

Abstract
Background and Purpose
Nebulized saline solutions are used in the treatment of multiple pulmonary diseases including cystic fibrosis (CF), asthma and chronic obstructive pulmonary disease (COPD). The benefits of these therapies include improved lung function, phlegm clearance and fewer lung infections. The thiocyanate anion (SCN) is a normal component of the airway epithelial lining fluid (ELF) secreted by pulmonary epithelia with antioxidant and host defence functions. We sought to test if SCN could be nebulized to combat lung infection by bolstering innate immune defence and antioxidant capacity.
Experimental Approach
We established an effective antioxidant concentration of SCN in vitro using a bronchiolar epithelial cell line. We then developed a nebulization method of SCN in mice that increased ELF SCN above this concentration up to 12 h and used this method in a prolonged Pseudomonas aeruginosa infection model to test if increasing SCN improved host defence and infection outcomes.
Key Results
SCN protected against cytotoxicity in vitro from acute and sustained exposure to inflammation-associated oxidative stress. Nebulized SCN effectively reduced bacterial load, infection-mediated morbidity and airway inflammation in mice infected with P. aeruginosa. SCN also sustained adaptive increases in reduced GSH in infected mice.
Conclusions and Implications
SCN is a dually protective molecule able to both enhance host defence and decrease tissue injury and inflammation as an antioxidant. Nebulized SCN could be developed to combat lung infections and inflammatory lung disease.
Keywords: thiocyanate, innate immunity, inhaled antioxidant, inflammation, neutrophil, lung infection, Pseudomonas aeruginosa, cystic fibrosis
Introduction
Thiocyanate (SCN) is an acidic pseudohalide thiolate ubiquitously found in the extracellular fluids of mammals (Thomas, 1981; Wijkstrom-Frei et al., 2003). SCN enters the body from the diet (Leung et al., 2011) or is synthesized from cyanide by sulfurtransferase enzymes including mitochondrial rhodanese and cytosolic mercaptopyruvate sulfurtransferase (Westley, 1981; Nagahara et al., 1995). SCN is an important molecule in the innate immune system of mammals where it is oxidized by hydrogen peroxide (H2O2), supplied by phagocytic NADPH oxidases and epithelial dual oxidases (Geiszt et al., 2003), to form hypothiocyanite (HOSCN) in a reaction catalysed by the haloperoxidases (myeloperoxidase, MPO; lactoperoxidase, LPO; eosinophil peroxidase; etc.) (Thomas, 1981; van Dalen et al., 1997; Wu et al., 1999). SCN is the preferred electron donor for each of these enzymes (e.g. the specificity constant of MPO for SCN is 730 times greater than for chloride) (van Dalen et al., 1997). HOSCN facilitates the killing and growth inhibition of bacteria, viruses and fungi (Lenander-Lumikari, 1992; Mikola et al., 1995; Wijkstrom-Frei et al., 2003). SCN also reacts directly with hypochlorite (HOCl), a neutrophil-associated oxidant (Thomas and Fishman, 1986), and HOCl metabolites such as chloramines (RNHCl), acting as an antioxidant and resulting in the formation of HOSCN (Xulu and Ashby, 2010).
Cystic fibrosis (CF) is caused by an inheritable mutation in the CF transmembrane conductance regulator (cftr) gene, leading to impaired or absent cystic fibrosis transmembrance conductance regulator (CFTR) protein at the apical surface of cells (Davis, 2006). CFTR transports chloride (Quinton, 1983), GSH (Gao et al., 1999; Velsor et al., 2001), bicarbonate (Garcia et al., 2009) and SCN (Fragoso et al., 2004). CFTR directly regulates the concentration of secreted SCN in the human airway (Conner et al., 2007; Moskwa et al., 2007) and the oral cavity (Minarowski et al., 2008). Epithelia that lack CFTR secrete SCN at lower concentrations and less rapidly than normal or genetically corrected epithelia (Moskwa et al., 2007; Gould et al., 2010). SCN protects epithelia from HOCl toxicity in a CFTR- and dose-dependent manner (Gould et al., 2010).
Multiple species of bacteria commonly colonize CF airways that are not readily removed by the impaired host defence system and require antibiotic intervention (Lyczak et al., 2002). Infection with Pseudomonas aeruginosa is the most prevalent bacterial airway infection and correlates with lung function decline. P. aeruginosa creates a biofilm in the airway, making it particularly difficult to eradicate (Davies and Bilton, 2009). In addition, CF patients also suffer from sustained airway inflammation characterized by chronic airway neutrophilia (Walker et al., 2005). Neutrophils rapidly activate and undergo cell death after entering the airway, contributing to an excess of purulent phlegm and leaving behind a number of damaging enzymes and pro-inflammatory factors (Henke et al., 2011). The combination of chronic infection and sustained inflammatory response results in progressive destruction of the airway, which is the leading cause of morbidity and mortality in CF patients. It is estimated that ultimately 80–95% of CF patients' morbidity and mortality results from these processes (Lyczak et al., 2002).
A number of lung diseases are treated with therapeutic nebulization of saline and/or drug excipients including CF (Eng et al., 1996), asthma (Fahy et al., 2001), chronic obstructive pulmonary disease (COPD) (Wilson et al., 2006) and pneumonia (Ioannidou et al., 2007). Hypertonic saline (HS) is notable because it improves lung function and decreases exacerbations in CF patients (Wark and McDonald, 2009). Furthermore, HS increases thiol antioxidants in the airway epithelial lining fluid (ELF), including SCN, which, in addition to airway surface hydration, may contribute to its beneficial effects (Gould et al., 2010). However, HS also appears to deliver an osmotic stress to airway epithelial cells (Eng et al., 1996) and it is possible that years of daily HS inhalation will have unanticipated consequences. In the present study, we examined the effects of SCN on airway epithelia exposed to a toxic dose of HOCl or exposed to MPO-mediated HOCl toxicity. In addition, we characterized the pharmacokinetics of normal (isotonic) saline with 0.5% SCN in C57BL/6J mice that were nebulized for 30 min. We then treated mice infected with P. aeruginosa with either isotonic saline or isotonic saline formulated with SCN and observed effects on infection, inflammation and oxidative stress. This study is the first to demonstrate the benefits of SCN supplementation on airway infection in vivo, including improved host defence and decreased inflammation and morbidity.
Methods
Chemicals
All chemicals and enzymes were purchased from Sigma-Aldrich (St. Louis, MO, USA), with the exceptions of LPO from bovine milk (EMD-Millipore, Billerica, MA, USA), MPO from human granulocytes (EMD, Millipore), bovine fibrinogen (MP-Biomedical, Santa Ana, CA, USA) and human thrombin (GenTrac, Bristol, TN, USA). Tissue culture media and components were purchased from CellGro.
Cell culture
Human bronchial epithelia (16HBE, American Type Culture Collection, Manassas, VA, USA) cells were grown in DMEM with 4.5 g·L−1 glucose, without sodium pyruvate supplemented with 10% FBS and penicillin-streptomycin. Cells were grown to approximately 85% confluence and plated at a density of 2.5 × 105 cells·mL−1 on 24-well tissue culture plates (Corning, NY, USA) and allowed to adhere overnight. Cytotoxicity was measured by LDH release into the medium 24 h after beginning of exposure, calculated as [LDHmedium/LDHtotal] as previously described (Kariya et al., 2008).
Hypochlorite oxidant injury model
The stock concentration of sodium hypochlorite (HOCl) was established by measuring absorbance at 290 nm (ε = 350·M−1·cm−1) and diluted in water to the proper working concentration (Gould et al., 2010). Cells were washed with PBS before being exposed to HOCl (400 μM) for 15 min in PBS at 37°C with or without co-administration of thiocyanate (SCN, 100 μM) at pH 7.4 and 6.8 to mimic the plasma and airway fluid respectively. Cells were placed in fresh medium after the exposure. Cytotoxicity was analysed using the LDH assay 24 h after the start of the HOCl exposure.
MPO oxidant injury model
Cells were washed with PBS before being exposed to 160 mU·mL−1 glucose oxidase (GOX), 5 U·mL−1 MPO and/or 400 μM SCN in PBS. Glucose (100 mM) was added to drive the formation of hydrogen peroxide (H2O2) by GOX for 2 h at 37°C. This system produces a steady-state exposure of HOCl or HOSCN dependent on the addition of SCN (van Dalen et al., 1997). Cells were placed in fresh medium after the exposure to the MPO system. Cytotoxicity was analysed using the LDH assay 24 h after the start of the exposure to the MPO system.
Animals
Adult male C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Five mice were used per experimental group, for a total of 50 mice (25 for pharmacokinetic study and 25 for infection model). In the infection study, mice were weighed daily at 07:00 h over the duration of the study. Mice were anaesthetized with a lethal dose of sodium pentobarbital by i.p. injection (100 mg·kg−1) and anaesthesia was confirmed by the absence of response to tactile pressure applied to each paw. Mice were exsanguinated by cardiac puncture from which plasma was isolated. Bronchoalveolar lavage (BAL) was performed by cannulating the trachea and instilling two 750 μL of aliquots of isotonic sodium phosphate buffer (310 mOsm, pH 6.8) and collecting by gentle aspiration. The BAL dilution factor used to express the undiluted ELF value was determined by urea analysis of the lavage fluid and plasma (Teco Diagnostics, Anaheim, CA, USA) as previously described (Gould et al., 2010). Whole lungs were perfused with saline and removed by excision. All animal studies received prior approval by the National Jewish Health Animal Care and Use Committee and were maintained in compliance with their guidelines including fresh food and water and a 12-hour light–dark cycle.
Fibrin plug model of retained Pseudomonas aeruginosa lung infection
A CF clinical mucoid P. aeruginosa was cultured from frozen glycerol stock for 6 h in 5 mL of LB broth containing 100 μg·mL−1 ampicillin (LB-amp) at 37°C with shaking and this was transferred to 100 mL LB-amp and cultured overnight at the same conditions. Bacteria were then pelleted, washed with 15 mL of sterile PBS and resuspended in 3 mL of PBS. This stock was diluted in sterile PBS to give an absorbance of 1.0 at 600 nm (l = 1 cm). Lyophilized fibrinogen was dissolved to saturation in sterile PBS and then diluted 1:2 with sterile PBS. Thrombin was prepared as 800 U·mL−1 in sterile PBS. Both fibrinogen and thrombin solutions were inoculated with P. aeruginosa at 1:50 v/v and kept on ice prior to use. Mice were anaesthetized with isoflurane, placed on a tilting rodent workstation (Harvard Apparatus, Holliston, MA, USA) and intubated with a curved 24 G gavage needle. Furthermore, 50 μL of fibrinogen and thrombin solutions containing P. aeruginosa was sequentially instilled intratracheally, followed by 150 μL of air to disperse and mix the solutions into the airways to form fibrin plugs. This process was repeated once for a total of 200 μL of inoculum per animal. Mice were given 3.0 × 107 colony forming units (cfu) of P. aeruginosa in total and allowed to recover 24 h before intervention with SCN. At the end of the study, cfu were determined on homogenized whole lung tissue diluted 10-fold and plated on LB agar at 37°C. After 24 h, plates were assessed for P. aeruginosa and scored according to the number of colonies formed and corrected for dilution.
Measurement of pyocyanin
Lung tissue homogenates were incubated in 0.2 N hydrochloric acid (HCl) for 10 min and centrifuged at 20 000× g for 10 min. The absorbance of the supernatant fraction at 520 nm was used to quantitate pyocyanin (ε = 2.46 × 103·M−1·cm−1) (O'Malley et al., 2004). Non-infected lung tissue samples were used as blanks. The amount of pyocyanin detected was normalized to the concentration of protein in the lung tissue homogenate.
Isotonic SCN formulation, nebulization and pharmacokinetics
Normal (0.9% w/v NaCl) PBS and normal PBS supplemented with SCN (0.5% w/v NaSCN and 0.54% w/v NaCl) were sterile-filtered and stored at 4°C prior to nebulization. Both solutions were at pH 7.4 prior to nebulization. A Plexiglas nebulizing chamber with dimensions 22 × 14 × 12 (L × H × W) cm was used to treat five mice at a time over 30 min with 20 mL of saline or SCN aerosolized by a RespiTouch CE-331 vibrating mesh nebulizer (MicroMed, Durban, South Africa). Mice were killed immediately and at 4 h intervals after nebulization for pharmacokinetic studies. Pharmacokinetic parameters were obtained from modelling the change of SCN concentrations over time using a one-compartment model (PK Analyst, MicroMath). In the lung infection study, mice were treated every 12 h starting 24 h after infection and ending 12 h before being killed at 72 h, for a total of 4 treatments at 24, 36, 48 and 60 h post-infection.
Airway cell analysis
A 50 μL of aliquot of BAL fluid was diluted 200 times in Isoton II and analysed by a calibrated Coulter Z1 (Beckman Coulter, Brea, CA, USA). Cells were gated at 10–30 μm (leukocytes) and 5–10 μm (erythrocytes) to determine absolute cell counts. After counting, cells were isolated from BAL fluid by centrifugation (20 000× g for 10 min) and red blood cells were removed with Buffer EL (Qiagen, Vengo, The Netherlands). Cells were resuspended in 200 μL of PBS and a 20 μL of aliquot was used to adhere cells to slides using a Cytospin centrifuge (2000× g). Cells were fixed with methanol and stained for leukocyte identification using the Hema 3 kit (Fisher, Hampton, NH, USA). The respective percentages of leukocytes were normalized to the absolute number of cells.
Cytokine measurement
Mouse KC (CXCL1) from BAL fluid was measured by elisa (ELISA Tech, Denver, CO, USA) according to manufacturer's instructions.
Peroxidase activity
Total peroxidase activity of lung tissue was measured kinetically by following the change in absorbance at 650 nm (ε = 3.9 × 104·M−1·cm−1) after adding 150 μM H2O2 to 1.3 mM 3,3′,5,5′-tetramethylbenzidine (TMB) incubated with lung tissue homogenates in 50 mM sodium acetate buffer, pH 5.2. Activity was normalized to the amount of protein in the lung tissue homogenate.
Measurement of SCN
Samples were incubated with 3% trichloroacetic acid for 10 min and centrifuged at 20 000× g for 10 min to precipitate protein. Samples were measured by HPLC with electrochemical detection (CoulArray, ESA Inc., Chelmsford, MA, USA). Electrodes were set at 650, 750 and 900 mV and SCN was separated using a Synergi 4 μ Hydro-RP 80A 150 × 4.6 mm column with a flow rate of 0.5 mL·min−1 and an elution buffer containing 50 mM sodium acetate, 1 mM tetrabutylammonium hydrogen sulfate, and 15% acetonitrile at pH 5. SCN elutes at 8 min under these conditions.
Measurement of glutathione
Samples were incubated with 1% metaphosphoric acid for 10 min and centrifuged at 20 000× g for 10 min to precipitate protein. Samples were measured by HPLC with electrochemical detection (CoulArray, ESA Inc.). Electrode potentials were set at 250, 525, 575 and 850 mV, and GSH was separated using a Synergi 4 μ Hydro-RP 80A 150 × 4.6 mm column with a flow rate of 0.5 mL·min−1 and an elution buffer containing 50 mM sodium phosphate and 0.7% methanol at pH 3. GSH elutes at 13 min under these conditions. Lung tissue homogenates and cell lysate GSH levels were normalized to protein concentrations.
Statistics
Graphical and tabular data are expressed as mean ± SEM. Prism 5 (GraphPad, La Jolla, CA, USA) was used to perform and evaluate one-way anova with Tukey's post-test after square root transform to minimize variance or (where specifically indicated in figure legends) parametric t-test with F-test for equal variances. A P-value of P < 0.05 was considered significant. Asterisks (*) denote significant difference of a treated group compared with control, while pound symbols (#) denote significant difference between specifically indicated treated groups.
Results
SCN is cytoprotective against HOCl- and MPO-mediated cell injury
Human bronchial epithelia (16HBE) cells were exposed to 400 μM HOCl in PBS at pH values associated with the plasma (7.4) or ELF (6.8) for 15 min. Cytotoxicity was assessed by LDH release 24 h later. HOCl elicited significant toxicity (P < 0.001) at both pHs, which was greatest at the mildly acidic pH of the airway fluid (Figure 1A). SCN (100 μM) completely abolished HOCl-mediated cytotoxicity (P < 0.001) at both pHs tested. 16HBE cells were also exposed to an oxidase-peroxidase enzyme system that mimics the elevated reactive oxygen species generation during infection (Majewska et al. 2004). The system included 5 U·mL−1 MPO, 160 mU·mL−1 GOX and 100 mM glucose in PBS for 2 h and cytotoxicity assessed by LDH release 24 h later. The complete system generates HOCl or HOSCN over the 2 h period dependent on the presence or absence of SCN. Both the GOX (which produces H2O2) and the MPO complete system produced significant cytotoxicity (P < 0.001) (Figure 1B). SCN completely ablated MPO-mediated cytotoxicity (P < 0.001), further demonstrating SCN's ability to act as an antioxidant against HOCl, and also demonstrating its ability to protect against toxic H2O2 accumulation through peroxidase metabolism.
Figure 1.
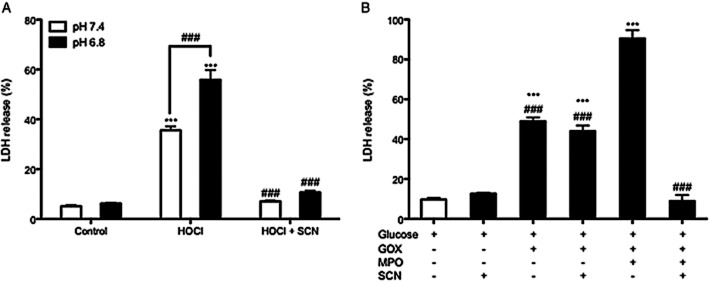
Thiocyanate (SCN) protects against HOCl- and MPO-mediated cytotoxicity. (A) Cells were washed with PBS and treated with 400 μM hypochlorite (HOCl) with or without 100 μM SCN in PBS for 15 min and assayed for cytotoxicity by LDH release 24 h later. (B) Cells were washed with PBS and exposed to 100 mM glucose, 160 mU·mL−1 glucose oxidase (GOX), 5 U·mL−1 myeloperoxidase (MPO) and 400 μM SCN in PBS for 2 h. Cells were assayed for cytotoxicity 24 h after the start of the exposure. ***P < 0.001 compared to respective pH value of control; ###P < 0.001 compared to respective pH value of HOCl, MPO + GOX + Glucose, or between HOCl (pH 7.4) and HOCl (pH 6.8) (by parametric t-test for this comparison only).
Nebulization of SCN efficiently raises ELF and plasma SCN levels
A buffered isotonic solution was formulated to contain 0.5% SCN at a pH of 7.4 to deliver SCN to the airway while minimizing airway irritation. Mice were nebulized with SCN for 30 min and subsets were killed immediately after nebulization (time 0) and at 4, 8 and 12 h post-exposure. Nebulization of 0.5% SCN increased airway ELF SCN levels 20-fold above basal levels to 2.3 mM (Figure 2A) and was well tolerated by mice. SCN nebulization also increased plasma SCN levels 18-fold above basal levels to almost 1.2 mM (Figure 2B). SCN cleared rapidly from both the ELF and the plasma compartments with half-lives (t1/2) of 3.9 and 4.4 h respectively (Table 1). Based on the pharmacokinetic data, mice were nebulized with SCN once every 12 h to maintain elevated levels of SCN.
Figure 2.
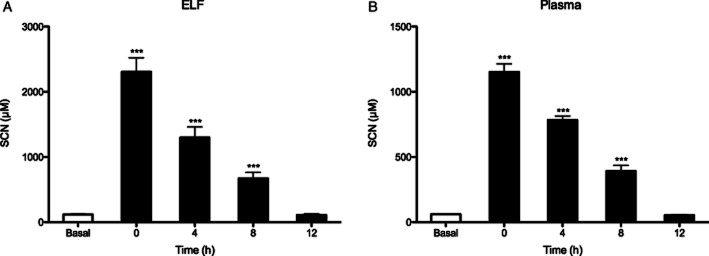
Pharmacokinetic analysis of nebulized thiocyanate (SCN). Mice were nebulized with 20 mL of isotonic saline containing 0.5% (w/v) NaSCN for 30 min. Basal (control) levels of (A) ELF and (B) plasma SCN levels are shown juxtaposed with SCN levels at times 0, 4, 8 and 12 h after SCN exposure. ***P < 0.001 compared with control.
Table 1.
Inhaled SCN pharmacokinetics in plasma and ELF of mice*
| t1/2 (h) | Co (μM) | AUC (h·μmol·L−1) | |
|---|---|---|---|
| Plasma | 4.4 ± 0.3 | 1153 ± 62 | 7 143 ± 399 |
| ELF | 3.9 ± 0.6 | 2308 ± 215 | 12 086 ± 1061 |
Modelled using one-compartment pharmacokinetic model (n = 5).
Nebulized SCN decreases P. aeruginosa infection and associated morbidity
Mice were instilled with 3 × 107 cfus of P. aeruginosa in a fibrin plug airway infection model. Mice were allowed to recover for 24 h before beginning treatment regimen with nebulized isotonic saline or isotonic saline containing 0.5% SCN every 12 h and weighed each morning prior to treatment. Mice were sacrificed 72 h post-infection and changes in body weight and markers of lung infection were assessed. Mice infected with fibrin plug (sham) or fibrin plug containing P. aeruginosa lost 5% of their body weight within the first 24 h period after infection (P < 0.001) and in the P. aeruginosa-infected saline treatment group continued to lose weight up to 72 h (P < 0.001) (Figure 3A). The P. aeruginosa-infected SCN treatment group responded quickly and significantly to treatment (P < 0.001), regaining body weight at a rate equal to the sham-treated group. P. aeruginosa-infected SCN-treated mice also had significantly less cfu·mL−1 in their lung tissue than saline control (P < 0.01) (Figure 3B). The P. aeruginosa virulence marker pyocyanin was also significantly decreased in the lung tissue of SCN-treated mice compared with saline control (P < 0.001) (Figure 3C). These data are consistent with SCN treatment improving lung host defence and resulting in decreased morbidity.
Figure 3.
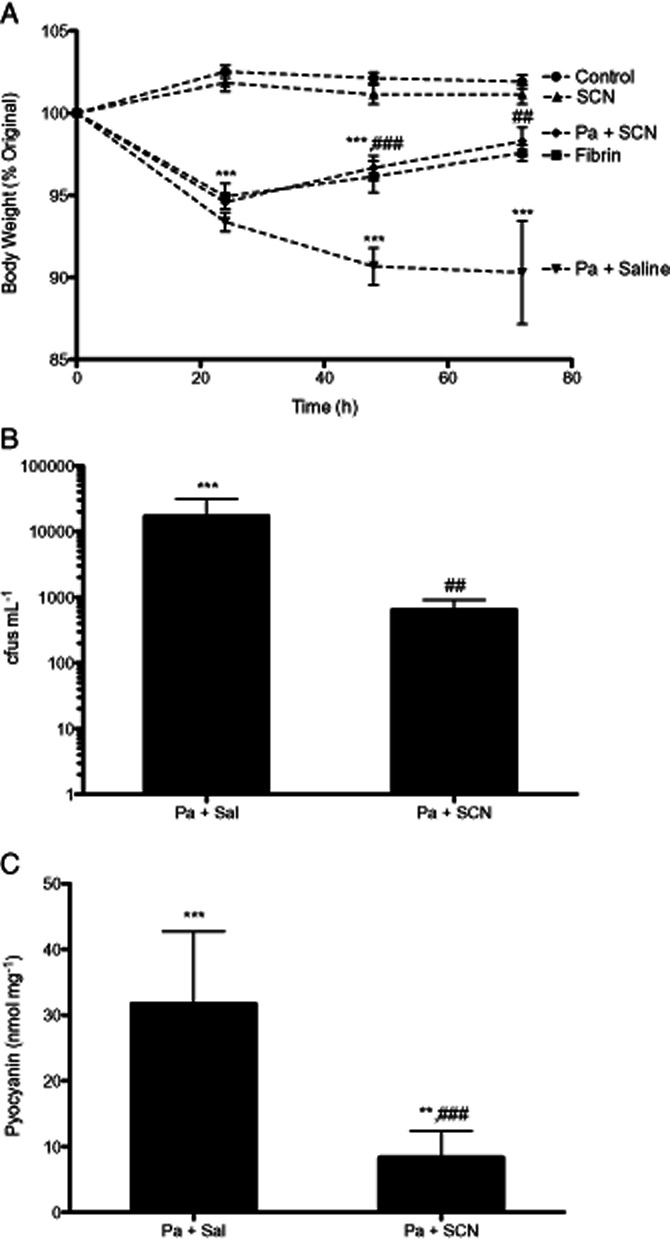
Nebulized thiocyanate (SCN) reduces morbidity and bacterial burden in infected mice. Mice were intratracheally instilled with 3.0 × 107 cfu·mL−1 Pseudomonas aeruginosa in fibrin, sterile fibrin sham or not treated. All treatments began 24 h after infection. (A) Body weight was tracked daily up to 72 h. The mice were killed at 72 h, at which point (B) lung tissue was homogenized and cultured for P. aeruginosa colony formation for 24 h at 37°C. (C) Pyocyanin, a virulence marker of P. aeruginosa, was assayed in lung tissue homogenate and normalized to total protein level. Mean absorbance from sterile mice was used as baseline. Data from uninfected controls are not shown in B or C. **P < 0.01, ***P < 0.001 compared with control; ##P < 0.01, ###P < 0.001 compared With Pa + saline.
Nebulized SCN decreases infection-mediated neutrophilic lung inflammation
Neutrophils play an important role in host defence and are a significant source of airway inflammation during P. aeruginosa infection. BAL cells were isolated, identified and quantitated. Neutrophils were significantly increased in P. aeruginosa-infected mice (P < 0.001), but those that received SCN had less neutrophil infiltrate compared with saline-treated mice (P < 0.05) while remaining higher than control (P < 0.05) (Figure 4A). Sterile fibrin sham also increased neutrophil infiltrate (P < 0.01). Infection also significantly increased airway lymphocytes (P < 0.01) with a non-significant decrease in infected animals that received SCN (Figure 4B). Interestingly, SCN treatment was also associated with an increase in the number of macrophages in the airway of infected mice (P < 0.05) (Figure 4C). Lastly, there was a non-significant increase in airway red blood cells associated with P. aeruginosa infection (Figure 4D). SCN treatment alone had no effect on airway infiltrates. Additionally, infection with P. aeruginosa significantly increased the levels of the neutrophil chemoattractant KC (mouse IL-8 analogue) (P < 0.001) in the BAL fluid and peroxidase activity (P < 0.05) in lung tissue, and both effects were abolished with SCN treatment (P < 0.01, P < 0.05, respectively) (Figure 5). The significant decrease in these inflammatory markers with SCN treatment appears consistent with decreases seen in airway neutrophils. IL-10 was investigated as a possible anti-inflammatory effect of SCN treatment but was not significantly different from control in any of the groups (data not shown). We also investigated if SCN treatment could block LPS-induced nuclear p65 translocation as another possible anti-inflammatory mechanism in a murine alveolar macrophage (J774) but observed no effect (data not shown). Finally, we measured the antioxidant and redox modulator GSH in plasma, ELF, BAL cells and lung tissue by HPLC with electrochemical detection. GSH levels in the plasma and lung tissue did not significantly change in any of the groups, but GSH did significantly increase (P < 0.05) in the ELF and BAL cells of infected mice that were treated with SCN (Figure 6).
Figure 4.

Nebulized SCN reduces infection-mediated airway inflammation. Airway cells isolated from bronchoalveolar lavage (BAL) fluid were counted (Coulter Z1) after 72 h. Cells were fixed to glass slides with methanol and counterstained for identification. The total number of (A) neutrophils, (B) lymphocytes, (C) macrophages and (D) erythrocytes were determined from the initial cell count for leukocytes (10–30 μm) or erythrocytes (5–10 μm), and in the case of leukocytes this value was multiplied by the percentage of each respective cell type in the airway. *P < 0.05, **P < 0.01, ***P < 0.001 compared with control; #P < 0.05 compared with Pseudomonas aeruginosa + saline.
Figure 5.
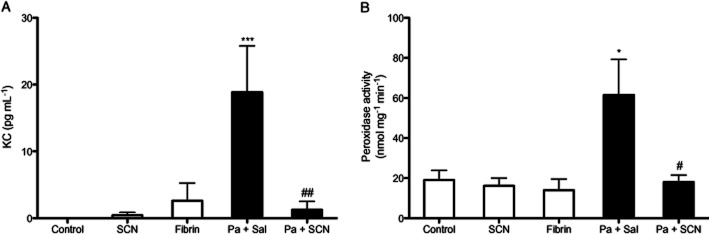
Nebulized thiocyanate (SCN) diminishes infection-mediated markers of lung inflammation. BAL fluid and lung tissue were analysed for markers of airway and tissue inflammation after 72 h. (A) The neutrophil chemoattractant KC (mouse IL-8 analogue) was quantified from BAL fluid by elisa. (B) Total lung tissue homogenate peroxidase activity was measured by TMB oxidation and normalized to total protein level. *P < 0.05, ***P < 0.001 compared with control; #P < 0.05, ##P < 0.01 compared with Pseudomonas aeruginosa + saline.
Figure 6.

Nebulized thiocyanate (SCN) preserves infection-mediated GSH adaptive responses in the airway. The concentration of GSH in experimental animals was quantified by electrochemical HPLC (EC-HPLC) after 72 h in (A) plasma, (B) ELF, (C) BAL cell lysates and (D) lung tissue homogenate. *P < 0.05 compared with control.
Consumption of SCN during lung infection and restoration with nebulized SCN
SCN levels were measured by HPLC with electrochemical detection in the plasma and ELF at the end of the study, 12 h after the last SCN nebulization. There were no significant differences in the SCN in the plasma or ELF of SCN-treated mice (Figure 7), in agreement with pharmacokinetic data showing that basal levels of SCN are restored 12 h post-treatment (Figure 2). Interestingly, infected mice treated with isotonic saline alone had significantly lower ELF SCN levels than control mice (P < 0.001) (Figure 7B). These data suggest that the lung actively utilizes SCN during infection resulting in the depletion of extracellular SCN steady-state levels that can be restored by intervention with SCN from an exogenous source.
Figure 7.
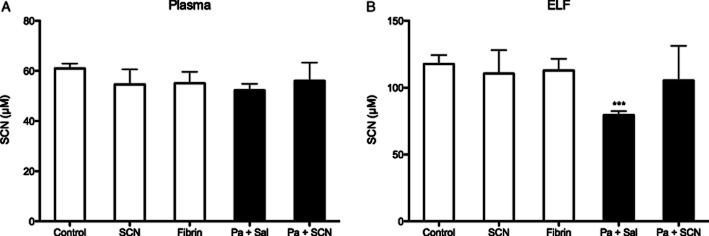
Thiocyanate (SCN) is depleted during infection. The concentration of SCN in experimental animals was quantified by EC-HPLC after 72 h in (A) plasma and (B) ELF. While SCN did not accumulate in animals augmented with nebulized SCN, it was significantly less than control in the airways of infected mice that were only given saline. ***P < 0.001 compared with control by parametric t-test.
Discussion
SCN has antioxidant properties in addition to its better known host defence properties (Chandler and Day, 2012). These findings show that SCN is cytoprotective against both purified HOCl and MPO/H2O2/saline-mediated lung epithelial cell injury, which is in agreement with other reports from mammalian cell lines (Xu et al., 2009; Gould et al., 2010). Some of SCN's antioxidant effects are due to its ability to directly react with HOCl to form HOSCN (Xulu and Ashby, 2010), which has been shown to be less toxic to mammalian tissue than HOCl and other hypohalous acids (HOX) (Wagner et al., 2004). In addition, SCN is the preferred substrate for MPO and other haloperoxidases, resulting in proportionately more HOSCN generated with increasing concentrations of SCN at the expense of more injurious oxidizing agents including HOX, nitrogen dioxide radical and urate radical (Wu et al., 1999; Wagner et al., 2004; Meotti et al., 2011). SCN can even repair some metabolites of HOCl: the monochloramines (RNHCl), which may be highly cytotoxic (Klamt and Shacter, 2005). SCN directly reacts with RNHCl resulting in the repair of the parent amino group and formation of HOSCN (Thomas and Fishman, 1986). Interestingly, HOSCN reacts with RNHCl at an even faster rate than SCN, suggesting it may also have direct antioxidant behaviour for some oxidizing agents (Xulu and Ashby, 2010).
We hypothesized that supplementation of exogenous SCN into the airways would result in the amplification of these antioxidant mechanisms as well as improving host defence (Figure 8). Indeed, our studies showed infected C57BL/6 mice nebulized with SCN had decreased inflammation and pro-inflammatory markers and rapidly regained body weight lost during the first 24 h of infection. Yet, the reason HOSCN is better tolerated by lung epithelium than HOX while remaining a potent antimicrobial agent is still unclear. This phenomenon may have developed in mammals to specifically attack invading pathogens while sparing local tissue, which would explain the specificity of many haloperoxidases for SCN over the halides.
Figure 8.
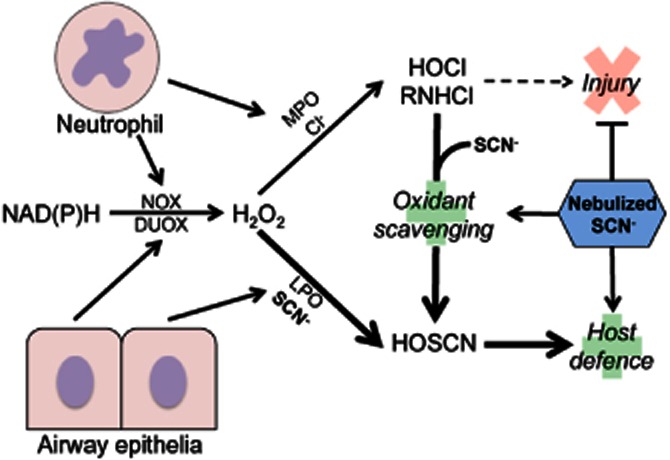
Nebulized thiocyanate (SCN) proposed mechanism of action on lung infection and inflammation outcomes in the airway. Infection results in the infiltration of inflammatory cells such as neutrophils to the lung tissue and airway. These cells, along with the epithelia, combat infection by releasing haloperoxidases (myeloperoxidase, MPO; lactoperoxidase, LPO) and hydrogen peroxide (H2O2) generated by cellular oxidases (NADPH oxidase, NOX; dual oxidase, DUOX). SCN and Cl− are secreted by epithelia into the ELF and are the major reductants of H2O2-oxidized compound I of MPO and LPO. SCN is the preferred substrate for LPO and MPO but is found at lower concentrations than Cl−. When compound I is reduced by Cl−, HOCl and monochloramines (RNHCl) are generated. These are highly toxic to the host tissue as well as pathogens. In contrast, SCN is oxidized by compound I to form HOSCN, which is specifically toxic to pathogens and spares lung tissue. SCN also scavenges HOCl and RNHCl directly, acting as a dual antioxidant and host defence agent. SCN nebulization into the airway greatly enhanced its concentration resulting in three major effects: (i) increased scavenging and prevented formation of HOCl and RNHCl; (ii) blocking of injury and enhancement of redox tone; and (iii) the bolstering of host defence, which enables tissue remodelling with less inflammation and oxidative stress.
Nebulization of saline solutions is common in the treatment of lung diseases associated with exacerbations and recurring infections, including CF (Wark and McDonald, 2009). HS has been shown to increase the concentration of antioxidants in the airway, including SCN and GSH (Gould et al., 2010), and improves lung function, but can also be irritating and induce bronchospasms. We designed a dose of isotonic 0.5% SCN that provided the ELF with a concentration of SCN in excess of 400 μM, which was the concentration we used to protect against MPO toxicity in lung epithelium and was originally reported in humans (Wijkstrom-Frei et al., 2003). Nebulization of SCN resulted in ELF concentrations 5.8-fold greater than the concentration of SCN from our cell culture studies, but was still well tolerated by our mice and readily cleared. Clinical data from early 20th century use of SCN as an antihypertensive demonstrate it is well tolerated in humans, up to steady-state plasma concentrations in excess of 1 mM (Chandler and Day, 2012). With nebulization, the plasma Cmax was only half that in the ELF due to the topical delivery method. SCN levels returned to basal values by 12 h post-nebulization, creating the rationale for the twice-daily dosing. The rapid clearance of SCN from the ELF and plasma suggests that accumulation should not be an issue with repeat dosing at proper intervals. Indeed, SCN-treated control mice did not show any sign of accumulation after a series of four treatments.
Our findings are the first to show that nebulization of SCN is well tolerated and efficacious in a sustained P. aeruginosa lung infection mouse model, both improving host defence and decreasing lung inflammation and infection-mediated morbidity. SCN was not given to the mice until 24 h after infection, yet there were dramatic improvements in the body weights of infected mice given SCN compared with the infected controls, which lost weight for the entire duration of the study. Infected mice treated with SCN had significantly less cultured P. aeruginosa and the virulence marker pyocyanin (Sorensen et al., 1983; Kanthakumar et al., 1993) in their lungs. Unlike other thiols such as GSH, SCN is capable of being endogenously converted into the antimicrobial agent HOSCN (Marshall and Reiter, 1980; Thomas et al., 1994). SCN therapy may prove to be more effective than previous attempts to use inhaled antioxidants such as GSH (Griese et al., 2004; Bishop et al., 2005) and N-acetylcysteine (NAC) (Nash et al., 2009) due to its dual antioxidant and host defence properties and its greater resistance to auto-oxidation.
Neutrophils play an important role in airway host defence, but in certain lung diseases, they contribute to pathophysiology (Meyer and Zimmerman, 1993). In the CF lung, neutrophils (as part of overall host defence) fail to clear bacteria and actually worsen conditions by releasing pro-inflammatory cytokines, injurious enzymes (including MPO) and their DNA. These factors contribute to thickened phlegm, bacterial biofilm formation and airway damage that promote progressive airway obstruction and respiratory failure in CF (Henke et al., 2011). We observed an increase in the neutrophil chemoattractant KC (IL-8 analogue), increased airway neutrophils and increased lung tissue peroxidase activity with P. aeruginosa infection. However, the inflammatory markers significantly decreased with SCN treatment and neutrophil infiltration decreased as well. Similar effects have been reported with HS therapy, which also elevates ELF SCN and reduces IL-8 (Reeves et al., 2011). There was also a significant increase in BAL lymphocytes in P. aeruginosa-infected mice that was decreased with SCN supplementation. Studies have linked lymphocytes in CF and release of the neutrophil chemokine IL-17 (Tan et al., 2011). P. aeruginosa also contributes to inflammatory tone through the synthesis of pyocyanin, which is redox active and stimulates IL-8 release by airway epithelia (Denning et al., 1998). SCN treatment decreased pyocyanin in infected mice, which may have indirectly reduced IL-8 signalling. In addition, antioxidants such as NAC decrease inflammatory signalling in the airway (Denning et al., 1998) and SCN treatment sustained increased GSH levels in P. aeruginosa-infected mice. Hence, SCN may act by multiple mechanisms to decrease inflammation and pro-inflammatory signals in infected tissue. Interestingly, we did not observe direct anti-inflammatory mechanisms of SCN in either IL-10 secretion or p65 translocation (data not shown) suggesting SCN's anti-inflammatory effects in this model could be purely secondary results of its dual extracellular antibacterial and oxidant-scavenging effects.
Apically expressed CFTR is the major identified constitutive pathway of SCN transport into the ELF. CFTR knockout mice have deficient basal ELF SCN (Gould et al., 2010) and exhibit enhanced inflammation and mortality in response to P. aeruginosa lung infection (Heeckeren et al., 1997). Benefits of nebulizing SCN in C57BL/6 mice suggest the potential for even more dramatic results in the CF airway where: (i) SCN is poorly maintained; (ii) inflammation is constitutively increased; and (iii) infections are not well cleared. Interestingly the fibrin plug used in our studies to retain bacterial infection mimics bacterial biofilms and thick airway phlegm, both of which contain large amounts of fibrin and are central to CF lung disease (Seear et al., 1997). However, caution is advised when comparing our effects to CF due to its complexity that extends beyond simple differences in anion transport.
SCN therapy was associated with elevated levels of GSH in infected mice in both ELF and in airway cells, which acquire GSH from the ELF (Gould et al., 2011). P. aeruginosa infection has been shown to dramatically increase GSH levels in ELF (Day et al., 2004). However, after 72 h post-infection, no adaptive response was observed in infected controls except with the intervention of SCN. SCN alone did not increase GSH in uninfected mice, but may sustain adaptive GSH increase by decreasing oxidative and inflammatory burdens. SCN can directly react with many oxidants that GSH also scavenges, including HOCl and RNHCl (Xulu and Ashby, 2010). A biologically relevant effect of SCN is to diminish the reactivity of HOCl and chloramines by reacting with them to form HOSCN (Thomas and Fishman, 1986), which has a redox potential less than half that of HOCl (Arnhold et al., 2006). Elevated levels of SCN drive the formation of HOSCN, which is a more selective oxidant that preferentially reacts with acidic thiols to form disulfides and sulfenic acids (Pattison et al., 2012) that can be readily reduced and repaired (Chandler and Day, 2012). Furthermore, HOCl reacts with ionized thiols in its protonated form, making SCN a better target than GSH in this regard at physiologic pH (Xulu and Ashby, 2010). The second-order rate constant of the reaction of SCN with HOCl is close to diffusion limiting at 2.34 × 107·M−1·s−1 (Ashby et al., 2004), whereas GSH is given as 1 × 107·M−1·s−1 (Winterbourn and Brennan, 1997). These rate constants clearly indicate that when SCN is in greater abundance than GSH it will be the predominant reductant of HOCl. The less reactive HOSCN produced may then preferentially react with more acidic thiols instead of GSH (Nagy et al., 2009).
ELF SCN in infected control mice was less than in control mice, suggesting that SCN is depleted faster than it can be replaced during infection. This effect may be even more pronounced in diseases of impaired SCN transport like CF. However, chronic infection and inflammation may result in the up-regulation of alternative SCN transport proteins such as the Ca2+-dependent chloride channel and pendrin (Pedemonte et al., 2007). The relative efficacy of these transporters versus CFTR in exporting SCN to the lumen during health or sickness is unclear, as is the phenotype associated with their expression that may override any beneficial increase in SCN. Clinical studies of CF frequently compare infected and/or symptomatic individuals to non-infected controls, limiting the ability to isolate CFTR as a variable (Lorentzen et al., 2011). Yet, sterile newborn pigs homozygous for CFTR-ΔF508 have significantly impaired transport of SCN to the airway than control littermates (Lorentzen et al., 2011).
The augmentation of ELF with nebulized SCN during lung infection directly impacts the processes of host defence and tissue protection and is markedly beneficial to the host, as summarized in Figure 8. In mice infected with P. aeruginosa and given nebulized SCN, we observed enhanced host defence, decreased morbidity and inflammation, as well as sustained adaptive antioxidant increases. These observations provide strong evidence supporting the rationale for nebulized SCN in the treatment of human lung infections. Because SCN has been shown to be deficient or depleted in CF and these patients suffer from chronic infection and inflammation, they may stand to benefit from nebulized SCN as well.
Acknowledgments
This work was supported by NIH grant RO1 HL084469 (B. J. D.) and by a Cystic Fibrosis Foundation Research Grant (B. J. D. and D. P. N.).
Glossary
- BAL
bronchoalveolar lavage
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrance conductance regulator
- COPD
chronic obstructive pulmonary disease
- ELF
epithelial lining fluid
- GOX
glucose oxidase
- HBE
human bronchial epithelia
- HOCl
hypochlorous acid/hypochlorite
- HOSCN
hypothiocyanous acid/hypothiocyanite
- HOX
[non-specific] hypohalous acid/hypohalite
- HS
hypertonic saline
- LPO
lactoperoxidase
- MPO
myeloperoxidase
- NAC
N-acetyl cysteine
- NOX
NADPH oxidase
- RNHCl
monochloramine
- SCN
thiocyanate
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Nuclear p65 translocation stimulated by LPS is not affected by SCN. Murine alveolar macrophage cells were incubated with 400 μM SCN overnight. 50 ng·mL−1 LPS was added to the media for 2 h before nuclear harvest and Western blotting. Representative image included.
Figure S2 GSH and GSSG as a redox pair in lung tissue and plasma.
Figure S3 Homozygous CFTR ΔF508 CuFi-1 cells were treated with thiocyanate (SCN, 400 uM), myeloperoxidase (MPO), glucose oxidase system (GOX) or combinations for 2 h in PBS and then washed and fresh media added. Cytotoxicity was measured by LDH release 24 h after exposures.
References
- Arnhold J, Monani E, Furtmuller PG, Zederbauer M, Casella L, Obinger C. Kinetics and thermadynamics of halide and nitrite oxidation by mammalian heme peroxidases. Eur J Inorg Chem. 2006;2006:3801–3811. [Google Scholar]
- Ashby MT, Carlson AC, Scott MJ. Redox buffering of hypochlorous acid by thiocyanate in physiologic fluids. J Am Chem Soc. 2004;126:15976–15977. doi: 10.1021/ja0438361. [DOI] [PubMed] [Google Scholar]
- Bishop C, Hudson VM, Hilton SC, Wilde C. A pilot study of the effect of inhaled buffered reduced glutathione on the clinical status of patients with cystic fibrosis. Chest. 2005;127:308–317. doi: 10.1378/chest.127.1.308. [DOI] [PubMed] [Google Scholar]
- Chandler JD, Day BJ. THIOCYANATE: a potentially useful therapeutic agent with host defense and antioxidant properties. Biochem Pharmacol. 2012;84:1381–1387. doi: 10.1016/j.bcp.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner GE, Wijkstrom-Frei C, Randell SH, Fernandez VE, Salathe M. The lactoperoxidase system links anion transport to host defense in cystic fibrosis. FEBS Lett. 2007;581:271–278. doi: 10.1016/j.febslet.2006.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dalen CJ, Whitehouse MW, Winterbourn CC, Kettle AJ. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem J. 1997;327(Pt 2):487–492. doi: 10.1042/bj3270487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JC, Bilton D. Bugs, biofilms, and resistance in cystic fibrosis. Respir Care. 2009;54:628–640. doi: 10.4187/aarc0492. [DOI] [PubMed] [Google Scholar]
- Davis PB. Cystic fibrosis since 1938. Am J Respir Crit Care Med. 2006;173:475–482. doi: 10.1164/rccm.200505-840OE. [DOI] [PubMed] [Google Scholar]
- Day BJ, van Heeckeren AM, Min E, Velsor LW. Role for cystic fibrosis transmembrane conductance regulator protein in a glutathione response to bronchopulmonary pseudomonas infection. Infect Immun. 2004;72:2045–2051. doi: 10.1128/IAI.72.4.2045-2051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning GM, Wollenweber LA, Railsback MA, Cox CD, Stoll LL, Britigan BE. Pseudomonas pyocyanin increases interleukin-8 expression by human airway epithelial cells. Infect Immun. 1998;66:5777–5784. doi: 10.1128/iai.66.12.5777-5784.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng PA, Morton J, Douglass JA, Riedler J, Wilson J, Robertson CF. Short-term efficacy of ultrasonically nebulized hypertonic saline in cystic fibrosis. Pediatr Pulmonol. 1996;21:77–83. doi: 10.1002/(SICI)1099-0496(199602)21:2<77::AID-PPUL3>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Fahy JV, Boushey HA, Lazarus SC, Mauger EA, Cherniack RM, Chinchilli VM, et al. Safety and reproducibility of sputum induction in asthmatic subjects in a multicenter study. Am J Respir Crit Care Med. 2001;163:1470–1475. doi: 10.1164/ajrccm.163.6.9901105. [DOI] [PubMed] [Google Scholar]
- Fragoso MA, Fernandez V, Forteza R, Randell SH, Salathe M, Conner GE. Transcellular thiocyanate transport by human airway epithelia. J Physiol. 2004;561(Pt 1):183–194. doi: 10.1113/jphysiol.2004.071548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Kim KJ, Yankaskas JR, Forman HJ. Abnormal glutathione transport in cystic fibrosis airway epithelia. Am J Physiol. 1999;277(1 Pt 1):L113–L118. doi: 10.1152/ajplung.1999.277.1.L113. [DOI] [PubMed] [Google Scholar]
- Garcia MA, Yang N, Quinton PM. Normal mouse intestinal mucus release requires cystic fibrosis transmembrane regulator-dependent bicarbonate secretion. J Clin Invest. 2009;119:2613–2622. doi: 10.1172/JCI38662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiszt M, Witta J, Baffi J, Lekstrom K, Leto TL. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J. 2003;17:1502–1504. doi: 10.1096/fj.02-1104fje. [DOI] [PubMed] [Google Scholar]
- Gould NS, Gauthier S, Kariya CT, Min E, Huang J, Day BJ. Hypertonic saline increases lung epithelial lining fluid glutathione and thiocyanate: two protective CFTR-dependent thiols against oxidative injury. Respir Res. 2010;11:119. doi: 10.1186/1465-9921-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould NS, Min E, Day BJ. Macropinocytosis of extracellular glutathione ameliorates tumor necrosis factor alpha release in activated macrophages. PLoS ONE. 2011;6:e25704. doi: 10.1371/journal.pone.0025704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griese M, Ramakers J, Krasselt A, Starosta V, Van Koningsbruggen S, Fischer R, et al. Improvement of alveolar glutathione and lung function but not oxidative state in cystic fibrosis. Am J Respir Crit Care Med. 2004;169:822–828. doi: 10.1164/rccm.200308-1104OC. [DOI] [PubMed] [Google Scholar]
- Heeckeren A, Walenga R, Konstan MW, Bonfield T, Davis PB, Ferkol T. Excessive inflammatory response of cystic fibrosis mice to bronchopulmonary infection with Pseudomonas aeruginosa. J Clin Invest. 1997;100:2810–2815. doi: 10.1172/JCI119828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke MO, John G, Rheineck C, Chillappagari S, Naehrlich L, Rubin BK. Serine proteases degrade airway mucins in cystic fibrosis. Infect Immun. 2011;79:3438–3444. doi: 10.1128/IAI.01252-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidou E, Siempos II, Falagas ME. Administration of antimicrobials via the respiratory tract for the treatment of patients with nosocomial pneumonia: a meta-analysis. J Antimicrob Chemother. 2007;60:1216–1226. doi: 10.1093/jac/dkm385. [DOI] [PubMed] [Google Scholar]
- Kanthakumar K, Taylor G, Tsang KW, Cundell DR, Rutman A, Smith S, et al. Mechanisms of action of Pseudomonas aeruginosa pyocyanin on human ciliary beat in vitro. Infect Immun. 1993;61:2848–2853. doi: 10.1128/iai.61.7.2848-2853.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariya C, Chu HW, Huang J, Leitner H, Martin RJ, Day BJ. Mycoplasma pneumoniae infection and environmental tobacco smoke inhibit lung glutathione adaptive responses and increase oxidative stress. Infect Immun. 2008;76:4455–4462. doi: 10.1128/IAI.00136-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klamt F, Shacter E. Taurine chloramine, an oxidant derived from neutrophils, induces apoptosis in human B lymphoma cells through mitochondrial damage. J Biol Chem. 2005;280:21346–21352. doi: 10.1074/jbc.M501170200. [DOI] [PubMed] [Google Scholar]
- Lenander-Lumikari M. Inhibition of Candida albicans by the peroxidase/SCN-/H2O2 system. Oral Microbiol Immunol. 1992;7:315–320. doi: 10.1111/j.1399-302x.1992.tb00595.x. [DOI] [PubMed] [Google Scholar]
- Leung AM, Lamar A, He X, Braverman LE, Pearce EN. Iodine status and thyroid function of Boston – area vegetarians and vegans. J Clin Endocrinol Metab. 2011;96:E1303–E1307. doi: 10.1210/jc.2011-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorentzen D, Durairaj L, Pezzulo AA, Nakano Y, Launspach J, Stoltz DA, et al. Concentration of the antibacterial precursor thiocyanate in cystic fibrosis airway secretions. Free Radic Biol Med. 2011;50:1144–1150. doi: 10.1016/j.freeradbiomed.2011.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyczak JB, Cannon CL, Pier GB. Lung infections associated with cystic fibrosis. Clin Microbiol Rev. 2002;15:194–222. doi: 10.1128/CMR.15.2.194-222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewska E, Kasielski M, Luczynski R, Bartosz G, Bialasiewicz P, Nowak D. Elevated exhalation of hydrogen peroxide and thiobarbituric acid reactive substances in patients with community acquired pneumonia. Respir Med. 2004;98:669–676. doi: 10.1016/j.rmed.2003.08.015. [DOI] [PubMed] [Google Scholar]
- Marshall VM, Reiter B. Comparison of the antibacterial activity of the hypothiocyanite anion towards Streptococcus lactis and Escherichia coli. J Gen Microbiol. 1980;120:513–516. doi: 10.1099/00221287-120-2-513. [DOI] [PubMed] [Google Scholar]
- Meotti FC, Jameson GN, Turner R, Harwood DT, Stockwell S, Rees MD, et al. Urate as a physiological substrate for myeloperoxidase: implications for hyperuricemia and inflammation. J Biol Chem. 2011;286:12901–12911. doi: 10.1074/jbc.M110.172460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KC, Zimmerman J. Neutrophil mediators, Pseudomonas, and pulmonary dysfunction in cystic fibrosis. J Lab Clin Med. 1993;121:654–661. [PubMed] [Google Scholar]
- Mikola H, Waris M, Tenovuo J. Inhibition of herpes simplex virus type 1, respiratory syncytial virus and echovirus type 11 by peroxidase-generated hypothiocyanite. Antiviral Res. 1995;26:161–171. doi: 10.1016/0166-3542(94)00073-h. [DOI] [PubMed] [Google Scholar]
- Minarowski L, Sands D, Minarowska A, Karwowska A, Sulewska A, Gacko M, et al. Thiocyanate concentration in saliva of cystic fibrosis patients. Folia Histochem Cytobiol. 2008;46:245–246. doi: 10.2478/v10042-008-0037-0. [DOI] [PubMed] [Google Scholar]
- Moskwa P, Lorentzen D, Excoffon KJ, Zabner J, McCray PB, Jr, Nauseef WM, et al. A novel host defense system of airways is defective in cystic fibrosis. Am J Respir Crit Care Med. 2007;175:174–183. doi: 10.1164/rccm.200607-1029OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara N, Okazaki T, Nishino T. Cytosolic mercaptopyruvate sulfurtransferase is evolutionarily related to mitochondrial rhodanese. Striking similarity in active site amino acid sequence and the increase in the mercaptopyruvate sulfurtransferase activity of rhodanese by site-directed mutagenesis. J Biol Chem. 1995;270:16230–16235. doi: 10.1074/jbc.270.27.16230. [DOI] [PubMed] [Google Scholar]
- Nagy P, Jameson GN, Winterbourn CC. Kinetics and mechanisms of the reaction of hypothiocyanous acid with 5-thio-2-nitrobenzoic acid and reduced glutathione. Chem Res Toxicol. 2009;22:1833–1840. doi: 10.1021/tx900249d. [DOI] [PubMed] [Google Scholar]
- Nash EF, Stephenson A, Ratjen F, Tullis E. Nebulized and oral thiol derivatives for pulmonary disease in cystic fibrosis. Cochrane Database Syst Rev. 2009;(1) doi: 10.1002/14651858.CD007168.pub2. CD007168. [DOI] [PubMed] [Google Scholar]
- O'Malley YQ, Reszka KJ, Spitz DR, Denning GM, Britigan BE. Pseudomonas aeruginosa pyocyanin directly oxidizes glutathione and decreases its levels in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L94–103. doi: 10.1152/ajplung.00025.2004. [DOI] [PubMed] [Google Scholar]
- Pattison DI, Davies MJ, Hawkins CL. Reactions and reactivity of myeloperoxidase-derived oxidants: differential biological effects of hypochlorous and hypothiocyanous acids. Free Radic Res. 2012;46:975–995. doi: 10.3109/10715762.2012.667566. [DOI] [PubMed] [Google Scholar]
- Pedemonte N, Caci E, Sondo E, Caputo A, Rhoden K, Pfeffer U, et al. Thiocyanate transport in resting and IL-4-stimulated human bronchial epithelial cells: role of pendrin and anion channels. J Immunol. 2007;178:5144–5153. doi: 10.4049/jimmunol.178.8.5144. [DOI] [PubMed] [Google Scholar]
- Quinton PM. Chloride impermeability in cystic fibrosis. Nature. 1983;301:421–422. doi: 10.1038/301421a0. [DOI] [PubMed] [Google Scholar]
- Reeves EP, Williamson M, O'Neill SJ, Greally P, McElvaney NG. Nebulized hypertonic saline decreases IL-8 in sputum of patients with cystic fibrosis. Am J Respir Crit Care Med. 2011;183:1517–1523. doi: 10.1164/rccm.201101-0072OC. [DOI] [PubMed] [Google Scholar]
- Seear M, Hui H, Magee F, Bohn D, Cutz E. Bronchial casts in children: a proposed classification based on nine cases and a review of the literature. Am J Respir Crit Care Med. 1997;155:364–370. doi: 10.1164/ajrccm.155.1.9001337. [DOI] [PubMed] [Google Scholar]
- Sorensen RU, Klinger JD, Cash HA, Chase PA, Dearborn DG. In vitro inhibition of lymphocyte proliferation by Pseudomonas aeruginosa phenazine pigments. Infect Immun. 1983;41:321–330. doi: 10.1128/iai.41.1.321-330.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HL, Regamey N, Brown S, Bush A, Lloyd CM, Davies JC. The Th17 pathway in cystic fibrosis lung disease. Am J Respir Crit Care Med. 2011;184:252–258. doi: 10.1164/rccm.201102-0236OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas EL. Lactoperoxidase-catalyzed oxidation of thiocyanate: equilibria between oxidized forms of thiocyanate. Biochemistry. 1981;20:3273–3280. doi: 10.1021/bi00514a045. [DOI] [PubMed] [Google Scholar]
- Thomas EL, Fishman M. Oxidation of chloride and thiocyanate by isolated leukocytes. J Biol Chem. 1986;261:9694–9702. [PubMed] [Google Scholar]
- Thomas EL, Milligan TW, Joyner RE, Jefferson MM. Antibacterial activity of hydrogen peroxide and the lactoperoxidase-hydrogen peroxide-thiocyanate system against oral streptococci. Infect Immun. 1994;62:529–535. doi: 10.1128/iai.62.2.529-535.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velsor LW, van Heeckeren A, Day BJ. Antioxidant imbalance in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am J Physiol Lung Cell Mol Physiol. 2001;281:L31–L38. doi: 10.1152/ajplung.2001.281.1.L31. [DOI] [PubMed] [Google Scholar]
- Wagner BA, Reszka KJ, McCormick ML, Britigan BE, Evig CB, Burns CP. Role of thiocyanate, bromide and hypobromous acid in hydrogen peroxide-induced apoptosis. Free Radic Res. 2004;38:167–175. doi: 10.1080/10715760310001643302. [DOI] [PubMed] [Google Scholar]
- Walker TS, Tomlin KL, Worthen GS, Poch KR, Lieber JG, Saavedra MT, et al. Enhanced Pseudomonas aeruginosa biofilm development mediated by human neutrophils. Infect Immun. 2005;73:3693–3701. doi: 10.1128/IAI.73.6.3693-3701.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wark P, McDonald VM. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database Syst Rev. 2009;(2) doi: 10.1002/14651858.CD001506.pub3. CD001506. [DOI] [PubMed] [Google Scholar]
- Westley J. Thiosulfate: cyanide sulfurtransferase (rhodanese) Methods Enzymol. 1981;77:285–291. doi: 10.1016/s0076-6879(81)77039-3. [DOI] [PubMed] [Google Scholar]
- Wijkstrom-Frei C, El-Chemaly S, Ali-Rachedi R, Gerson C, Cobas MA, Forteza R, et al. Lactoperoxidase and human airway host defense. Am J Respir Cell Mol Biol. 2003;29:206–212. doi: 10.1165/rcmb.2002-0152OC. [DOI] [PubMed] [Google Scholar]
- Wilson AM, Leigh R, Hargreave FE, Pizzichini MM, Pizzichini E. Safety of sputum induction in moderate-to-severe smoking-related chronic obstructive pulmonary disease. COPD. 2006;3:89–93. doi: 10.1080/15412550600651339. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC, Brennan SO. Characterization of the oxidation products of the reaction between reduced glutathione and hypochlorous acid. Biochem J. 1997;326(Pt 1):87–92. doi: 10.1042/bj3260087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Chen Y, Hazen SL. Eosinophil peroxidase nitrates protein tyrosyl residues. Implications for oxidative damage by nitrating intermediates in eosinophilic inflammatory disorders. J Biol Chem. 1999;274:25933–25944. doi: 10.1074/jbc.274.36.25933. [DOI] [PubMed] [Google Scholar]
- Xu Y, Szep S, Lu Z. The antioxidant role of thiocyanate in the pathogenesis of cystic fibrosis and other inflammation-related diseases. Proc Natl Acad Sci U S A. 2009;106:20515–20519. doi: 10.1073/pnas.0911412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xulu BA, Ashby MT. Small molecular, macromolecular, and cellular chloramines react with thiocyanate to give the human defense factor hypothiocyanite. Biochemistry. 2010;49:2068–2074. doi: 10.1021/bi902089w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.