

Abstract
Localized surface plasmon resonance (LSPR) spectroscopy has been widely used for label-free, highly-sensitive measurements of interactions at a surface. LSPR imaging (LSPRi) has the full advantages of LSPR, but enables high-throughput, multiplexed measurements by simultaneously probing multiple individually addressable sensors on a single sample surface. Each spatially distinct sensor can be tailored to provide data regarding different surface functionalities or reaction environments. Previously, LSPRi has focused on single particle sensing where the size scale is very small. Here, we create defined macroscale arrays of nanoparticles that are compatible with common patterning methods such as dip-pen nanolithography and multi-channel microfluidic delivery devices. With this new LSPR sensing format, we report the first demonstration of multiplexed LSPR imaging and show that the increased throughput of our instrument enables the collection of a complete Langmuir binding curve on a single sensor surface. In addition, the multiplexed LSPR sensor is highly selective, as demonstrated by the hybridization of single-stranded DNA to complementary sequences immobilized on the sensor surface. The LSPR arrays described in this work exhibit uniform sensitivity and tailorable optical properties, making them an ideal platform for high-throughput, label-free analysis of a variety of molecular binding interactions.
INTRODUCTION
Sensor arrays comprise multiple individual sensors operating in parallel in a defined geographic space, and allow simultaneous detection of multiple signals because each sensor component can be individually tailored to achieve a unique specificity or response to the environment. Thus, sensor arrays enhance the analysis of complex systems compared to single sensors by creating a more complete picture of the environment. Sensor arrays are frequently used for the detection of pressure variations,1 gasses,2 vapors,3, 4 biomolecule interactions,5 redox reactions,6 and ions.7 The application of sensor arrays to new technologies and new problems will drastically enhance our ability to study and monitor complex systems.
Due to their unique complexity, biological systems are an ideal candidate for study with sensor arrays. These arrays can utilize optically-active components such as fluorophores, quantum dots, and plasmonic materials, which allow for easy, optical readout. Among these, surface plasmon resonance (SPR) has emerged as an ideal candidate for array-based sensing because it negates the need for analyte labeling. Plasmon resonance occurs when incident light impinges upon nanoscale metallic films, causing the resonant oscillation of conduction electrons and wavelength-selective absorption. This resonant absorption is dependent upon the refractive index at the metal-dielectric interface, so that biomolecule binding to the metal surface results in shifts in the absorption spectrum of the film. Commercial SPR sensors can incorporate 400 unique receptors on a single chip.8 In academic studies, SPR arrays are being developed to analyze RNA,9 viruses,10 proteins,11 carbohydrates,12 and small molecules.13
Localized SPR (LSPR) is a detection technique closely related to SPR that capitalizes on the unique properties of nanoscale metal particles. In LSPR, the planar noble metal film of the SPR sensor is exchanged for arrays of noble metal nanoparticles. Due to the discrete nature of these nanoparticle arrays, light impinging on the sensor surface results not in surface propagating plasmons, but rather in localized surface plasmons with short decay lengths on the order of 5-10 nm.14, 15 Molecular binding events on the surface that cause refractive index changes within the plasmon sensing volume result in spectral shifts in the plasmon resonance. The localized nature of these plasmons leads to reduced bulk refractive index sensitivity and increased sensitivity to molecules binding to the LSPR sensor surface, thus drastically reducing the impact of background signals that result from changes in the surrounding medium.16 As a result, LSPR instrumentation is unencumbered by the bulky temperature-controlled housing used in SPR instruments, and can easily be made portable.17 These advantages make LSPR an attractive analytical technique, especially for field applications involving small molecule detection.16, 18, 19
Despite its advantages, few attempts to advance LSPR sensing by developing array-type sensors have been made.20 This remains a major challenge to the commercial implementation of LSPR sensing. LSPR sensing is typically performed with low throughput, where readout is accomplished using either single spot readout from nanoparticle arrays, or individual, optically isolated nanoparticles using dark field imaging.21, 22 Attempts to increase the throughput from LSPR sensors have involved imaging nanoparticle arrays but without collecting spectra, thus decreasing the analytical information obtained.23 We report the first demonstration of full spectral imaging of macroscale LSPR sensor arrays based on discrete LSPR sensors composed of surface confined nanoparticle arrays. The LSPR sensor arrays are illuminated with white light filtered through a liquid crystal tunable filter (LCTF), which allows full access to either the visible or near-IR spectral range. As the LCTF scans through a wavelength sequence, a camera takes a series of images which are later analyzed for intensity information. These intensity maps are then correlated back to illumination wavelength in order to construct a spectrum for each nanoparticle array region in the image. We show that this technique allows for simultaneous refractive index detection from multiple LSPR sensors, with a spatial resolution that is theoretically only diffraction limited. Imaging an LSPR sensor array functionalized with biotin and exposed to varying concentrations of anti-biotin enabled the determination of the biotin/anti-biotin binding constant in a single exposure experiment. In addition, an LSPR sensor array patterned with single stranded (ss) DNA was able to specifically detect the binding of complementary ssDNA while rejecting non-specific binding.
EXPERIMENTAL
Instrument Design
The sample chamber of a GWC SPRimager II instrument was modified to accommodate a liquid crystal tunable filter (LCTF, CRi, VariSpec) and a flow cell. The LCTF is tunable with a 0.7 nm resolution and can accommodate a wavelength range of either 650 – 1100 nm or 480 – 720 nm. The light polarization was set to s-polarization to maximize intensity. While selecting a polarization is unnecessary, the polarizer is a permanent part of the GWC instrument and is kept in place so that the instrument could be switched between LSPR and SPR measurement modes easily. The camera (with the GWC installed band pass filter removed) was positioned to align with the illumination beam path and a DFG/SV1 frame grabber (The Imaging Source) was installed on the computer to interface the camera with LabVIEW software. A custom written LabVIEW program automatically synchronized the LCTF wavelength selection and image acquisition to speed data collection. Three mirrors redirected the light through the sample and LCTF into the camera. A schematic of the instrument design is shown in Figure 1.
Figure 1.
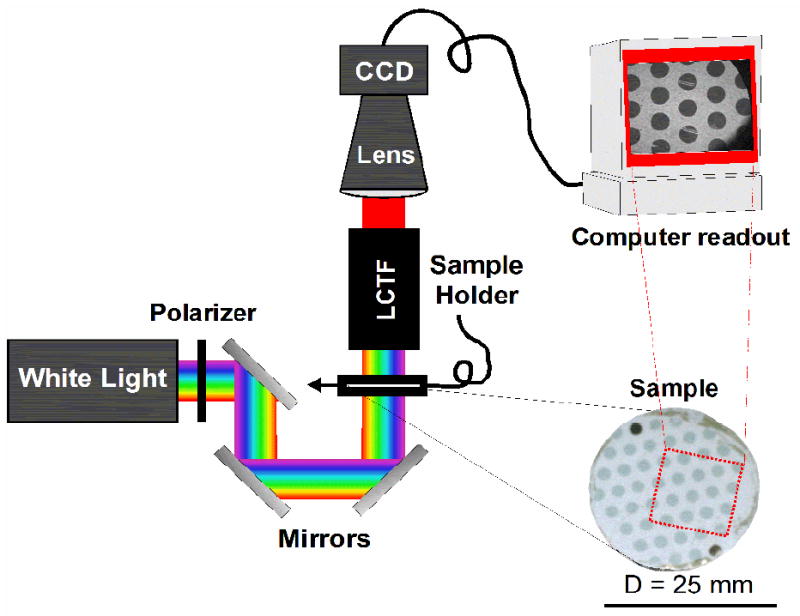
Schematic of LSPR imaging instrument. Collimated white light from the light source is sent through a polarizer and then directed via three mirrors through the sample into a liquid crystal tunable filter (LCTF). A single selected wavelength is sent through a lens to the CCD camera, which collects a single-wavelength black and white image. The computer cycles the LCTF through all the wavelengths of interest and triggers the CCD to take an image at each wavelength.
Intensities of given regions of interest (ROIs) were extracted from images with ImageJ and further data analysis was performed in Origin. The extinction at each wavelength was calculated using Equation 1, where IPA is the intensity of the particle array, IREF is the intensity of bare glass, and IDARK is the background light intensity. A single wavelength scan was used to collect IPA and IREF from substrates
| Equation 1 |
comprising regions of both particle arrays and bare glass. Background intensities (IDARK) were obtained in a separate wavelength scan with the beam blocked. Each wavelength scan only took a few minutes, with the exact time depending on scanning conditions.
Substrate Preparation
Glass cover slips (25 mm diameter, #2 thickness, VWR) were cleaned with fresh piranha for 10 minutes before rinsing and drying. Nanoparticle arrays were created using hole-mask colloidal lithography (HCL). Full details of the HCL technique have been published so only a brief description is provided here.24 A sacrificial layer of poly(methyl methacrylate) (PMMA) (MicroChem) was spin coated on the glass, soft baked, and exposed to oxygen plasma for 30 seconds. The sample was exposed to a 0.2% solution of poly(dialyldimethyl-ammonium chloride) (Sigma), rinsed, and dried and then exposed to a 0.2% solution of 190 nm diameter polystyrene beads (Thermo Fisher), rinsed, and dried. Physical vapor deposition of a 10 nm Au film was followed by removal of the polystyrene beads via tape-stripping, creating an etch mask comprising holes with the diameter and former positions of the beads. Reactive ion etching with O2 plasma removed the PMMA directly beneath the holes in the etch mask and created a gold-coated PMMA mask for physical vapor deposition of a 2 nm titanium adhesion layer and a gold or silver plasmonic layer (of varied thickness). The mask was then removed by sonication in acetone followed by water rinsing and drying, leaving behind an array of nanodiscs with a 190 nm diameter and a height that corresponds to the plasmonic layer that was deposited. A schematic of this process and an SEM image of the resulting nanodisc arrays is shown in the supplementary information (Figure S1). The figure also shows 1 in2 samples which have been homogeneously coated with nanodisc arrays.
For multiplexing, substrates were patterned with individually addressable regions of nanodisc arrays by using a shadow mask during the deposition of the plasmonic layer to create many isolated, circular regions of nanodisc arrays with the same metal thickness (Figure 2). These isolated regions could then be individually functionalized by hand-spotting solutions of reagents on each region. In another format, a movable shutter was used to expose different regions of the substrate to the plasmonic metal deposition for different periods of time, allowing deposition of different thicknesses of plasmonic metal in each region.
Figure 2.
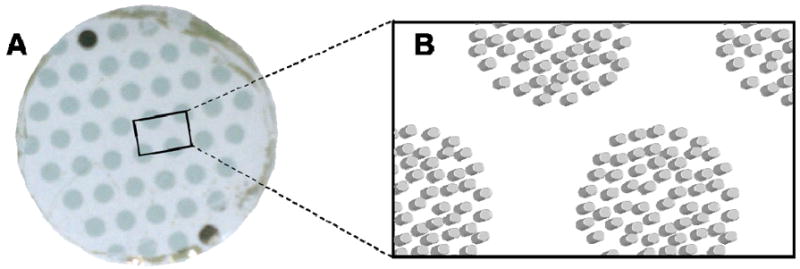
(A) Camera image of a 25 mm glass slide patterned with discrete circular regions of randomly ordered cylindrical nanoparticle arrays using a shadow mask. (B) Schematic showing randomly ordered nano-cylinder arrays within each circular region on the glass slide (not to scale).
Glycerol Solutions
Aqueous solutions of glycerol (0, 10, 20, 30, 40, 50, and 60 %) were prepared with milliQ water (18 MΩ) and used to measure the refractive index sensitivity of HCL samples. The refractive index range of these samples was approximately 1.33 – 1.41 refractive index units (RIU) as determined by Hoyt.25
Biotin/Anti-Biotin Binding
A self-assembled monolayer (SAM) was formed from a 13.6:1 mixture of hydroxyl:biotin terminated oligo(ethylene glycol) thiols (Sensopath), at a total concentration 2 mM in ethanol. HCL substrates comprising 38 nm Au nanodiscs were incubated in this solution for 16-24 hours. The substrate was rinsed thoroughly with water and ethanol then dried with nitrogen. The samples were exposed to solutions of varying concentrations of Anti-Biotin (Abcam) in phosphate buffered saline (PBS) (Sigma) for 1 - 2 hours. For each solution of a different Anti-Biotin concentration, a 0.5 μL drop was spotted on a circular region of the sensor by hand using a pipette.
DNA Immobilization and Hybridization
A SAM of mercaptoundecanoic acid (MUA, Sigma Aldrich) was formed from incubation in a 2 mM ethanolic solution for 2 days. The sample was removed from the MUA solution, rinsed with ethanol and water and sonicated in 10% acetic acid for 8 minutes, rinsed with water and dried with nitrogen. Half of the regions of the sample chip were then spotted with a mixture of 50 mM N-(3-dimethylamineoporpyl)-N’-ethylcarbodiimide hydrochloride (Sigma) and 1 mM aminated probe DNA in 0.1 M 2-(N-morpholino)ethanesulfonic acid (MES) buffer solution, pH ~ 4.5. The spotted sample was allowed to incubate for 2 hours in a hydration chamber. The probe sequence for immobilization ([AminoC6]ATCTAACTGCTGCGCCGCCGGGAAAATACTGTACGGTTAAG) and its perfect complement were ordered from Eurofins, MWG Operon. The substrate was rinsed thoroughly with water and ethanol then dried with nitrogen. The sample was then exposed to 1 μM complementary DNA in Tris-EDTA buffer (10 mM Tris Base, 1 M NaCl, and 1 mM ethylenediaminetetraacetic acid pH adjusted to 7.6) for 1.5 hr. The substrate was rinsed thoroughly with water and ethanol then dried with nitrogen.
RESULTS
Monodisperse Sensor Arrays
A sample with dimensions of ~ 2 cm by 1.5 cm was covered with a uniform array of gold nanodiscs that were 190 nm in diameter and 20 nm tall. A shadow mask was used to create 9 individually addressable circular regions of the nanodisc array with diameters of ~ 2 mm. An LSPR image of this surface at a wavelength of 850 nm is shown in Figure 3A. The LSPR spectrum of each region was obtained and fit with a Lorentzian model to determine the peak position (Figure 3B); all regions displayed a remarkably reproducible maximum resonance at 864.6 ± 2.7 nm. The shift in this maximum was measured as a function of the bulk refractive index using aqueous glycerol solutions (Figure 3C). All regions also showed a very similar refractive index sensitivity of 263 ± 15 nm/RIU.
Figure 3.
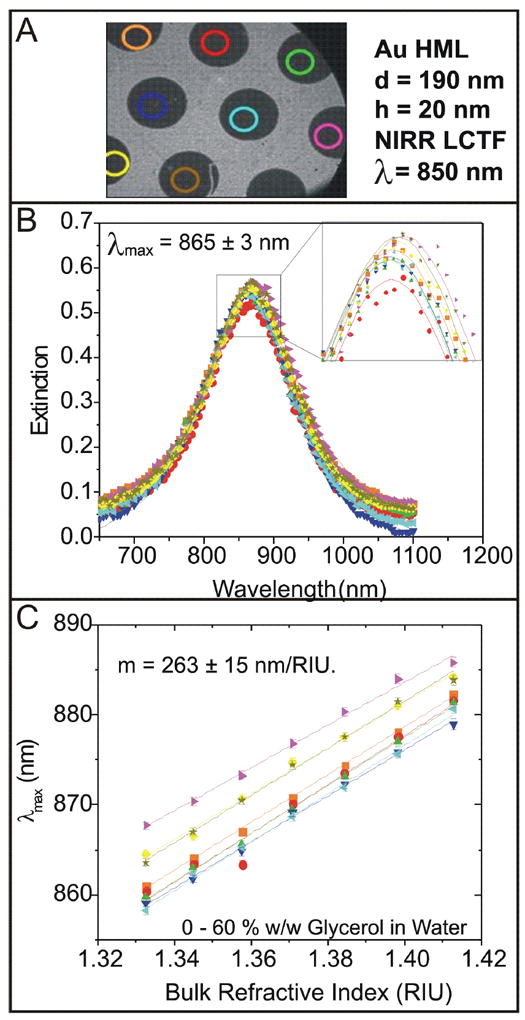
Uniform sensitivity. (A) A defined region of an LSPR sensor array was imaged at 850 nm. Gold nanodisc arrays appear as dark regions while glass appears as a light gray background. Regions of the array that were analyzed are highlighted with colored circles. (B) LSPR spectra from selected regions were analyzed by correlating image intensity with wavelength. (C) The sensitivity of each individual sensor spot was determined by exposing the sensor surface to varying concentrations of an aqueous glycerol solution. The peak position as a function of bulk dielectric constant is shown. Lines are fits to the data where the slope is the sensitivity or m-value.
Simultaneous Evaluation of Multiple Plasmonic Substrates
Arrays of silver nanoparticles with the same footprints but different heights were examined. Figure 4 shows the spectra of 5 unique plasmonic regions, each displaying distinct optical properties, patterned on a single substrate. These substrates comprise arrays of silver nanodiscs with a diameter of 80 nm and heights that vary from 10 to 30 nm. Because the resonance position of these Ag nanodiscs lies in the visible wavelength region, variations in resonance position can be discerned by eye in the color photograph at the bottom of Figure 4. The black and white series of images at the bottom show a progression of LSPR images taken at different points during a wavelength scan. The resonance of each area is the wavelength at which it appears lightest in the image series. As the height of the silver nanodisc increases, the aspect ratio decreases, giving rise to an increase in the resonance intensity and a blue shift, as expected.26-29
Figure 4.
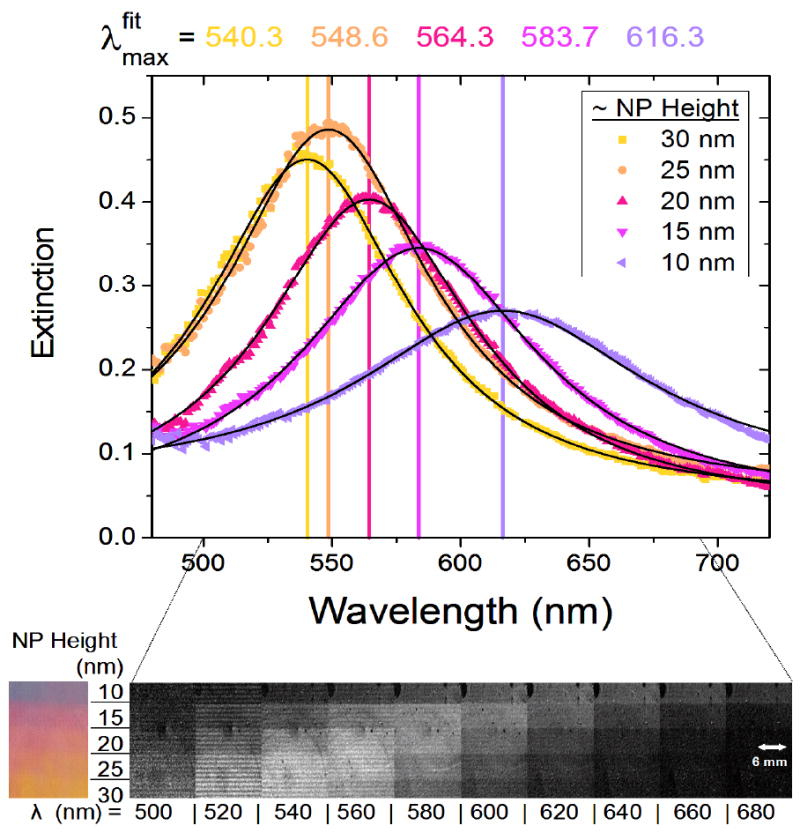
Tunable optical properties. LSPR spectra from 5 different regions of a silver nanoparticle array were collected by measuring the grayscale intensity of single-wavelength images collected from 450 to 750 nm. Single-wavelength intensity measurements are shown as data points, and the fits to the data are shown as black lines. The fit results for the peak position are listed above the spectra. Underneath the spectra is a color picture of the sample, and on the right are a series of black and white images of the same sample taken at selected wavelengths. High extinction is represented as white and low extinction is black.
A similar study was performed to screen gold nanodisc arrays (Figure 5A) with diameters of 190 nm and heights varying from 20 to 100 nm in order to determine the most effective substrate for biosensing. The large diameter gold nanodisc arrays all have resonances in the near infrared; similar to the silver nanodisc arrays discussed above, the peak positions shift to the blue as the height increases (Figure 5B). It has been shown that plasmonic sensors with redder resonances display greater sensitivity than those with resonances in the blue region of the spectrum.26, 30 The data we observed agrees with this expectation; all substrates exhibit refractive index sensitivities above 200 nm/RIU (Figure 5C). The 40 nm high substrate was chosen for biosensing applications based on a compromise between high sensitivity, high extinction intensity, and the ability to accommodate the substantial red shifting that will occur during these applications (within the range of the LCTF).
Figure 5.
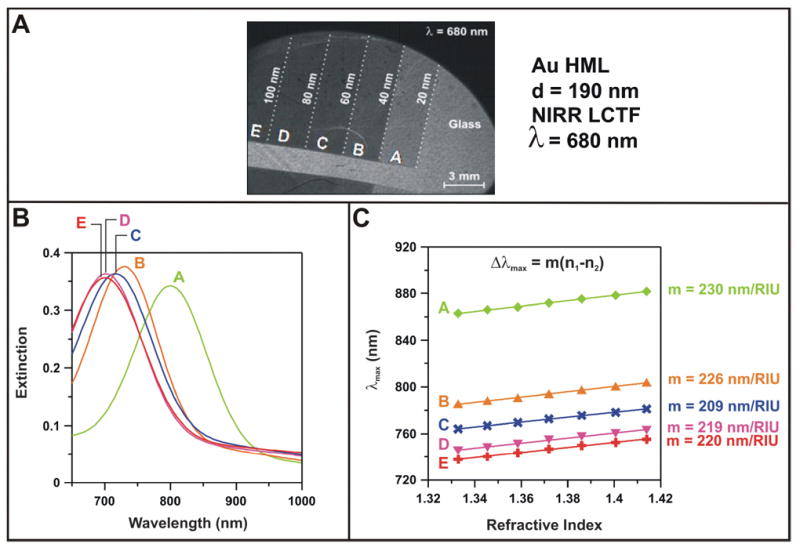
(A) LSPR image of a sample comprising 5 regions of different gold nanodisc arrays taken at 680 nm. The height of each area’s nanodiscs are indicated on the image. (B) LSPR spectra for each region taken in water. An LSPR spectrum was collected while the substrate was exposed to various concentrations of an aqueous glycerol solution. (C) The peak position as a function of bulk dielectric constant is shown. Fits to the data are shown as lines, and the slope of the line is the refractive index sensitivity, or m-value.
Single Chip Binding Curve
A binding curve of anti-biotin with biotin was obtained by simultaneously exposing different regions of the single biotin-functionalized sample depicted in Figure 6A and 6B to varying concentrations of anti-biotin. Anti-biotin concentrations ranged from 5 × 10-10 to 5 × 10-6 M; control spots were left bare during incubation. Due to the large imaging area of this instrument it was possible to obtain 15 data points from a single experiment. Two LSPRi scans were taken, one before and one after incubation with anti-biotin; each required about 1 minute to complete (~500 ms/image). The shift in the resonance peak position for each region as a result of incubation was measured and plotted against the anti-biotin incubation concentration (Figure 6C). The data was fit to a Langmuir model to give a binding constant of 2.4 ± 5.8 × 10-6 M, which is reasonable for an unoptimized system where biotin is tethered to the surface.31
Figure 6.
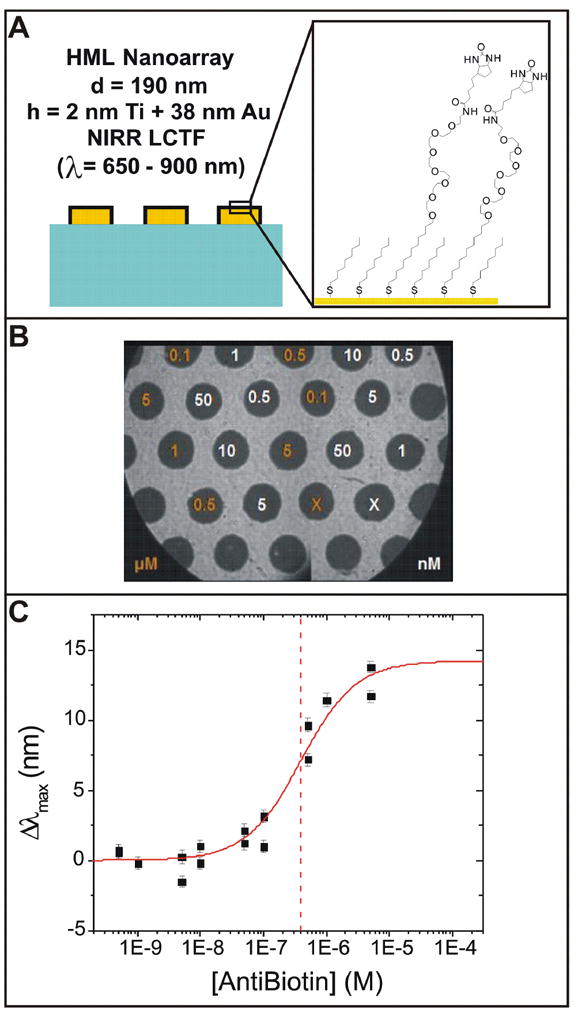
Binding curve of anti-biotin to surface bound biotin. (A) A schematic of the surface functionalization. (B) An LSPR image of the surface with the concentrations of anti-biotin solutions spotted on each region. (C) The shift in the peak position as a function of anti-biotin concentration. The points are data with error bars from the Lorentzian fit to the LSPR curves, and the red line is a Langmuir fit to the data. The vertical dashed line indicates the value of the dissociation constant.
Simultaneous Evaluation of Multiple Reactions on One Chip
A substrate containing individually addressable regions of gold nanodisc arrays 190 nm in diameter and ~40 nm in height was functionalized with a monolayer of mercaptoundecanoic acid. Specific regions were then modified with amine terminated DNA via covalent attachment to the carboxyl terminated SAM. The resonance peak shift from tethering the DNA to the surface is shown in Figure 7 along with the responses after the entire substrate was exposed to a complementary strand of DNA. The shifts seen are in agreement with the literature regarding both the amount of DNA that was tethered to the surface as well as the ~ 50 % hybridization efficiency with complementary sequences.
Figure 7.
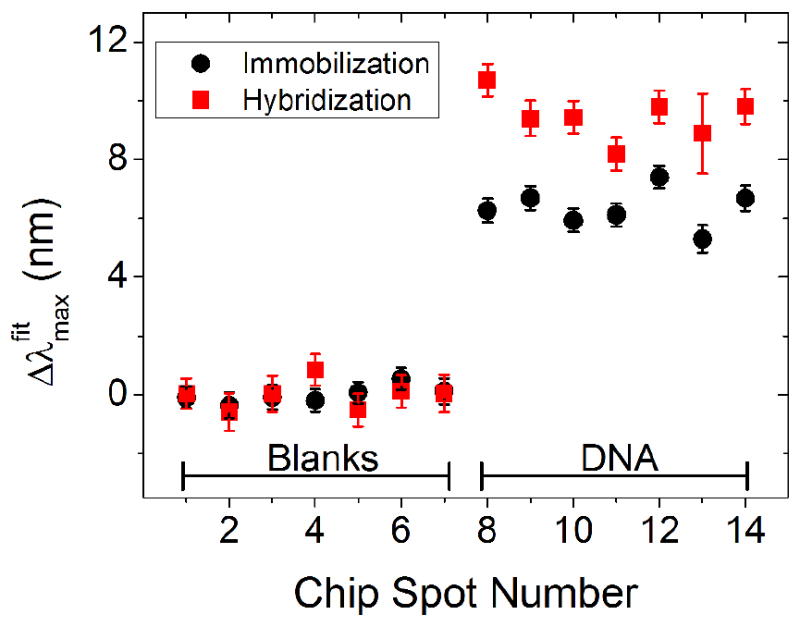
LSPR shift results from immobilization and hybridization of surface tethered DNA. All data was obtained from a single sample. The black circles are data from immobilization of DNA to chip spot numbers 8-14 giving rise to a ~ 7 nm shift. The red squares are the response when the entire chip was exposed to a solution of complementary DNA. Hybridization resulting in an additional ~ 3.5 nm shift (~50 % efficiency) is only observed on the spots where DNA was immobilized.
DISCUSSION
We have built an instrument capable of taking multiplexed, spatially-resolved LSPR measurements by using a CCD camera to monitor the transmittance of a ~ 2 cm diameter collimated light beam through an LSPR sample cell. We used the HCL method to fabricate a homogeneous array of nanodiscs across 6.45 cm2 substrates to create LSPR sensor surfaces that could take advantage of the large imaging area of our instrument. The performance of the combined system gave a uniform resonance peak position and sensitivity across the entire sample surface. This sensing uniformity demonstrates that the system can reliably be used for high-throughput heterogeneous analysis. We demonstrated the power of this system by screening plasmonic surfaces for sensor optimization as well as running common multiplexed bioassays.
In LSPR analysis, it is advantageous to engineer the plasmonic substrate to have a specific resonance. Given the diversity of nanostructured surfaces that are available, engineering an optimized sensor surface typically requires significant time to screen nanoparticles of different shapes, sizes, or materials. Here we demonstrated the ability to screen multiple plasmonic substrates simultaneously. In one example, the absorbance of silver nanodisc arrays was screened to evaluate how LSPR peak position changes with nanodisc height. This type of screen is useful when designing a resonance-enhanced LSPR assay, where the resonance of a chromophoric analyte overlaps with the resonance of a nanoparticle. It has been demonstrated that tuning the LSPR peak position to match the absorbance spectrum of an analyte can significantly enhance the observed signal for a binding reaction. 32-34 In a second example, the height of a gold, large diameter nanodisc array was optimized for biosensing. Though all of the arrays exhibited high (>200 nm/RIU) sensitivity, we determined that 40 nm high gold nanodiscs provided the greatest combination of sensitivity and resonance peak position to develop a high-throughput biosensing substrate.
Binding curve measurements are ubiquitously used throughout the biochemical and pharmaceutical industries to characterize binding interactions with proteins. This assay requires measuring the equilibrium binding level of an analyte at a variety of concentrations. The concentrations must span the range from where no binding is detectable up to where probe binding sites are completely saturated with analyte, and this range is frequently multiple orders of magnitude. Hence, it can be necessary to monitor binding at 10 to 20 different analyte concentrations. When measurements are performed in sequence, binding curve assays are time consuming and require large quantities of the analyte molecule. Running such assays in a parallel format where all concentrations are monitored simultaneously significantly decreases the time required to complete the assay, and spotting only 0.5 μL of solution per sensor region minimizes the analyte quantity needed. Though multiplexed formats have been used before, typically they employ analyte labeling; LSPRi creates a multiplexed format where there is no need to label the analyte. We present the first reported binding curve of anti-biotin to biotin obtained with a single multiplexed LSPR measurement.
The ability to measure a “single-chip” binding curve is highly advantageous, but the results presented here are simply proof-of-principle and greater levels of parallelization are easily realizable. Some factors that limited the number of simultaneous measurements our instrument can take are the substrate size, field of view, and ease of fluid delivery. For our demonstration, these parameters were chosen to be convenient given the existing materials and components available in the laboratory. By reconfiguring the instrument to have a larger field of view and using a larger glass substrate, a larger sensing area can be achieved. Also, by taking advantage of more advanced fluidic delivery like robotic spotters or massively parallelized microfluidics, the number of individually addressable regions within a given area can be increased.35 A commercial microspotter can pattern surfaces with 26 spots/mm2, theoretically enabling our 2 × 1.5 cm sensor to contain 7800 individually addressable regions. This would provide the ability to run 7800 experiments simultaneously, further increasing the benefits of an LSPR imaging instrument like the one described here. To achieve such multiplexing, all regions must be spatially resolvable; the resolution of the instrument presented here is limited by a combination of the camera resolution and the focus lens which are both trivial to change. None of these properties were fully optimized in the instrument presented here and further improvements in this technology can readily be achieved.
A second type of high throughput sensing involves detection of many analytes simultaneously from a single solution by immobilizing different probes on defined regions on a single surface. Here we show a simple proof-of-principle experiment where we observe DNA hybridizing to complementary sequences. The 50% hybridization efficiency observed is in agreement with the literature on hybridization of surface tethered DNA.36-39 The signal observed for immobilization indicates that DNA was tethered to the surface at a density of ~ 2 × 1012 molecules/cm2, which is in agreement with the literature for amine immobilized DNA.36 Though not demonstrated here, these studies could be expanded by immobilizing different strands of DNA at different places on the sensor substrate to create a single sample capable of detecting many distinct analytes. Such studies have been performed before using other detection techniques,36, 38, 40, 41 and the results presented here are sufficient to prove that the same can now be done with LSPRi. Being able to directly detect analytes without the need for labeling or employing a sandwich assay has significant benefits for disease diagnostics. For example, diagnostics for Alzheimer’s disease have been particularly difficult to develop. Recent research indicates that a panel of many different biomarkers show a distinct pattern of over and under expression in Alzheimer’s patients.42 A sensor that could detect the levels of all biomarkers in the panel at once could easily be created by immobilizing probes for each biomarker at a different place on an LSPRi sensor. Since there is no need to label the analytes, exposing the sample to blood and monitoring the LSPR shift would provide a quick test that would be the first to definitively diagnose Alzheimer’s.
CONCLUSIONS
In this paper we described an LSPR imaging instrument created through modification of a commercial SPRi instrument (GWC Technologies). Proof-of-principle studies were presented to demonstrate homogeneity of sensing across a large field of view (~ 1.1 cm2), simultaneous measurement of the wavelength-dependent resonance of many nanoparticle array substrates patterned on a single surface, and the ability to perform multiple biosensing experiments simultaneously. This type of multiplexed patterning and imaging of nanoparticle arrays will dramatically expand the way that LSPR sensors can be used. LSPR is already recognized as a highly sensitive, label-free technique that can be easily made portable because of the simple detection equipment needed. By moving to a multiplexed modality, these advantages can be exploited to create truly powerful sensors. For example, one can easily imagine methods by which to fabricate a single device to detect a wide variety of chemical warfare agents, to increase the screening efficiency of therapeutics in pharmaceutical development, and to improve the diagnoses of disease. The work described in this paper is a collection of studies demonstrating the feasibility of using label-free LSPR sensors to meet these aims in the near future.
Supplementary Material
Acknowledgments
This research was supported by the National Science Foundation (Grants EEC-0647560 and DMR-1121262), the National Institutes of Health (Grant 5R56DK078691–02), the Defense Advanced Research Projects Agency (Grants FA9550–08–1–0221/P00003 and N66001-11-1-4179) and a Ryan Fellowship to W.P.H.
Footnotes
Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Someya T, Sekitani T, Iba S, Kato Y, Kawaguchi H, Sakurai T. Proc Natl Acad Sci U S A. 2004;101(27):9966–9970. doi: 10.1073/pnas.0401918101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin X, Huang Y, Mason A, Zeng X. Anal Chem. 2009;81(2):595–603. doi: 10.1021/ac8018697. [DOI] [PubMed] [Google Scholar]
- 3.Lin HW, Jang M, Suslick KS. J Am Chem Soc. 2011;133(42):16786–16789. doi: 10.1021/ja207718t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin HW, Suslick KS. J Am Chem Soc. 2010;132(44):15519–15521. doi: 10.1021/ja107419t. [DOI] [PubMed] [Google Scholar]
- 5.Washburn AL, Luchansky MS, Bowman AL, Bailey RC. Anal Chem. 2010;82(1):69–72. doi: 10.1021/ac902451b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quinton D, Girard A, Kim LTT, Raimbault V, Griscom L, Razan F, Griveau S, Bedioui F. Lab on a Chip. 2011;11(7):1342–1350. doi: 10.1039/c0lc00585a. [DOI] [PubMed] [Google Scholar]
- 7.Wang Z, Palacios MA, Anzenbacher P., Jr Anal Chem. 2008;80(19):7451–9. doi: 10.1021/ac801165v. [DOI] [PubMed] [Google Scholar]
- 8.Piliarik M, Homola J. Ser Chem Sens Bio Sens. 2006;4:95–116. [Google Scholar]
- 9.Nelson BP, Liles MR, Frederick KB, Corn RM, Goodman RM. Environ Microbiol. 2002;4(11):735–43. doi: 10.1046/j.1462-2920.2002.00350.x. [DOI] [PubMed] [Google Scholar]
- 10.Yuk JS, Ha KS. Methods Mol Cell Biol. 2009;503:37–47. doi: 10.1007/978-1-60327-567-5_3. [DOI] [PubMed] [Google Scholar]
- 11.Nedelkov D. Anal Chem. 2007;79(15):5987–90. doi: 10.1021/ac070608r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith EA, Thomas WD, Kiessling LL, Corn RM. J Am Chem Soc. 2003;125(20):6140–8. doi: 10.1021/ja034165u. [DOI] [PubMed] [Google Scholar]
- 13.Kanoh N, Kyo M, Inamori K, Ando A, Asami A, Nakao A, Osada H. Anal Chem. 2006;78(7):2226–30. doi: 10.1021/ac051777j. [DOI] [PubMed] [Google Scholar]
- 14.Haes AJ, Zou SL, Schatz GC, Van Duyne RP. J Phys Chem B. 2004;108(1):109–116. [Google Scholar]
- 15.Haes AJ, Zou SL, Schatz GC, Van Duyne RP. J Phys Chem B. 2004;108(22):6961–6968. [Google Scholar]
- 16.Yonzon CR, Jeoungf E, Zou SL, Schatz GC, Mrksich M, Van Duyne RP. J Am Chem Soc. 2004;126(39):12669–12676. doi: 10.1021/ja047118q. [DOI] [PubMed] [Google Scholar]
- 17.Neuzil P, Reboud J. Anal Chem. 2008;80(15):6100–3. doi: 10.1021/ac800335q. [DOI] [PubMed] [Google Scholar]
- 18.Svedendahl M, Chen S, Dmitriev A, Kall M. Nano Lett. 2009;9(12):4428–33. doi: 10.1021/nl902721z. [DOI] [PubMed] [Google Scholar]
- 19.Hall WP, Modica J, Anker J, Lin Y, Mrksich M, Van Duynet RP. Nano Lett. 2011;11(3):1098–1105. doi: 10.1021/nl103994w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen KJ, Lu CJ. Talanta. 2010;81(4-5):1670–1675. doi: 10.1016/j.talanta.2010.03.023. [DOI] [PubMed] [Google Scholar]
- 21.Bingham JM, Willets KA, Shah NC, Andrews DQ, Van Duyne RP. J Phys Chem C. 2009;113(39):16839–16842. doi: 10.1021/jp907377h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang HY, Rong GX, Yan B, Yang LL, Reinhard BM. Nano Lett. 2011;11(2):498–504. doi: 10.1021/nl103315t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raphael MP, Christodoulides JA, Mulvaney SP, Miller MM, Long JP, Byers JM. Anal Chem. 2012;84(3):1367–1373. doi: 10.1021/ac2023266. [DOI] [PubMed] [Google Scholar]
- 24.Fredriksson H, Alaverdyan Y, Dmitriev A, Langhammer C, Sutherland DS, Zaech M, Kasemo B. Adv Mater. 2007;19(23):4297. [Google Scholar]
- 25.Hoyt LF. Journal of Industrial and Engineering Chemistry. 1934;26(3):329–332. [Google Scholar]
- 26.Hanarp P, Kall M, Sutherland DS. J Phys Chem B. 2003;107(24):5768–5772. [Google Scholar]
- 27.Jensen TR, Malinsky MD, Haynes CL, Van Duyne RP. J Phys Chem B. 2000;104(45):10549–10556. [Google Scholar]
- 28.Kelly KL, Coronado E, Zhao LL, Schatz GC. J Phys Chem B. 2003;107(3):668–677. [Google Scholar]
- 29.Kang SK, Kim Y, Hahn MS, Choi I, Lee J, Yi J. Curr Appl Phys. 2006;6(Supplement 1 (0)):e114–e120. [Google Scholar]
- 30.Willets KA, Van Duyne RP. Annu Rev Phys Chem. 2007;58:267–297. doi: 10.1146/annurev.physchem.58.032806.104607. [DOI] [PubMed] [Google Scholar]
- 31.Adamczyk M, Mattingly PG, Shreder K, Yu ZG. Bioconjugate Chem. 1999;10(6):1032–1037. doi: 10.1021/bc990057e. [DOI] [PubMed] [Google Scholar]
- 32.Haes AJ, Zou SL, Zhao J, Schatz GC, Van Duyne RP. J Am Chem Soc. 2006;128(33):10905–10914. doi: 10.1021/ja063575q. [DOI] [PubMed] [Google Scholar]
- 33.Zhao J, Das A, Schatz GC, Sligar SG, Van Duyne RP. J Phys Chem C. 2008;112(34):13084–13088. [Google Scholar]
- 34.Das A, Zhao J, Schatz GC, Sligar SG, Van Duyne RP. Anal Chem. 2009;81(10):3754–3759. doi: 10.1021/ac802612z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu JP, Eddings MA, Miles AR, Bukasov R, Gale BK, Shumaker-Parry JS. Anal Chem. 2009;81(11):4296–4301. doi: 10.1021/ac900181f. [DOI] [PubMed] [Google Scholar]
- 36.Wolf LK, Fullenkamp DE, Georgiadis RM. J Am Chem Soc. 2005;127(49):17453–17459. doi: 10.1021/ja056422w. [DOI] [PubMed] [Google Scholar]
- 37.Peterson AW, Heaton RJ, Georgiadis RM. Nucleic Acids Res. 2001;29(24):5163–5168. doi: 10.1093/nar/29.24.5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jordan CE, Frutos AG, Thiel AJ, Corn RM. Anal Chem. 1997;69(24):4939–4947. [Google Scholar]
- 39.Gao Y, Wolf LK, Georgiadis RM. Nucleic Acids Res. 2006;34(11):3370–3377. doi: 10.1093/nar/gkl422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heyduk T, Heyduk E. Nat Biotechnol. 2002;20(2):171–176. doi: 10.1038/nbt0202-171. [DOI] [PubMed] [Google Scholar]
- 41.Xu XY, Zhao Z, Qin LD, Wei W, Levine JE, Mirkin CA. Anal Chem. 2008;80(14):5616–5621. doi: 10.1021/ac8007016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, Boxer A, Blennow K, Friedman LF, Galasko DR, Jutel M, Karydas A, Kaye JA, Leszek J, Miller BL, Minthon L, Quinn JF, Rabinovici GD, Robinson WH, Sabbagh MN, So YT, Sparks DL, Tabaton M, Tinklenberg J, Yesavage JA, Tibshirani R, Wyss-Coray T. Nat Med. 2007;13(11):1359–1362. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.