

Abstract
Background
Desired serotonin 5HT2 receptor pharmacology for treatment of psychoses is 5HT2A antagonism and/or 5HT2C agonism. No selective 5HT2A antagonist has been approved for psychosis and the only approved 5HT2C agonist (for obesity) also activates 5HT2A and 5HT2B receptors, which can lead to clinical complications. Studies herein tested the hypothesis that a dual-function 5HT2A antagonist/5HT2C agonist that does not activate 5HT2B receptors would be suitable for development as an antipsychotic drug, without liability for weight gain.
Methods
The novel compounds (+)- and (−)-trans-4-(4′-chlorophenyl)-N,N-dimethyl-2-aminotetralin (p-Cl-PAT) were synthesized, characterized in vitro for affinity and functional activity at human 5HT2 receptors, and administered by intraperitoneal (i.p.) and oral (gavage) routes to mice in behavioral paradigms that assessed antipsychotic efficacy and effects on feeding behavior.
Results
(+)- and (−)-p-Cl-PAT activated 5HT2C receptors, with (+)-p-Cl-PAT being 12-times more potent, consistent with its higher affinity across 5HT2 receptors. Neither p-Cl-PAT enantiomer activated 5HT2A or 5HT2B receptors at concentrations up to 300-times greater than their respective affinity (Ki), and (+)-p-Cl-PAT was shown to be a 5HT2A competitive antagonist. When administered i.p. or orally, (+)- and (−)-p-Cl-PAT attenuated the head-twitch response (HTR) in mice elicited by the 5HT2 agonist (−)-2,5-dimethoxy-4-iodoamphetamine (DOI) and reduced intake of a highly palatable food in non-food-deprived mice, with (+)-p-Cl-PAT being more potent across behavioral assays.
Conclusions
The novel in vitro pharmacology of (+)-p-Cl-PAT (5HT2A antagonism/5HT2C agonism without activation of 5HT2B) translated in vivo to an orally-active drug candidate with preclinical efficacy to treat psychoses without liability for weight gain.
Keywords: anorectic response, antipsychotic drug, binge eating DOI head-twitch response, schizophrenia, serotonin (5HT) 2
1 INTRODUCTION
Brain dopamine and serotonin systems have long been thought to be involved in the pathophysiology and pharmacotherapy of schizophrenia and other psychoses. For example, the dopamine hypothesis of schizophrenia arose from observations that the first relatively safe and effective antipsychotic drugs, the phenothiazines (e.g., chlorpromazine) in the early 1950s, affect brain dopamine metabolism (Carlsson and Lindquist, 1963). Neuroleptics such as chlorpromazine and the butyrophenone haloperidol “take hold” (lepsis) of the central nervous system (CNS) to suppress movement as well as other behavior, and resulting debilitating extrapyramidal movement side effects are implicit in the clinical definition of neuroleptic antipsychotic drugs. Newer antipsychotic drugs, without substantial risk of extrapyramidal effects, are referred to as “atypical” and have a mechanism other than (or in addition to) postsynaptic D2-type receptor blockade. Such second generation antipsychotics include the dibenzodiazepines (e.g., clozapine, olanzapine) that have less potential for extrapyramidal side effects and have significant activity at brain serotonin 5HT2-type receptors (as well as adrenergic α1/α2, muscarinic, and/or histamine H1 receptors) in addition to activity at D2-type receptors (Meltzer, 1996; Meltzer et al., 2003; Seeman, 2002; Tandon and Fleischhacker, 2005). Speculatively, a relatively selective 5HT2A antagonist might function as a useful antipsychotic medication. Several new potential medications have been developed as antipsychotic drugs that are selective 5HT2A antagonists/inverse agonists, however little success in the clinic has been observed (clinicaltrials.gov reveals no ongoing trials using selective 5HT2A compounds) suggesting that selective blockade of 5HT2A alone may not represent the ideal pharmacological profile.
One side effect of atypical antipsychotics is weight gain (Coccurello and Moles, 2010; Lieberman et al., 2005; Maayan and Correll, 2010; McCloughen and Foster, 2011). In addition to H1 antagonism (Kroeze et al., 2003), it has been proposed that the weight gain effects of many atypical antipsychotic drugs is due to concurrent antagonism of the 5HT2C receptor along with the 5HT2A receptor (Balt et al., 2011; Kirk et al., 2009; Meltzer and Massay, 2011; Roerig et al., 2011). In fact, most atypical antipsychotic drugs that are 5HT2A antagonists/inverse agonists are also 5HT2C receptor antagonists/inverse agonists (Herrick-Davis et al., 2000; Rauser et al., 2001). Preclinical data suggest that activation of 5HT2C receptors is associated with decreased eating and weight loss (Hayashi et al., 2002; Hewitt et al., 2002; Rowland et al., 2008). In addition, one neuropharmacological consequence of 5HT2C activation is preferential decreases in mesolimbic compared to nigrostriatal dopamine levels (Di Giovanni et al., 2000; Marquis et al., 2007) which may suggest an improved antipsychotic effect without motor-related side effects. Thus, the desirable serotonergic pharmacology for an antipsychotic medication is 5HT2A antagonism and 5HT2C agonism. It should be noted that there is no clinical tolerance for activation of 5HT2B receptors as this is associated with severe cardiac valvulopathy and other cardiotoxic effects (Elangbam, 2010; Roth, 2007).
We previously reported that a novel phenylaminotetralin, (−)-(2S, 4R)-trans-4-phenyl-N,N-dimethyl-2-aminotetralin (PAT; Fig. 1), was a 5HT2C agonist with 5HT2A and 5HT2B inverse agonist/antagonist activity (Booth et al., 2009) that demonstrated preclinical antipsychotic activity (Booth et al., 2008) and decreased intake of a highly palatable food in non-food-deprived mice (a binge eating model) (Rowland et al., 2008). The PAT molecular scaffold (Figure 1) has two chiral centers, thus, there are a pair of cis- and trans-enantiomers, for a total of four stereoisomers. For PAT, the (−)-(2S, 4R)-trans enantiomer is most potent and efficacious regarding binding and function across 5HT2 receptor subtypes (Booth et al., 2009). Here we report on two novel analogs that contain a chlorine moiety at 4′- or para-position of the pendant C(4) phenyl group on the PAT parent compound, (+)-(2R, 4S)- and (−)-(2S, 4R)-trans-4-(4′-chlorophenyl)-N,N-dimethyl-2-aminotetralin (p-Cl-PAT; Fig. 1). Computational chemistry studies suggest that the electronegative chlorine atom at the para-position of the PAT phenyl ring provides an electronic and steric symmetry that modulates affinity and function at 5HT2 receptors (Cordova-Sinjago et al., 2011, 2012). Here we report the in vitro 5HT2 affinity and function of (+)- and (−)-p-Cl-PAT and assess its in vivo 5HT2 activity after intraperitoneal (i.p.) and oral (p.o.) administration in behavioral paradigms sensitive to the antipsychotic and “anti-binge-eating” effects of potential therapeutics.
Fig. 1.
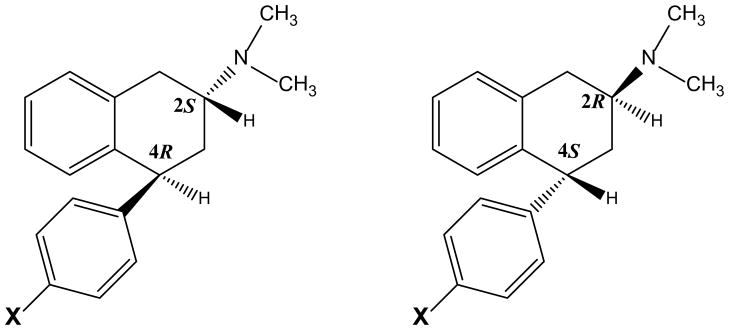
Structures: (+)-(2R,4S) and (−)-(2S, 4R)-trans-4-phenyl-N,N-dimethyl-2-aminotetralin (X=H; PAT); (+)-(2R, 4S)- and (−)-(2S, 4R)-trans-4-(4′-chlorophenyl)-N,N-dimethyl-2-aminotetralin (X=Cl; p-Cl-PAT)
2 METHODS
2.1 Chemicals
The radioligands [3H]-mesulergine and [3H]-ketanserin were purchased from Perkin-Elmer Life Science (Boston, MA). The 5HT2 agonist (−)-2,5-dimethoxy-4-iodoamphetamine hydrochloride (DOI) was generously supplied by the National Institute on Drug Abuse. The (+)-(2R, 4S)- and (−)-(2S, 4R)-trans enantiomers of 4-(4′-chlorophenyl)-N,N-dimethyl-2-aminotetralin (p-Cl-PAT) hydrochloride were synthesized by Dr. Rajeev Sakhuja at the University of Florida Department of Medicinal Chemistry (Morgan et al., 2012) and a full description of the methods and physical characterization will be reported elsewhere. Briefly, 4-(4′-chlorophenyl)-tetralen-2-ol phenylacetate was synthesized from 4-chlorostyrene and trifluoroacetyl phenylacetyl anhydride via cascade Friedel–Crafts cycli-acylalkylation, enolization, and O-acylation following our published procedures (Vincek and Booth, 2009). The tetralen-2-ol phenylacetate was reduced with sodium borohydride at 50°C in methanol to yield the cis-tetralol (major) that was converted to the corresponding cis-tosylate using p-tosyl chloride in pyridine at room temperature. The cis-tosylate on amination with aqueous dimethylamine solution in a sealed tube at 80°C for 24 h yielded racemic trans-4-(4′-chlorophenyl)-N,N-dimethyl-2-aminotetralin (high resolution mass spectrometry results for C18H21ClN [M+H]+ : 286.1364; calculated: 286.1363). The trans racemic p-Cl-PAT mixture was resolved to the (+)- and (−)-enantiomers using chiral stationary-phase preparative HPLC (MeOH:EtOH:1-PrOH:2-PrOH:Hexanes [5:5:5:5:80] + 0.2% TEA modifier; flow rate = 1.5 mL/min; t1 = 10.9, t2 =12.8). The water-soluble (+)- and (−)-p-Cl-PAT hydrochloride (HCl) salts were prepared ([α]25D = +45.5 and −46.0 in CH2Cl2, respectively) and used in all in vitro and in vivo pharmacological experiments.
2.2 Clonal cell culture and transfection
Human embryonic kidney 293 cells (HEK, ATCC CRL-1573) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 5% fetal bovine serum and 1% penicillin – streptomycin. Cells were grown in a humidified incubator at 37 °C with 5% carbon dioxide. The cDNAs encoding the human 5HT2A, 5HT2B and 5HT2CINI (unedited wild type isoform) wild type receptors were obtained from UMR cDNA Resource Center (Rolla, MO). HEK-293 cells were grown to 90% confluency in DMEM (10–013-CV, Mediatech, Manassas, VA), supplemented with 5% dialyzed fetal bovine serum in 10 cm plates. Cells were washed then transfected with 24 μg of 5HT2 receptor subtype cDNA mixed with 40 μl Lipofectamine 2000 reagent in Opti-MEM and placed in an incubator for 24 to 48 hr. Membranes were then collected in 50 mM Tris, 10 mM MgCl2-6H2O, and 0.1 mM EDTA (assay buffer) using previous methods (Booth et al., 2009) and stored at −80°C until binding assays were performed.
2.3 In vitro molecular pharmacology
2.3.1 Radioreceptor competition binding assays
Radioligand competitive displacement binding assays were performed in 96-well plates, using 3–5 μg of protein from membrane samples per well, similar to laboratory methods used previously (Booth et al.,2009). Radioligands were included in assay mixtures at ~Kd concentration, i.e., 2.0 nM [3H]-ketanserin (5HT2A receptors), 1.95nM [3H]-mesulergine (5HT2B receptors), or 1.4 nM [3H]-mesulergine (5HT2C receptors). Non-specific binding was determined in the presence of 10 μM mianserin for all 5HT2 receptors. Incubation of radioreceptor binding assay mixtures was for 1.0 h at 37°C, with termination by rapid filtration through Whatman GF/B filters using a 96-well cell harvester (Tomtec, Hamden, CT) and subsequently washed five times with 50 mM Tris-HCl at room temperature. Filters containing bound [3H]-radioligand were dried, placed in vials containing 2 mL scintillation cocktail (ScintiVerse), allowed to equilibrate overnight, and then were counted for 3H-induced scintillation using a Beckman-Coulter LS6500 counter. Binding experimental conditions were performed in triplicates, and each experiment was performed a minimum of three times. Data were analyzed using nonlinear regression curve-fitting algorithms in GraphPad Prism, 5.03 for Windows (San Diego, CA). Data points were limited to eight, thus Hill slopes were not calculated (Motulsky and Christopoulos, 2003); data were fit using the “one site fit-Ki” model that constrains the Hill slope to 1.0. Ligand affinity is expressed as Ki values by conversion of the IC50 data using the equation Ki = IC50/1 + L/KD where L is the concentration of radioligand (Cheng and Prusoff, 1973). Comparisons of Ki values were performed using two-way ANOVA with Bonferroni’s post-hoc test. Differences were considered statistically significant when the p-value was less than 0.05.
2.3.2 Measurement of PLC Activation and [3H]-IP Formation
Functional activity was measured as PLC activation and [3H]-IP formation in HEK cells transiently expressing 5HT2A, 5HT2B or 5HT2C receptors (Booth et al., 2009; Canal et al., 2011). HEK cells grown to approximately 80% confluency in DMEM containing 10% fetal bovine serum and 1% antibiotic in 10 cm plates at 37°C, 5% CO2 (incubator) were washed one time with PBS, then transfected with 20 μg 5HT2 receptor subtype cDNA and 40 μl lipofectamine 2000 reagent in 5 mL DMEM containing 5% dialyzed fetal bovine serum and 5 mL Opti-MEM (transfection media). Cells were placed in an incubator, and approximately 16 hr later, transfection media was removed and replaced with 16 mL inositol-free DMEM containing 2.5% dialyzed fetal bovine serum. Cells were then detached by vigorous pipetting, 0.1 μ Ci/mL [3H]-myo-inositol was added to the mixture (labeling media), and cells were seeded 300 μl per well into 48 well CellBind® plates (Costar), and placed in an incubator. 24 hr later, plates were centrifuged at 2500 rpm for 10 min at room temperature, labeling media was discarded, and 450 μl inositol-free, serum-free DMEM was added to each well. Cells were placed in an incubator for one hour. Cells were then incubated for 30 min with the desired concentration of test compound diluted in inositol-free, serum-free DMEM containing a final concentration of 50 mM LiCl and 10 μM pargyline per well. Plates were again centrifuged at 2500 rpm for 10 min at room temperature, drug incubation media was discarded, and 400 μl of 50 mM formic acid was added to each well to lyse cells. One hour later, 200 μl of 150 mM NH4OH was added to each well to neutralize cells, and plates were stored at −20°C overnight. After thawing, 500 μl of solution from each well was added to individual anion-exchange columns to separate [3H]-inositol phosphates formed from [3H]-myo-inositol. Following a 10 mL wash with deionized ultra-purified water, bound [3H]-inositol phosphates were eluted with 4 mL 800 mM ammonium formate into vials. To 1.0 mL of eluate was added 10 mL scintillation fluid and counted, as above. Functional assay experimental conditions were performed in triplicate, and each independent experiment was performed a minimum of three times. Resulting data were analyzed using the nonlinear regression algorithms in Prism, with the one-site model providing the best fit. Data is expressed as mean percentage of basal control [3H]-IP formation, with potency expressed as EC50 (test ligand concentration required to stimulate [3H]-IP formation by 50% over basal) or pA2(testligand concentration required to increase 5HT EC50 value two-times, as calculated from Schild analysis), with SEM determined from at least three independent experiments; comparisons of potency values were performed using Student’s t-test.
2.4 In vivo behavioral pharmacology
2.4.1 Subjects
C57Bl/6J male mice were obtained from Harlan (food studies) or Jackson (HTR) Laboratories at approximately 8 weeks of age, and allowed to acclimate to the temperature- and humidity-controlled colony room for at least 1 week prior to testing. Mice were singly- (food studies) or pair- (HTR) housed in standard cages and allowed unlimited access to laboratory chow and water. Experiments were conducted at approximately the middle of the light phase (lights on at 6 am, and lights off at 6 pm). All compounds were dissolved in sterile saline prior to behavioral testing, and administered in a volume of 0.01 ml/g body weight. All experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals, as promulgated by the National Institutes of Health, and were approved by the University of Florida’s Institutional Animal Care and Use Committee.
2.4.2 DOI-elicited head-twitch response and locomotor activity
On the day of testing, mice were habituated to the testing room for approximately 30 minutes. Testing consisted of administration (i.p.) of sterile saline or a particular dose of (+) or (−)-p-Cl-PAT. Ten minutes later, mice were administered (−)-DOI or sterile saline. Ten minutes later, mice were individually placed into a clear plexiglas chamber (17″ × 17″ × 12″). Following oral administration of the PAT analog or saline, 20 minutes elapsed before administration of (−)-DOI. Head-twitch responses (HTRs), defined as a clear, rapid, and discrete, back and forth rotation of the head, were counted by a trained observer (A.K.) who was blind to drug treatment conditions, during a 10-min observation session. An overhead camera videotaped the session, and activity (distance travelled in cm) was analyzed and calculated by Ethovision software (Noldus Information Technology Inc.).
2.4.3 Palatable meal eating
Mice were tested in their home cages with Purina 5001 chow and water available at all times. Five days/week, mice were given a measured number of Fruit Crunchies (Bio-Serv, Frenchtown, NJ) in a glass beaker inside the cage. Crunchies, in 3 flavors, are purified ingredient and sweet spherical pellets approximately 190 mg and 3.5 kcal/g. Access was rapidly tapered to 30 min/day and intake was measured daily. After ~2 weeks when day-to-day intake was stable, mice received the test compound by i.p. injection (2 ml/kg) 15 min before access to Crunchies. Intake was expressed as % of the baseline averaged across the previous 4 days.
2.4.4 Statistics
Differences in dependent measures were analyzed by 2-way ANOVA and post-hoc Newman-Keuls tests, or Student’s t-test, as appropriate using commercially available statistical software (Sigmastat 3.1). Differences were considered statistically significant when the p-value was less than 0.05. ED50 values and 95% confidence limits were determined using log-linear interpolation from the descending limb of the dose-effect curves.
3 RESULTS
3.1 In vitro molecular pharmacology of (+) and (−)-p-Cl-PAT at 5HT2 receptors
3.1.1 Affinity
Both (+) and (−)-p-Cl-PAT potently displaced [3H]-ketanserin and [3H]-mesulergine from 5HT2A and 5HT2B or 2C receptors, respectively (Figure 2, data summarized in Table 1), with (+)-p-Cl-PAT having significantly higher affinity (Ki) than (−)-p-Cl-PAT at 5HT2A, 5HT2B, and 5HT2C receptors (F2,22=5.23, F1,22=92.04, and F2,22=12.17, respectively; p < 0.05 for all subtypes). In contrast to its function (see below), the affinity of (+)-p-Cl-PAT was not different (p > 0.05) at the 5HT2 receptor subtypes, whereas, (−)-p-Cl-PAT binds with higher affinity (p < 0.05) at 5HT2C compared to 5HT2A and 5HT2B receptors.
Fig. 2.
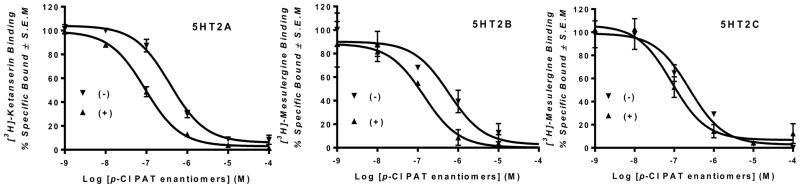
Representative radioligand competition binding curves for (+) and (−)-p-Cl-PAT enantiomers at human 5HT2 receptors expressed in human embryonic kidney cells (HEK) cells. Data are represented as mean ± SEM of at least three independent experiments each performed in triplicate.
Table 1.
In vitro pharmacology: Affinity and function of (+)- and (−)-p-Cl-PAT at 5HT2A, 5HT2B and 5HT2C receptors.
| p-Cl PAT | 5HT2A | 5HT2B | 5HT2C | |||
|---|---|---|---|---|---|---|
| Ki ± SEM (nM) | Function | Ki ± SEM (nM) | Function | Ki ± SEM (nM) | EC50±SEM (nM) | |
| (+)-trans | 42 ± 6.0 | No Activation1 pA2=6.21 ± 0.55 |
80 ± 10 | No Activation1 | 45 ± 7.0 | 140 ± 20 |
| (−)-trans | 240 ± 32 | No Activation1 | 300 ± 30 | No Activation1 | 130 ± 16 | 1650 ± 149 |
No activation of PLC/IP formation at 10 μM
3.1.2 Function
Figure 3 shows functional activity of 5HT in comparison to (+) and (−)-p-Cl-PAT at 5HT2A, 5HT2B and 5HT2C receptors, as measured by activation of PLC and [3H]-IP formation. Consistent with the literature (e.g., Booth et al., 2009), the endogenous agonist serotonin (5HT) had an EC50 value of 106 ± 19 nM, 50 ± 10nM and 10 ± 3 nM at 5HT2A, 5HT2B and 5HT2C receptors, respectively. Neither (+) nor (−)-p-Cl-PAT activated 5HT2A and 5HT2B receptors, even at 10 μM concentration (125– to 300-times Ki values summarized in Table 1). In contrast, both (+) and (−)-p-Cl-PAT were partial agonists (compared to 5HT) at the 5HT2C receptor. Consistent with its higher affinity (Table 1), the (+)-p-Cl-PAT enantiomer was about 12-times more potent (EC50 = 140 ± 20nM) (p < 0.0001) than (−)-p-Cl-PAT (EC50 = 1650 ± 149nM), however, there was no difference (p > 0.05) in efficacy (Emax). Additional functional studies undertaken with (+)-p-Cl-PAT confirmed competitive antagonism (pA2 = 6.21 ± 0.55) with respect to 5HT activation of 5HT2A-mediated [3H]-IP formation.
Fig. 3.

A–C: Representative concentration–response data for 5HT and (+)- and (−)-p-Cl-PAT activation of 5HT2A, 5HT2B and 5HT2C receptors; D: Representative 5HT concentration–response data at 5HT2A receptors in presence of increasing concentrations of (+)-p-Cl-PAT.
3.2 In vivo behavioral pharmacology
3.2.1 Head-twitch responses
When (−)-DOI was administered (preceded by a saline injection), there was a dose-dependent increase in the number of head-twitch responses (HTR) observed during the 10-minute session (Figure 4, left panel). The number of head-twitches increased from an average of 1.25 following saline administration, to 6, 22, and 45 head-twitches following 0.1, 0.3, and 1.0 mg/kg (−)-DOI (F9,28=42.3; p<0.001; significant increases at 0.3 and 1.0 mg/kg DOI). Both the (+) and (−) enantiomers of p-Cl-PAT failed to alter the number of HTRs when administered with saline (range from 0 to 2.5 across doses; Figure 4, left panel). Subsequent experiments examined the effects of p-Cl-PAT pretreatment before administration of 1 mg/kg (−)-DOI (Figure 4, right panel). Both isomers produced a dose-dependent attenuation of the DOI-elicited HTR such that following pretreatment with 30 mg/kg of the (+) and (−) enantiomers, there were 3.3 and 13 responses, respectively. ANOVA (F6,27=15.66; p<0.001) revealed that 10 and 30 mg/kg doses of (+) and 30 mg/kg of (−)-p-Cl-PAT resulted in significantly lower levels of HTRs relative to saline pretreatment. The dose required to produce a 50% attenuation (ED50 value ±95% CL) of the DOI-elicited response was 8.2 (5.4–12.4) and 20.1 (14.0–28.8) mg/kg for the (+) and (−) enantiomers, and there was a significant difference between the dose-effect curves of (+) and (−)-p-Cl-PAT (F1,23=17.49; p<0.001). The effects of 30 mg/kg of each enantiomer and the racemic combination were assessed following oral administration (via gavage). Following oral saline administration, (−)-DOI (1.0 mg/kg) elicited an average of 37.5 HTRs (Figure 5). Oral administration of (+), (−), and (±) p-Cl-PAT (30 mg/kg) resulted in an attenuated response to DOI: 8.5, 18.25, and 13.25 HTRs respectively. Each dose combination resulted in fewer HTRs (F3,16=31.1; p<0.001) relative to saline pretreatment, and there was a statistically significant difference between the (+)- and (−)-enantiomers (p=0.038).
Fig. 4.

Head-twich responses in C57BL/6J mice. (−)-DOI elicits a dose-dependent increase in head-twitch responses (left panel). Both the (+) and (−) enantiomers of p-Cl-PAT fail to elicit head-twitches when administered with saline (left panel). When administered as a pretreatment to (−)-DOI (1.0 mg/kg), both enantiomers resulted in a dose-dependent attenuation of the elicited head twitches (right panel). The (+) enantiomer was more potent in producing this effect. * indicates a difference from saline.
Fig. 5.

Effects of oral administration of p-Cl-PAT. The figure shows the effects of 30 mg/kg of the (+) and (−) enantiomers, and the racemic combination, administered orally (p.o. route via gavage) 20 min before DOI administration. The (+) enantiomer was more effective than the (−) enantiomer in attenuating the DOI-elicited HTR. When the racemic mixture of p-Cl-PAT was administered orally, there was intermediate level of attenuation. * indicates a difference from saline pretreatment, and ** indicates differences from saline and each other
3.2.2 Locomotor activity
During all behavioral sessions, levels of overall activity were recorded and are shown in Table 2. Although, there appeared to be a slight increase in activity following administration of 1.0 mg/kg (−)-DOI, there were no statistically significant differences following any drug alone, relative to saline. The highest doses of (+) and (−) p-Cl-PAT administered as a pretreatment to 1.0 mg/kg (−)-DOI resulted in statistically significant lower levels of activity relative to saline pretreatment (p values <0.05). Oral administration of p-Cl-PAT failed to alter locomotor activity when compared to oral saline pretreatment.
Table 2.
Activity during 10-min test sessions. Doses are expressed as mg/kg.
| Distance travelled (mean ± s.e.m.) | ||
|---|---|---|
| Saline pretreatment | ||
|
| ||
| Saline | 2770 ± 248 | |
|
| ||
| (−)-DOI | 0.1 | 2829 ± 481 |
| 0.3 | 2804 ± 353 | |
| 1.0 | 3626 ± 392 | |
|
| ||
| (−) p-Cl-PAT | 3 | 2863 ± 3.8 |
| 10 | 3097 ± 937 | |
| 30 | 1994 ± 165 | |
|
| ||
| (+) p-Cl-PAT | 3 | 2216 ± 98 |
| 10 | 2142 ± 124 | |
| 30 | 870 ± 502 | |
|
| ||
| DOI (1 mg/kg) pretreatment | ||
|
| ||
| Saline | 3531 ± 508 | |
|
| ||
| (−) p-Cl-PAT | 3 | 3386 ± 150 |
| 10 | 3064 ± 309 | |
| 30 | 1889 ± 191 * | |
|
| ||
| (+) p-Cl-PAT | 3 | 3523 ± 518 |
| 10 | 2557 ± 99 | |
| 30 | 1329 ± 434 * | |
|
| ||
| Oral pretreatment before 1.0 mg/kg DOI | ||
|
| ||
| Saline | 3413 ± 263 | |
| (−) p-Cl-PAT | 30 | 1944 ± 654 |
| (+) p-Cl-PAT | 30 | 1586 ± 398 |
| (±) p-Cl-PAT | 30 | 1662 ± 468 |
indicates statistically significant difference from DOI (with saline) administration.
3.2.3 Palatable meal eating
Mice were provided 10 Fruit Crunchies daily, and ate on average, 5.2 (± 0.1) pellets during the baseline 30-minute sessions and 5.0 (± 0.25) pellets following administration of vehicle. Both enantiomers produced a dose-dependent attenuation of the number of pellets consumed such that following pretreatment with 24 mg/kg of the (+) and (−) enantiomers, eating was statistically significantly decreased to 15.9% (± 6.4) and 36.4% (± 9.1) of baseline levels, respectively (Figure 6; F6,58=33.6, p<0.001). The doses required to produce a 50% attenuation (ED50 value ±95% CL) of consumption was 14.8 (11.8–18.7) and 21.3 (18.5–24.4) mg/kg for the (+) and (−) enantiomers, and there was a significant difference between the dose-effect curves of (+) and (−)-p-Cl-PAT (F1,42=10.2, p<0.003).
Fig. 6.

“Crunchies” (highly-palatable, nutritionally-complete treats) consumed by C57Bl/6J mice. Mice eat approximately 5 pellets during baseline and following vehicle administration. Both the (+) and (−) enantiomers of p-Cl-PAT produced a dose-dependent decrease in pellets consumed (displayed as % baseline). The (+) enantiomer was more potent in producing this effect. * indicates a difference from saline.
4 DISCUSSION
It is complicit from preclinical and clinical literature and practice that optimal 5HT2 pharmacology desired for antipsychotic drug efficacy without liability for weight-gain or other untoward side effects (such as cardiopulmonary toxicity associated with 5HT2B activation) is absolutely-selective 5HT2A antagonism and/or 5HT2C agonism. Accordingly, the unique multifunctional 5HT2 pharmacology (i.e., 5HT2A/2B antagonism together with 5HT2C agonism) demonstrated by (−)-(2S, 4R)-trans-PAT (Booth et al., 2009) appears to be optimal for development of 5HT2-based antipsychotic drugs without weight gain and other deleterious side effects. Based on computational and molecular modeling structure–activity studies, we predicted that the novel analog p-Cl-PAT would demonstrate 5HT2 pharmacology similar to PAT, i.e. (pre)clinical relevant affinity at 5HT2A and 5HT2C receptors but specifically activate only 5HT2C receptors. Unexpectedly, stereoselectivity regarding 5HT2 affinity and functional activity was reversed for p-Cl-PAT in comparison to PAT. Thus, the (+)-(2R, 4S)-trans-p-Cl-PAT enantiomer had (pre)clinically-relevant high affinity across 5HT2 receptors, in comparison to the (−)-enantiomer, which had several-fold lower affinity. Likewise, functionally, the (+)-p-Cl-PAT enantiomer was the more potent 5HT2C (partial) agonist. Importantly, however, (+)-p-Cl-PAT did not activate 5HT2A or 5HT2B receptors (nor did the [−]-enantiomer) and was shown to competitively antagonize 5HT activation of 5HT2A-mediated activation of PLC and IP formation. These unexpected results concerning stereochemistry impact on 5HT2 ligand affinity and function provide important information for computational and modeling studies regarding the 3-dimensional arrangement of 5HT2 receptor amino acids in the binding pocket. Results will be used to refine current 5HT2 receptor models based on homology to the β-adrenergic receptors (Cordova-Sinjago et al., 2011, 2012) for accurate prediction of ligand interactions for drug discovery purposes. Nevertheless, here we identified (+)-p-Cl-PAT as a new high affinity 5HT2 receptor ligand that potently and specifically activates 5HT2C and not 5HT2A or 5HT2B receptors, precisely the 5HT2-based pharmacology desired for antipsychotic activity, without liability for 5HT2-mediated untoward side-effects such enhanced feeding behavior or cardiopulmonary toxicity.
To determine if the unique multifunctional 5HT2 receptor pharmacology of p-Cl-PAT would translate in vivo to preclinical antipsychotic activity, we examined their ability to modulate the DOI-elicited HTR in mice. DOI-elicited HTRs have been purported to model a number of behavioral and pathological conditions including schizophrenia/psychosis, obsessive-compulsive disorders, tics associated with Tourette’s syndrome, and hallucinogenesis (Canal and Morgan, 2012). Similarities in the subjective effects of psychedelic hallucinogens and the perceptual and subjective effects associated with psychosis-related disorders (e.g. schizophrenia) suggested that the behavioral effects following administration of hallucinogenic compounds (e.g. DOI-elicited HTRs) could be a model of the “positive” symptoms (in particular, hallucinations) of schizophrenia (González-Maseso et al, 2008). In this regard, every effective antipsychotic medication used in humans is effective in attenuating the HTR elicited by DOI administration, including “typical” (primarily D2 antagonists) and “atypical” (5HT2/D2 antagonists) antipsychotics, as well as novel medications being investigated in clinical trials (e.g. glutamatergic compounds; clinicaltrials.gov). Although there are few “false negatives” associated with this model as a “screen” for antipsychotic activity, there are examples of “false positives” (i.e. drugs that work in the animal model and do not translate into effective antipsychotic compounds in humans). An important example is the relatively selective 5HT2A antagonist/inverse agonist M100907 which apparently failed in clinical trials for treating schizophrenia.
In fact, the in vitro 5HT2 molecular pharmacology of p-Cl-PAT translated congruently in vivo, with the (+)-enantiomer being more potent than the (−)-enantiomer in the DOI-elicited HTR assay, and with the (+/−)-racemic mixture being intermediate between the two in potency. It is proposed here that activation of 5HT2C receptors (5HT2A antagonism is complicit) associated with the lead (+)-p-Cl-PAT analog may enhance its potential as an antipsychotic drug given that 5HT2C activation preferentially decreases mesolimbic dopamine levels while sparing nigrostriatal dopamine cell activity and extracellular levels (Di Giovanni et al., 2000), a pharmacological profile suggesting antipsychotic effects without debilitating extrapyramidal side effects.
Another primary side effect of currently available antipsychotic medications is weight gain (Coccurello and Moles, 2010; Lieberman et al., 2005; Maayan and Correll, 2010; McCloughen and Koster, 2011). In addition to the adverse health effects associated with weight gain itself (National Task Force on the Prevention and Treatment of Obesity, 2000), patient weight gain dramatically decreases antipsychotic medication compliance (Weiden et al., 2004). There are conflicting data regarding the physiological mechanisms by which weight gain associated antipsychotic use occurs (Balt et al., 2011; Correll et al., 2011; Roerig et al., 2011), however it has been suggested that antagonism of 5HT2C receptor systems may be partially responsible. For example, atypical antipsychotics generally have clinically-relevant affinity at several neurotransmitter receptor types, including, adrenergic (α1 and α2), dopamine (D1, D3 and D4), histamine (H1), muscarinic, and serotonin (5HT1A, 5HT2A, 5HT2C, 5HT6 and 5HT7). While 5HT2A receptor antagonism is thought to contribute to the efficacy of atypical antipsychotic drugs, most, such as olanzapine, also are about equi-potent at blocking 5HT2C receptors, which could contribute to their propensity to cause weight gain. In contrast, compounds that activate 5HT2C receptors are thought to enhance satiety, decrease meal size, and decrease caloric intake by stimulating pro-opiomelanocortin secretion from arcuate nucleus neurons (and subsequent activation of melanocortin systems) (Heisler et al., 2003; Hewitt et al., 2002). Based on these and similar findings, there are drug discovery efforts toward a 5HT2C-specific agonist as a weight-loss medication (Halford et al., 2011), but, for now, only a 5HT2C-preferring agonist (lorcaserin, Belviq) is available (Thomsen et al., 2008).
Preliminary off-target binding studies with (+)- and (−)-p-Cl-PAT have been undertaken and initial results suggest that, as for (−)-trans-PAT (Booth et al., 2009), these compounds have comparatively low (Ki > 0.5 μM) or very low (Ki > 1 μM) affinity for adenosine, adrenergic, benzodiazepine, cholinergic, dopamine, GABAA, histamine H2, H3, H4, NMDA/PCP, opiate, and serotonin 5-HT1A, 5-HT1B, receptors (PDSP, 2012). Like (−)-trans-PAT, however, previous studies with racemic p-Cl-PAT indicate high affinity at histamine H1 GPCRs (Bucholtz et al., 2009)—this is important because antagonist activity at the H1 GPCR correlates with atypical antipsychotic-induced weight gain (Kroeze et al., 2003). Nevertheless, (−)-trans-PAT negatively modulates food intake in the mouse model of compulsive or binge eating (Robertson and Rowland, 2005; Rowland et al., 2008; Rowland, 2012), and it was predicted that p-Cl-PAT also would decrease consumption of highly palatable “treats” based on the ability of p-Cl-PAT to activate 5HT2C receptors in vitro, similar to (−)-trans-PAT (Booth et al., 2009). In the current studies, mice ate ~40% of their daily caloric intake during a 30-minute session, and both enantiomers of p-Cl-PAT decreased consumption. Consistent with the in vitro pharmacology, and, similar to the HTR studies, the (+)-p-Cl-PAT enantiomer was more potent and effective than the (−)-isomer, and these effects occurred at doses similar to those that produced an “antipsychotic” effect. We propose that weight gain would not be a problem during chronic administration of (+)-p-Cl-PAT as we have previously demonstrated that this acute effect on compulsive eating generalizes from acute to chronic protocols with (−)-trans-PAT (Rowland et al., 2008).
In summary, the data presented here suggest that especially the (+)-enantiomer of p-Cl-PAT shows promise for development as an orally effective antipsychotic medication that would not have liability for 5HT2-mediated weight gain or cardiovascular toxicities, and, likely would not demonstrate extrapyramidal motor side effects. Such an antipsychotic drug may help to overcome patient compliance issues commonly associated with adverse weight gain and motor side effects commonly observed with currently available antipsychotic drugs.
Highlights.
Two 5HT2C agonists with 2A antagonist/inverse agonist activity were characterized
These 5HT2 modulators demonstrated preclinical antipsychotic activity
These novel compounds decreased consumption in a model of “binge-eating”
Both compounds were behaviorally active when administered orally
Acknowledgments
This research was supported by the National Institutes of Health grants R01 DA023928, DA030989, MH081193. The NIH did not have a role in the study design, in the collection, analysis, and interpretation of the data, in the writing of the manuscript, or in the decision to submit the paper for publication.
Footnotes
Financial Disclosures
The authors declare no conflicts of interest or financial disclosures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balt SL, Galloway GP, Baggott MJ, Schwartz Z, Mendelson J. Mechanisms and genetics of antipsychotic-associated weight gain. Clin Pharmacol Ther. 2011;90:179–183. doi: 10.1038/clpt.2011.97. [DOI] [PubMed] [Google Scholar]
- Booth RG, Fang L, Huang Y, Wilczynski A, Sivendran S. (1R, 3S)-(−)-trans-PAT: a novel full-efficacy serotonin 5-HT2C receptor agonist with 5-HT2A and 5-HT2B receptor inverse agonist/antagonist activity. Eur J Pharmacol. 2009;615:1–9. doi: 10.1016/j.ejphar.2009.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth RG, Rowland N, Gingrich JA. A Novel Serotonin 5-HT2C agonist with 5-HT2A/5-HT2B inverse agonist activity demonstrates antipsychotic efficacy without weigh-gain liability. Drugs Future. 2008;33 (Suppl A):68–69. [Google Scholar]
- Bucholtz EC, Brown RL, Tropsha A, Booth RG, Wyrick SD. Synthesis, evaluation, and comparative molecular field analysis of 1-phenyl-3-amino-1,2,3,4-tetrahydronaphthalenes as ligands for histamine H(1) receptors. J Med Chem. 1999;42:3041–3054. doi: 10.1021/jm980428x. [DOI] [PubMed] [Google Scholar]
- Canal CE, Cordova-Sintjago TC, Villa NY, Fang LJ, Booth RG. Drug discovery targeting human 5-HT(2C) receptors: residues S3.36 and Y7.43 impact ligand-binding pocket structure via hydrogen bond formation. Eur J Pharmacol. 2011;673:1–12. doi: 10.1016/j.ejphar.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canal CE, Morgan D. Head-twitch response in rodents induced by the hallucinogen 2,5-dimethoxy-4-iodoamphetamine: a comprehensive history, a re-evaluation of mechanisms, and its utility as a model. Drug Test Anal. 2012;4:556–576. doi: 10.1002/dta.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson A, Lindquist M. Effect of chlorpromazine and haloperidol on formation of 3-methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol Toxicol. 1963;20:140–144. doi: 10.1111/j.1600-0773.1963.tb01730.x. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Coccurello R, Moles A. Potential mechanisms of atypical antipsychotic-induced metabolic derangement: clues for understanding obesity and novel drug design. Pharmacol Ther. 2010;127:210–51. doi: 10.1016/j.pharmthera.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Cordova-Sintjago T, Sakhuja R, Kondabolu K, Canal CE, Booth RG. Molecular determinants for ligand binding at serotonin 5-HT2A and 5-HT2C GPCRs: Experimental affinity results analyzed by molecular modeling and ligand docking studies. Int J Quant Chem. 2012;112:3807–3814. doi: 10.1002/qua.24237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordova-Sintjago T, Villa N, Canal CE, Booth RG. Human serotonin 5-HT2C G protein-coupled receptor homology model from the β2 adrenoceptor structure: Ligand docking and mutagenesis studies. Int J Quant Chem. 2011;112:140–149. doi: 10.1002/qua.23231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correll CU, Lencz T, Malhotra AK. Antipsychotic drugs and obesity. Trends Mol Med. 2011;17:97–107. doi: 10.1016/j.molmed.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giovanni G, Di Matteo V, Di Mascio M, Esposito E. Preferential modulation of mesolimbic vs. nigrostriatal dopaminergic function by serotonin(2C/2B) receptor agonists: a combined in vivo electrophysiological and microdialysis study. Synapse. 2000;35:53–61. doi: 10.1002/(SICI)1098-2396(200001)35:1<53::AID-SYN7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Elangbam CS. Drug-induced valvulopathy: an update. Toxicol Pathol. 2010;38:837–848. doi: 10.1177/0192623310378027. [DOI] [PubMed] [Google Scholar]
- González-Maeso J, Ang RL, Yuen T, Chan P, Weisstaub NV, Lopez-Gimenez JF, Zhou M, Okawa Y, Callado LF, Milligan G, Gingrich JA, Filizola M, Meana JJ, Sealfon SC. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature. 2008;452:93–99. doi: 10.1038/nature06612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford JCG, Boyland EJ, Lawton CL, Blundell JE, Harrold JA. Serotonergic anti-obesity agents. Past experience and future prospects. Drugs. 2011;71:2247–2255. doi: 10.2165/11596680-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Hayashi A, Suzuki M, Sasamata M, Miyata K. Agonist diversity in 5-HT(2C) receptor-mediated weight control in rats. Psychopharmacology. 2005;178:241–249. doi: 10.1007/s00213-004-2019-z. [DOI] [PubMed] [Google Scholar]
- Heisler LK, Cowley MA, Kishi T, Tecott LH, Fan W, Low MJ, Smart JL, Rubinstein M, Tatro JB, Zigman JM, Cone RD, Elmquist JK. Central serotonin and melanocortin pathways regulating energy homeostasis. Ann N Y Acad Sci. 2003;994:169–174. doi: 10.1111/j.1749-6632.2003.tb03177.x. [DOI] [PubMed] [Google Scholar]
- Herrick-Davis K, Grinde E, Teitler M. Inverse agonist activity of atypical antipsychotic drugs at human 5-hydroxytryptamine2C receptors. J Pharmacol Exp Ther. 2000;295:226–232. [PubMed] [Google Scholar]
- Hewitt KN, Lee MD, Dourish CT, Clifton PG. Serotonin 2C receptor agonists and the behavioural satiety sequence in mice. Pharmacol Biochem Beh. 2002;71:691–700. doi: 10.1016/s0091-3057(01)00709-2. [DOI] [PubMed] [Google Scholar]
- Kirk SL, Glazebrook J, Grayson B, Neill JC, Reynolds GP. Olanzapine-induced weight gain in the rat: role of 5-HT2C and histamine H1 receptors. Psychopharmacology. 2009;207:119–25. doi: 10.1007/s00213-009-1639-8. [DOI] [PubMed] [Google Scholar]
- Kroeze WK, Hufeisen SJ, Popadak BA, Renock SM, Steinberg S, Ernsberger P, Jayathilake K, Meltzer HY, Roth BL. H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology. 2003;28:519–526. doi: 10.1038/sj.npp.1300027. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RS, Davis SM, Davis CE, Lebowitz BD, Severe J, Hsiao JK Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- Maayan L, Correll CU. Management of antipsychotic-related weight gain. Expert Rev Neurother. 2010;10:1175–1200. doi: 10.1586/ern.10.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquis LK, Sabb AL, Logue SF, Brennan JA, Piesla MJ, Comery TA, Grauer SM, Ashby CR, Jr, Nguyen HQ, Dawson LA, Barrett JE, Stack G, Meltzer HY, Harrison BL, Rosenzweig-Lipson S. WAY-163909 [(7bR,10aR)-1,2,3,4,8,9,10,10a-Octahydro-7bH-cyclopenta-[b][1,4]diazepino[6,7,1hi]indole]: a novel 5-hydroxytryptamine 2C receptor-selective agonist with preclinical antipsychotic–like activity. J Pharmacol Exp Ther. 2007;320:486–496. doi: 10.1124/jpet.106.106989. [DOI] [PubMed] [Google Scholar]
- McCloughen A, Foster K. Weight gain associated with taking psychotropic medication: an integrated review. Int J Ment Health Nurs. 2011;20:202–222. doi: 10.1111/j.1447-0349.2010.00721.x. [DOI] [PubMed] [Google Scholar]
- Meltzer HY. Pre-clinical pharmacology of atypical antipsychotic drugs: a selective review. Br J Psychiatry. 1996;168:23–31. [PubMed] [Google Scholar]
- Meltzer HY, Li Z, Kaneda Y, Ichikawa J. Serotonin receptors: their key role in drugs to treat schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:1159–1172. doi: 10.1016/j.pnpbp.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Meltzer HY, Massey BW. The role of serotonin receptors in the action of atypical antipsychotic drugs. Curr Opin Pharmacol. 2011;11:59–67. doi: 10.1016/j.coph.2011.02.007. [DOI] [PubMed] [Google Scholar]
- Morgan D, Canal CE, Kondabolu K, Sakhuja R, Robertson K, Rowland NE, Booth RG. A novel serotonin-2 (5-HT2) modulator as a candidate drug to treat impulsive behavioral disorders and psychoses without weight gain as a side effect. Neuropsychopharmacology. 2012;38:S104–105. [Google Scholar]
- Motulsky HJ, Christopoulos A. A practical guide to curve-fitting. GraphPad Software, Inc; San Diego, CA: 2003. Fitting models to biological data using linear and nonlinear regression. [Google Scholar]
- National Task Force on the Prevention and Treatment of Obesity. Overweight, obesity, and health risk. Arch Intern Med. 2000;160:898–904. doi: 10.1001/archinte.160.7.898. [DOI] [PubMed] [Google Scholar]
- PDSP; Psychoactive Drug Screening Program. B.L Roth, Director. NIMH Contract NO2MH80002. University of North Carolina; Chapel Hill, NC: 2012. http://pdsp.med.unc.edu/ [Google Scholar]
- Rauser L, Savage JE, Meltzer HY, Roth BL. Inverse agonist actions of typical and atypical antipsychotic drugs at the human 5-hydroxytryptamine(2C) receptor. J Pharmacol Exp Ther. 2001;299:83–9. [PubMed] [Google Scholar]
- Robertson KL, Rowland NE. Effect of two types of environmental enrichment for singly housed mice on food intake and weight gain. Lab Anim (NY) 2005;34:29–32. doi: 10.1038/laban1005-29. [DOI] [PubMed] [Google Scholar]
- Roerig JL, Steffen KJ, Mitchell JE. Atypical antipsychotic-induced weight gain: insights into mechanisms of action. CNS Drugs. 2011;25:1035–1059. doi: 10.2165/11596300-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Roth BL. Drugs and valvular heart disease. N Engl J Med. 2007;356:6–9. doi: 10.1056/NEJMp068265. [DOI] [PubMed] [Google Scholar]
- Rowland NE. Animal models of overeating. Methods Mol Biol. 2012;829:367–375. doi: 10.1007/978-1-61779-458-2_24. [DOI] [PubMed] [Google Scholar]
- Rowland NE, Crump EM, Nguyen N, Robertson K, Sun Z, Booth RG. Effect of (−)-trans-PAT, a novel 5-HT2C receptor agonist, on intake of palatable food in mice. Pharmacol Biochem Behav. 2008;91:176–180. doi: 10.1016/j.pbb.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman P. Atypical antipsychotics: mechanism of action. Can J Psychiatry. 2002;47:27–38. [PubMed] [Google Scholar]
- Tandon R, Fleischhacker W. Comparative efficacy of antipsychotics in the treatment of schizophrenia: a critical assessment. Schizophr Res. 2005;79:145–155. doi: 10.1016/j.schres.2005.07.025. [DOI] [PubMed] [Google Scholar]
- Thomsen WJ, Grottick AJ, Menzaghi F, Reyes-Saldana H, Espitia S, Yuskin D, Whelan K, Martin M, Morgan M, Chen W, Al-Shamma H, Smith B, Chalmers D, Behan D. Lorcaserin, a novel selective human 5-hydroxytryptamine2C agonist: in vitro and in vivo pharmacological characterization. J Pharmacol Exp Ther. 2008;325:577–587. doi: 10.1124/jpet.107.133348. [DOI] [PubMed] [Google Scholar]
- Vincek AS, Booth RG. New approach to 4-phenyl-β-aminotetralin from 4-(3-halophenyl)tetralen-2-ol Phenylacetate. Tetrahedron Let. 2009;50:5107–5109. doi: 10.1016/j.tetlet.2009.06.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiden PJ, Mackell JA, McDonnell DD. Obesity as a risk factor for antipsychotic noncompliance. Schizophrenia Res. 2004;66:51–57. doi: 10.1016/s0920-9964(02)00498-x. [DOI] [PubMed] [Google Scholar]