estrogenic pollutants induce acute vascular relaxation by inhibiting L-type Ca2+ channels in smooth muscle cells. FASEB J. 1998;12:613–619. doi: 10.1096/fasebj.12.7.613. [DOI] [PubMed] [Google Scholar]
- Sahebgharani M, Hardy SP, Lloyd AW, Hunter AC, Allen MC. Volume – activated chloride currents in HeLa cells are blocked by tamoxifen but not by a membrane impermeant quaternary analogue. Cell Physiol Biochem. 2001;11:99–104. doi: 10.1159/000047797. [DOI] [PubMed] [Google Scholar]
- Schmidt K, Andrew P, Schrammel A, Groschner K, Schmitz V, Kojda G, et al. Comparison of neuronal and endothelial isoforms of nitric oxide synthase in stably transfected HEK 293 cells. Am J Physiol Heart Circ Physiol. 2001;281:H2053–H2061. doi: 10.1152/ajpheart.2001.281.5.H2053. [DOI] [PubMed] [Google Scholar]
- Tang XD, Garcia ML, Heinemann SH, Hoshi T. Reactive oxygen species impair Slo1 BK channel function by altering cysteine-mediated calcium sensing. Nat Struct Mol Biol. 2004;11:171–178. doi: 10.1038/nsmb725. [DOI] [PubMed] [Google Scholar]
- Tep-areenan P, Kendall DA, Randall MD. Mechanisms of vasorelaxation to 17beta-oestradiol in rat arteries. Eur J Pharmacol. 2003;476:139–149. doi: 10.1016/s0014-2999(03)02152-6. [DOI] [PubMed] [Google Scholar]
- Valverde MA, Rojas P, Amigo J, Cosmelli D, Orio P, Bahamonde MI, et al. Acute activation of Maxi-K channels (hSlo) by estradiol binding to the beta subunit. Science. 1999a;285:1929–1931. doi: 10.1126/science.285.5435.1929. [DOI] [PubMed] [Google Scholar]
- Valverde MA, Rojas P, Amigo J, Cosmelli D, Orio P, Mann GE, et al. Activation of Maxi-K channel (hslo) by 17 beta-estradiol requires coexpression of alpha and beta subunits. Biophys J. 1999b;76:A186–A186. doi: 10.1126/science.285.5435.1929. [DOI] [PubMed] [Google Scholar]
- Werner ME, Meredith AL, Aldrich RW, Nelson MT. Hypercontractility and impaired sildenafil relaxations in the BKCa channel deletion model of erectile dysfunction. Am J Physiol Regul Integr Comp Physiol. 2008;295:R181–R188. doi: 10.1152/ajpregu.00173.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wet H, Lippiat JD, Allen M. Analysing steroid modulation of BK(Ca) channels reconstituted into planar lipid bilayers. Methods Mol Biol. 2009;491:177–186. doi: 10.1007/978-1-59745-526-8_14. [DOI] [PubMed] [Google Scholar]
- White RE, Darkow DJ, Lang JLF. Estrogen relaxes coronary-arteries by opening bkca channels through a cgmp-dependent mechanism. Circ Res. 1995;77:936–942. doi: 10.1161/01.res.77.5.936. [DOI] [PubMed] [Google Scholar]
- White RE, Han G, Maunz M, Dimitropoulou C, El-Mowafy AM, Barlow RS, et al. Endothelium-independent effect of estrogen on Ca(2+)-activated K(+) channels in human coronary artery smooth muscle cells. Cardiovasc Res. 2002;53:650–661. doi: 10.1016/s0008-6363(01)00428-x. [DOI] [PubMed] [Google Scholar]
- Xu H, Watts SW, Rondetli CM, Fink GD, Galligan JJ. Tempol activates BK channels to cause mesenteric arterial vasodilation in normotensive and deoxycorticosterone acetate (DOCA)-salt hypertensive rats. FASEB J. 2005;19:A1306–A1306. [Google Scholar]
- Xu H, Jackson WF, Fink GD, Galligan JJ. Activation of potassium channels by tempol in arterial smooth muscle cells from normotensive and deoxycorticosterone acetate-salt hypertensive rats. Hypertension. 2006;48:1080–1087. doi: 10.1161/01.HYP.0000249511.96555.57. [DOI] [PubMed] [Google Scholar]
- Zeng XH, Xia XM, Lingle CJ. Redox-sensitive extracellular gates formed by auxiliary beta subunits of calcium-activated potassium channels. Nat Struct Biol. 2003;10:448–454. doi: 10.1038/nsb932. [DOI] [PubMed] [Google Scholar]
- Zhang G, Xu R, Heinemann SH, Hoshi T. Cysteine oxidation and rundown of large-conductance Ca2+-dependent K+ channels. Biochem Biophys Res Commun. 2006;342:1389–1395. doi: 10.1016/j.bbrc.2006.02.079. [DOI] [PubMed] [Google Scholar]
- Zhang HT, Wang Y, Deng XL, Dong MQ, Zhao LM, Wang YW. Daidzein relaxes rat cerebral basilar artery via activation of large-conductance Ca2+-activated K+ channels in vascular smooth muscle cells. Eur J Pharmacol. 2010;630:100–106. doi: 10.1016/j.ejphar.2009.12.032. [DOI] [PubMed] [Google Scholar]
- Zhou XB, Arntz C, Kamm S, Motejlek K, Sausbier U, Wang GX, et al. A molecular switch for specific stimulation of the BKCa channel by cGMP and cAMP kinase. J Biol Chem. 2001;276:43239–43245. doi: 10.1074/jbc.M104202200. [DOI] [PubMed] [Google Scholar]