

Abstract
Objective
Isopeptidase T is a cysteine protease deubiquitinating enzyme that hydrolyzes unanchored polyubiquitin chains to free monoubiquitin. Nitric oxide (NO) decreases 26S proteasome activity in vascular smooth muscle cells (VSMCs) and inhibits neointimal hyperplasia in animal models. As NO can cause S-nitrosylation of active-site cysteines, we hypothesize that NO inhibits isopeptidase T activity through S-nitrosylation. Because accumulation of polyubiquitin chains inhibits the 26S proteasome, this may be one mechanism through which NO prevents neointimal hyperplasia.
Methods
To investigate our hypothesis, we examined the effect of NO on isopeptidase T activity, levels, and localization in VSMCs in vitro and in a rat carotid balloon injury model in vivo.
Results
NO inhibited recombinant isopeptidase T activity by 82.8% (t = 60 minutes, P < .001 vs control). Dithiothreitol and glutathione (5 mmol/L) both significantly reversed NO-mediated inhibition of isopeptidase T activity (P < .001). NO caused a time-dependent increase in S-nitrosylated isopeptidase T levels in VSMCs, which was reversible with dithiothreitol, indicating that isopeptidase T undergoes reversible S-nitrosylation on exposure to NO in vitro. Although NO did not affect isopeptidase T levels or subcellular localization in VSMCs in vitro, it decreased isopeptidase T levels and increased ubiquitinated proteins after balloon injury in vivo.
Conclusions
Local administration of NO may prevent neointimal hyperplasia by inhibiting isopeptidase T levels and activity in the vasculature, thereby inhibiting the 26S proteasome in VSMCs. These data provide additional mechanistic insights into the ability of NO to prevent neointimal hyperplasia after vascular interventions. (J Vasc Surg 2013;■:1-8.)
Clinical Relevance
The 26S proteasome is responsible for degrading polyubiquitinated proteins. Isopeptidase T is a deubiquitinating enzyme that recycles polyubiquitin chains to monoubiquitin. Nitric oxide (NO) decreases formation of neointimal hyperplasia in animal models and decreases 26S proteasome activity in vascular smooth muscle cells. We investigated the effects of NO on isopeptidase T and showed that NO inhibits recombinant isopeptidase T activity, increases S-nitrosylated isopeptidase T levels in vascular smooth muscle cells, and, after balloon injury in vivo, decreases isopeptidase T levels and increases ubiquitinated proteins. Local administration of NO may prevent neointimal hyperplasia by targeting isopeptidase T and inhibiting the 26S proteasome.
Nitric oxide (NO) is a known potent inhibitor of neointimal hyperplasia in large and small animal models of vein bypass grafting and arterial injury.1-3 We have shown previously that external administration of the NO donor disodium 1-[(2-carboxylato)pyrrolidin-1-yl]diazen-1-ium-1,2-diolate (PROLI/NO) to the adventitia causes a 90% decrease in neointimal hyperplasia in a rat carotid artery balloon injury model.2 Furthermore, we demonstrated that one mechanism by which NO may inhibit cell cycle progression is through inhibition of 26S proteasome catalytic activity,4 which prevents vascular smooth muscle cell (VSMC) proliferation and, thus, neointimal hyperplasia. Given that unanchored polyubiquitin chains are known to inhibit the 26S proteasome,5,6 and isopeptidase T is known to hydrolyze predominantly unanchored polyubiquitin chains into free ubiquitin monomers,5-9 we hypothesized that direct inhibition of isopeptidase T may be another mechanism by which NO inhibits VSMC proliferation. Furthermore, it is possible that NO may prevent protein breakdown by affecting the activity of isopeptidase T through S-nitrosylation of active site cysteines.
Proteins in eukaryotic cells are mainly degraded by the ubiquitin-proteasome pathway. A protein targeted for destruction is marked by ligation of a polyubiquitin strand, which is recognized and removed by the 26S proteasome.10 Polyubiquitin chains are formed by first linking ubiquitin via a peptide bond from its C-terminal glycine (G76) to the ε-amino group of a lysine on the target protein.7 Subsequent ubiquitins are added to the chain by conjugation to G76 through an isopeptide bond to lysine 48 (K48), which marks the protein for proteasomal degradation.11 As there are seven lysines in ubiquitin, several polyubiquitin linkages, including monoubiquitination, that have other functions in eukaryotic cells are possible (see reviews).12,13 Here, we focus on K48-linked polyubiquitin chains, which are the preferred substrate for isopeptidase T, are sufficient to send tagged proteins to the 26S proteasome for destruction,14 and are the principal signal for proteasomal degradation in cells.15
Although isopeptidase T can bind several ubiquitin configurations, it has great affinity for unanchored polyubiquitin chains.6,16 Thus, isopeptidase T functions not only to recycle ubiquitin for reuse by ubiquitin enzymes, but also to promote proper function of the 26S proteasome. Although much is known about the mechanism of isopeptidase T activity in vitro, little is known about its activity in the vasculature. Nor is much known about how NO, an important molecule constitutively expressed in the vasculature, affects isopeptidase T activity. To investigate our hypothesis, the aim of this study was to evaluate the effect of exogenous NO on isopeptidase T activity and levels in the vasculature in vitro and in vivo.
METHODS
Isopeptidase T activity assay
Human recombinant isopeptidase T (5 nmol/L; Boston Biochem, Cambridge, Mass) was combined with reaction buffer (100 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.8, 1 mmol/L EDTA, 2 mmol/L dithiothreitol [DTT], 0.2 mg/mL bovine serum albumin [BSA; Sigma, St. Louis, Mo]). Next, the fluorogenic peptide substrate ubiquitin-AMC (0.5 μmol/L [Boston Biochem] in 50 mmol/L HEPES, 150 mmol/L NaCl, 1 mmol/L DTT, pH 8.0) was added to each treatment group. The final reaction volume was 100 μL. Isopeptidase T activity, determined by cleavage of the fluorescent substrate, was measured over 60 minutes using an excitation wavelength of 380 nm and an emission wavelength of 460 nm.
Cell culture
The previously described explant method was used to culture VSMCs from the abdominal aortas of male C57BL/6J mice (Jackson Laboratory; Bar Harbor, Me).17,18 Cells were maintained, and their purity was assessed, as previously described.19
Western blot analysis
VSMCs (0.5 × 106/plate) in 10-cm dishes were treated for 24 hours in the presence or absence of the S-nitrosothiol NO donor S-nitroso-N-acetylpenicillamine (SNAP). Cells were collected, protein concentration was determined, and proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transfer as previously described.19 Membranes were incubated with USP5 antibody (AP2134b; Abgent, San Diego, Calif) diluted 1:1000 in 5% BSA or ubiquitin antibody (Boston Biochem) diluted 1:1000 in 1× phosphate-buffered saline (PBS). Proteins were visualized and equal loading was verified as previously described.19 β-actin expression was used to normalize quantitations of isopeptidase T levels (ImageJ; National Institutes of Health, Bethesda, Md). Recombinant human isopeptidase T (E-322; Boston Biochem) was loaded as a positive control.
Biotin switch assay
VSMCs (0.8 × 106/plate) were treated with or without SNAP (500 μmol/L) for 2, 4, or 6 hours. For groups treated with DTT and SNAP, 5 mmol/L DTT was added 30 minutes before SNAP. Cells were washed with 3 mL cold 1× PBS, scraped in lysis buffer (50 mmol/L Tris, pH 7.5, 150 mmol/L NaCl, 0.5% deoxycholic acid, 0.1% SDS, 1% Nonidet P-40, 0.1 mmol/L neocuproine, and protease cocktail; Pierce, Rockford, Ill), incubated on ice for 30 minutes, and pelleted at 4°C. Protein concentration of the supernatant was measured by bicinchoninic acid (BCA) assay (Pierce). Blocking buffer—2.5% SDS and 20 mmol/L methyl methanethiosulfonate (MMTS) in HEN buffer (250 mmol/L HEPES-NaOH, pH 7.7, 1 mmol/L ethylenediaminetetra-acetic acid [EDTA], 0.1 mmol/L neocuproine)—was added to 500 μg of protein and incubated at 50°C for 20 minutes to block free thiol groups. MMTS was removed by addition of cold acetone and centrifugation, followed by resuspension of the pellet in HENS buffer (HEN plus 1% SDS).
Nitrosothiols were labeled by incubation in labeling solution (50 mmol/L biotin-HPDP (N-[6-(biotinamido) hexyl] 3′-(2′-pyridyldithio)propionamide; Pierce) in dimethylformamide dissolved to 4 mmol/L in dimethylsulfoxide) and ascorbate solution (50 mmol/L sodium ascorbate in water) for 1 hour at 25°C. Biotin-HPDP was removed by addition of cold acetone (−20°C) for 20 minutes, followed by centrifugation. This was repeated to completely remove biotin-HPDP. After resuspension in HENS buffer, neutralization buffer (20 mmol/L HEPES-NaOH, pH 7.7, 100 mmol/L NaCl, 1 mmol/L EDTA, and 0.5% Triton X-100 in water) was added, followed by incubation with streptavidin-agarose beads for 1 hour at room temperature. After five washes with neutralization buffer plus 600 mmol/L NaCl, with beads pelleted after each wash, bound biotinylated proteins were recovered by incubation for 20 minutes on ice in elution buffer (20 mmol/L HEPES-NaOH, pH 7.7, 100 mmol/L NaCl, 1 mmol/L EDTA, and 100 mmol/L 2-mercaptoethanol in water). Beads were resuspended in 3× SDS-PAGE sample buffer, boiled for 5 minutes at 100°C, and stored at −80°C until SDS-PAGE and Western blot analysis.
Animal surgery
Animal procedures were all approved by the Institutional Animal Care and Use Committee of Northwestern University and performed in accordance with National Institutes of Health guidelines outlined in the Guide for the Care and Use of Laboratory Animals (Publication 85-23, 1996). Surgeries were performed as previously described on male Sprague-Dawley rats weighing between 350 and 400 g.19 Rats anesthetized with inhaled isoflurane (0.5-3%) were given subcutaneous atropine (0.1 mg/kg) to decrease airway secretions. A midline neck incision allowed for dissection of the left common, external, and internal carotid arteries. Microclips were used to obtain proximal and distal control. A transverse arteriotomy on the external carotid allowed for insertion of a 2F Fogarty catheter (generously provided by Edwards Lifesciences) into the common carotid, which was injured by inflating the balloon for 5 minutes (5 atm). The balloon was removed, the external carotid ligated, and flow restored as previously described.19 After injury, the NO donor PROLI/NO was applied as a powder on the external adventitial surface of the injured common carotid artery as previously described.19 Controls included injury alone and no injury (n = 3/group).
Histological tissue processing
Carotid arteries harvested 3 or 14 days after balloon injury were frozen and cut into sections (5 μm) as previously described.19 The injury + NO group was treated with 10 mg of PROLI/NO.
Lysate tissue processing
Three days after balloon injury, carotid arteries were harvested and homogenized as previously described.19 After a BCA assay to determine protein concentration, samples were held at −80°C until analysis by SDS-PAGE and Western blotting. The injury + NO group was treated with 20 mg PROLI/NO.
Immunofluorescence staining
VSMC (1 × 104 cells/well) grown on glass coverslips (VWR International, West Chester, Penn) in 24-well plates were exposed to medium ± SNAP for 24 hours. Cells fixed with 10% buffered formalin (Fisher Scientific, Fair Lawn, NJ) were permeabilized with 0.3% Triton X-100 in PBS (Fisher Scientific). Next, cells were blocked in 5% BSA or donkey serum (Sigma) diluted 1:20 in 0.5% BSA. Cells were incubated in USP5 antibody (sc-79811; Santa Cruz Biotechnology, Santa Cruz, Calif) diluted 1:500 in 0.5% BSA, followed by donkey anti-goat AlexaFluor 555 (Invitrogen) diluted 1:3000 in PBS. Negative controls were incubated without primary antibody. 4′,6-Diamidino-2-phenylindole (DAPI, 60 nmol/L; Invitrogen) was used to stain nuclei. Coverslips were adhered to slides using ProLong Gold Antifade Reagent (Invitrogen), and digital images acquired on a Zeiss Imager.A2 microscope (Hallbergmoos, Germany) using the 20× objective.
Carotid artery sections from uninjured rats, injured rats, and rats subjected to injury + NO were fixed with paraformaldehyde (2%), then permeabilized in Triton X-100 (0.3% in PBS). After being blocked in 5% BSA, sections were incubated with USP5 antibody (1:100 in BSA; Santa Cruz Biotechnology). Negative controls were incubated without primary antibody. After incubation with donkey anti-goat AlexaFluor 555 antibody (1:1000 in PBS; Invitrogen), 60 nmol/L DAPI was used to stain nuclei, and slides were coverslipped as above. Digital images were acquired as above, except using the 10× objective.
Statistical analysis
Numerical results are given as means ± the standard errors of the mean. SigmaStat (SPSS, Chicago, Ill) was used to perform one-way analysis of variance (ANOVA) on differences between groups, and all pairwise comparisons were assessed with the Student-Newmann-Keuls post hoc test. P < .05 was used to indicate statistical significance.
RESULTS
NO inhibits isopeptidase T activity in a concentration-dependent manner
To evaluate isopeptidase T activity in vitro, an assay was performed using recombinant isopeptidase T enzyme ± SNAP (125-1000 μmol/L). Baseline activity for isopeptidase T at 60 minutes was 1001 ± 16 s−1. Increasing concentrations of SNAP led to significantly increased inhibition of isopeptidase T activity (P < .001 at SNAP ≥ 500 μmol/L) (Fig 1, A). SNAP (1000 μmol/L) inhibited isopeptidase T activity by 82.8% after 60 minutes. The parent compound NAP (1000 μmol/L), used as a negative control, only slightly inhibited isopeptidase T activity (17%, P < .02) compared with baseline. A kinetics assay showed that isopeptidase T activity plateaued after 60 minutes, and SNAP concentrations above 250 μmol/L significantly inhibited isopeptidase T activity (Fig 1, B).
Fig 1.

Nitric oxide (NO) inhibits isopeptidase T activity. Isopeptidase T activity was assayed with and without S-nitroso-N-acetylpenicillamine (SNAP) (A) at 60 minutes and (B) every 5 minutes for 60 minutes. N-acetylpenicillamine (NAP, 1000 μmmol/L) is a negative control. n = 3 per treatment group. Data are representative of four separate experiments. *P < .001 vs control, **P < .02 vs control.
Dithiothreitol and glutathione prevent the inhibition of isopeptidase T activity by NO
To determine if NO inhibits isopeptidase T activity via modification of cysteine thiol groups, DTT (5 mmol/L) was added to the activity assay before SNAP (1000 μmol/L). DTT significantly prevented SNAP-mediated inhibition of isopeptidase T activity, restoring activity to 77.8% of control (*P < .001 vs control; **P < .001 vs SNAP) (Fig 2). To further investigate the specificity of this interaction, glutathione (GSH, 5 mmol/L) was added to the activity assay before SNAP (1000 μmol/L). GSH significantly prevented inhibition of isopeptidase T activity mediated by SNAP, with activity levels observed at 77.8% of control (*P < .001 vs control; **P < .001 vs SNAP) (Fig 2). Neither reducing agent alone significantly affected isopeptidase T activity. Thus, isopeptidase T activity appears susceptible to regulation via modification of thiol groups.
Fig 2.
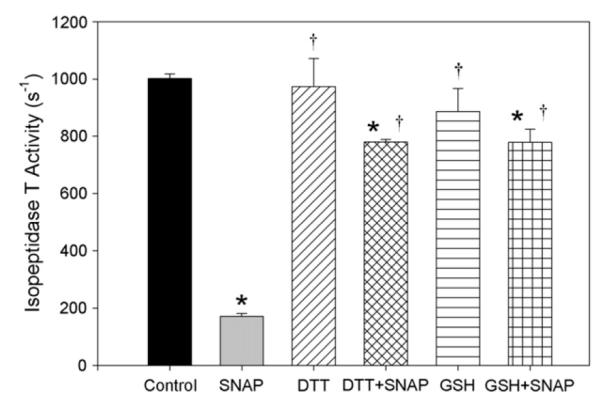
Nitric oxide (NO)-mediated inhibition of isopeptidase T activity is prevented by reducing agents. Isopeptidase T activity was assessed with S-nitroso-N-acetylpenicillamine (SNAP, 1000 μmol/L) ± dithiothreitol (DTT, 5 mmol/L) or glutathione (GSH, 5 mmol/L). t = 60 minutes, n = 3 per each treatment group. Data are representative of four separate experiments. *P < .001 vs control, †P < .001 vs SNAP.
NO causes S-nitrosylation of isopeptidase T in a time-dependent manner
Because isopeptidase T activity was affected by reducing agents, we sought to ascertain whether NO S-nitrosylates isopeptidase T. A biotin switch assay was performed after treatment of VSMCs with SNAP (500 μmol/L) ± DTT (5 mmol/L) for 2, 4, or 6 hours. As shown in Fig 3, longer SNAP (500 μmol/L) exposure times caused elevated levels of S-nitrosylated isopeptidase T (SNO-IsoT), seen most prominently at 4 hours. Though levels of SNO-IsoT decreased by 6 hours (not shown), this action of NO was prevented at all time points by addition of 5 mmol/L DTT.
Fig 3.
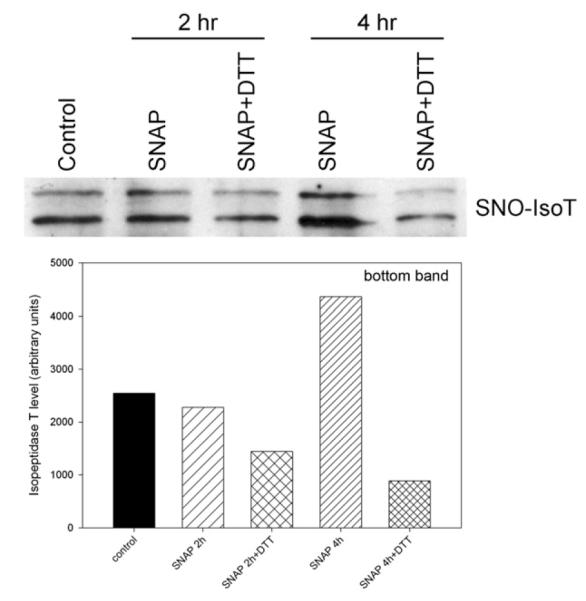
Nitric oxide (NO) causes S-nitrosylation of isopeptidase T (SNO-IsoT) in a time-dependent manner. After treatment of vascular smooth muscle cells (VSMCs) with S-nitroso-N-acetylpenicillamine (SNAP, 500 μmol/L) ± dithiothreitol (DTT, 5 mmol/L) for the times indicated, SNO-isopeptidase T was enriched from cell lysates via biotin switch assay, then subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis. Data are representative of three separate experiments.
NO does not affect isopeptidase T levels or intracellular localization in VSMCs
The effect of NO on isopeptidase T levels was assessed by Western blot analysis of VSMC exposed to SNAP (125-1000 μmol/L) for 24 hours. No effect of NO on isopeptidase T levels was observed by densitometry analysis of Western blots (Fig 4, A). Of note, the antibody used recognized two isopeptidase T isoforms, as previously described.6,20,21 We focused on the bottom band, as this is the most prominent band observed in the recombinant isopeptidase T (rIsoT) lane (Fig 4, A). To determine whether NO exposure influenced localization of isopeptidase T within the cell, immunofluorescence staining for isopeptidase T was performed in VSMC ± SNAP (1000 μmol/L, 24 hours). Isopeptidase T was located mostly in the cytoplasm (Fig 4, B), and addition of NO did not affect intracellular localization of isopeptidase T.
Fig 4.

Nitric oxide (NO) does not affect isopeptidase T (isoT) levels or intracellular localization in vascular smooth muscle cells (VSMCs). A, Western blot analysis of isopeptidase T levels in VSMC exposed to S-nitroso-N-acetylpenicillamine (SNAP, 125-1000 μmol/L) for 24 hours showed no effect of NO. B, Immunofluorescence staining performed on VSMC for isopeptidase T (red) after exposure to SNAP (1000 μmol/L) for 24 hours showed mostly cytoplasmic staining and no change in levels or localization with NO. Blue represents nuclei stained with 4′,6-diamidino-2-phenylindole. Data and images are representative of at least two separate experiments. rIsoT, Recombinant isopeptidase T.
NO decreases isopeptidase T levels in vivo
Although NO had no effect on isopeptidase T levels in VSMC in vitro, because there are many cell types in the arterial wall before and after injury, we wanted to investigate whether NO affected isopeptidase T levels in vivo. After injury, carotid arteries from male Sprague-Dawley rats were treated with or without 10 mg PROLI/NO, removed at 3 or 14 days, sectioned, and immunofluorescence stained for isopeptidase T. We observed an increase in isopeptidase T levels throughout the arterial wall at both time points after injury, with the biggest increase at 3 days observed in the adventitia and the biggest increase at 14 days seen in the neointima (Fig 5, A). Although NO decreased the overall levels of isopeptidase T throughout the arterial wall at both time points after injury, the most prominent decrease was seen at 14 days.
Fig 5.

Nitric oxide (NO) decreases levels of isopeptidase T (isoT) and free ubiquitin in vivo, with a concomitant increase in ubiquitinated proteins. A, Immunofluorescence staining for isopeptidase T (red) performed on balloon-injured rat carotid arteries treated with and without 10 mg PROLI/NO for 3 or 14 days showed that isopeptidase T increased after injury and decreased with NO. Nuclei are shown in blue (4’,6-diamidino-2-phenylindole), and internal elastic lamina autofluorescence is visible in green. The negative control (ie, no primary antibody) revealed no red staining. Data are representative of two separate stains. B, Western blot analysis performed on balloon-injured rat carotid artery lysates treated with and without 20 mg PROLI/NO for 3 days showed a significant decrease in isopeptidase T levels (left) in the injury + NO group. Free ubiquitin levels also decreased, with a concomitant increase in ubiquitinated proteins (right) observed in the injury + NO group. rIsoT, Recombinant isopeptidase T. *P < .03 vs control. n = 3/group.
To further assess isopeptidase T in our animal model, balloon-injured carotid arteries treated with or without 20 mg PROLI/NO were harvested at 3 days, homogenized, and analyzed by Western blotting for isopeptidase T or ubiquitinated protein levels. As seen in Fig 5, B, although there was a decrease in isopeptidase T expression after balloon injury, there was a marked decrease in isopeptidase T levels in the injury + NO vessels. Additionally, we observed an increase in ubiquitinated proteins in the injury + NO group which corresponded with decreases in free ubiquitin and isopeptidase T (Fig 5, B). Of note, the data in Fig 5, B, represent all cells present throughout the arterial wall.
DISCUSSION
We demonstrate here that NO inhibits the activity, but not levels, of isopeptidase T in VSMC in vitro, while decreasing its levels and increasing ubiquitinated proteins in the vasculature in vivo. This correlation between the effect of NO on isopeptidase T activity and the presence of S-nitrosylated isopeptidase T is further strengthened by evidence that NO inhibits isopeptidase T activity in a concentration-dependent fashion in vitro. S-Nitrosylation is likely responsible for mediating this effect, which is reversed by reducing agents. Immunofluorescent staining and Western blot analysis of VSMC in vitro illustrate that NO had no effect on the levels or intracellular location of isopeptidase T at short or long time points. Finally, we show that periadventitial administration of NO decreases levels of isopeptidase T in an animal model of neointimal hyperplasia, which correlates with increased ubiquitinated protein levels.
Although much is known about the biochemistry, structure, function, and mechanism of isopeptidase T, little is known about its role in the vasculature. Decreased isopeptidase T function can lead to accumulation of ubiquitinated proteins in cardiac myocytes, which can cause cell death and heart failure in humans.22 Kostin et al observed twofold increases in ubiquitin mRNA and ubiquitin-conjugating (E2) enzyme levels and a twofold reduction in isopeptidase T expression in failing human hearts.22 Chronic proteasomal inhibition also contributes to development of coronary atherosclerosis.23 Herrmann et al observed reductions in vasorelaxation, increased intima-to-media ratios, and increased oxidative stress in female pigs fed a high-cholesterol diet and given MLN-273 (a proteasome inhibitor).23 Interestingly, localized proteasomal inhibition was also linked to decreased development of restenosis in a rat carotid artery balloon injury model.24 Meiners et al administered the proteasome inhibitor MG132 for 5 minutes at the site of injury and saw decreased inflammation, increased apoptosis, and decreased intima-to-media area ratios up to 14 days after treatment.24 Neointimal hyperplasia has also been linked to proteasome activity. Faries et al stained biopsy specimens of atherosclerotic and normal vessels, as well as patent and occluded grafts, for 11S proteasome activator (PA28) expression and observed that areas of vessels with less PA28 staining (lower proteasome activity) had increased levels of atherosclerosis and neointimal hyperplasia.25 Finally, Stone et al observed reduced expression of ubiquitin, E2 enzymes, ubiquitin ligase enzymes, and proteasomal subunits at the anastomoses of vein grafts in the carotid arteries of mongrel dogs.26 Although increased ubiquitinated protein levels were described in some of these studies, and proteasome activity reductions were described in all of them, only Kostin et al specifically investigated isopeptidase T, and their study did not address isopeptidase T in blood vessels.22 Thus, although these studies make clear that 26S proteasome activity is linked to neointimal hyperplasia, much remains unknown about isopeptidase T in the vasculature. The work presented here adds valuable information to a nascent field of promising research.
We show, for the first time, that NO inhibits isopeptidase T activity in the vasculature, most likely by S-nitrosylation of a critical cysteine residue, a common mechanism by which NO regulates the activity of cysteine proteases.27 Previous work in our laboratory established that NO donors applied to the external surface of the rat carotid artery after balloon angioplasty result in significant inhibition of neointimal hyperplasia in several different animal models.2,3,28 Because we have used the same NO donor and amounts in this work, our data should be directly translatable. Thus, inhibiting isopeptidase T activity may allow unanchored polyubiquitin chains to accumulate and inhibit the 26S proteasome, resulting in the buildup of cell cycle proteins, ultimately leading to less proliferation, and less neointima formation. In support of this theory, NO is known to increase expression of cell cycle inhibitors like p21 and p27, as well as p53, and these increases are associated with inhibition of neointimal hyperplasia.17,29 Further, Dayal et al reported that isopeptidase T inhibition causes both p53 activation and unanchored polyubiquitin chain accumulation.8 Our current data also indicate a buildup of ubiquitinated proteins, and a concomitant loss of free ubiquitin, in the injury + NO treatment group. Thus, a causal link has been established between isopeptidase T activity and neointimal hyperplasia. Indeed, because NO affects such a varied number of cell cycle proteins and cyclin-dependent kinases, it makes sense that NO is acting on a central regulator, such as the ubiquitin-proteasome pathway, to exert these effects.
Although our data reveal a number of new discoveries about the actions of isopeptidase T in the vasculature and the relationship between NO and isopeptidase T, there are limitations to this study. For instance, we did not assess levels of SNO-isopeptidase T in balloon-injured vessels because of the limitations in currently available reagents specific to isopeptidase T or SNO-isopeptidase T. On the basis of previous studies, we would expect levels of SNO-isopeptidase T in NO-treated balloon-injured vessels to increase relative to both injury alone and uninjured vessels. Another limitation is that we did not directly assess isopeptidase T activity in VSMC or carotid artery lysates. These studies are challenging because of the abundance of deubiquitinating enzymes present in cells and the lack of reagents specific to isopeptidase T. We expect that these experiments would indicate that NO decreases isopeptidase T activity in vivo, which would lead to ubiquitin-proteasome pathway inhibition as a result of accumulation of proteins with unprocessed K48-linked polyubiquitin chains. We also did not stain carotid artery sections for the presence of SNO-isopeptidase T because of reagent limitations, but would expect it would increase in NO-treated vessels. As this field of research expands, new reagents will become available, allowing for these additional studies in the future.
Lastly, to shed light on the possible involvement of other cells in the vessel wall, we performed immunofluorescence staining for isopeptidase T on NIH-3T3 fibroblasts and primary rat VSMC. We observed no changes in either the levels of isopeptidase T or its subcellular localization in any of the cell types, or at any of the SNAP concentrations (125-1000 μmol/L), tested (data not shown). Because several other cell types are present in the arterial wall, including endothelial cells, macrophages, leukocytes, and stem cells, it is possible that some of these other cell types may be the cells in which NO has its effect. Furthermore, simple in vitro cell culture models cannot accurately mimic the environment in the arterial wall after injury. Thus, we are not surprised that we did not detect differences in the two cell types we evaluated in vitro.
In summary, we demonstrate a strong correlation between the time- and concentration-dependent inhibitory effects of NO on isopeptidase T activity in vascular cells and the presence of SNO-isopeptidase T in these cells. We also show a correlation between decreased isopeptidase T levels in the vasculature after administration of NO and a decrease in neointimal hyperplasia formation, which may be achieved via the formation of SNO-isopeptidase T. This effect on isopeptidase T activity can be prevented in cells by reducing agents, which prevent the formation of SNO on isopeptidase T. We provide evidence that, although administration of NO in vitro had no effect on isopeptidase T levels or intracellular localization in VSMC, external administration of NO in vivo in a carotid artery model of neointimal hyperplasia decreased isopeptidase T levels, while increasing levels of ubiquitinated proteins. We conclude that local administration of NO may prevent the formation of neointimal hyperplasia by inhibiting isopeptidase T activity via S-nitrosylation, resulting in accumulation of unanchored polyubiquitin chains and further inhibiting the 26S proteasome. As others have shown, the ubiquitin-proteasome pathway is a rich vein that can be mined for therapeutic interventions for a number of diseases. The data we present here add one more fine-scale tool that may be useful in preventing restenosis after vascular interventions.
The authors thank the Institute for BioNanotechnology in Medicine at Northwestern University, the Feinberg Cardiovascular Research Institute, Edwards Lifesciences for providing Fogarty balloon catheters and Lynnette Dangerfield for administrative support.
Acknowledgments
Funded by the National Institutes of Health (1K08HL0842-03 to M.R.K.), the Society for Vascular Surgery Foundation (Mentored Clinical Scientist Development Award to M.R.K.), the American Heart Association (0725766Z and 09POST2230028 to N.D.T.), and the generosity of Mrs Eleanor Baldwin and Mrs Hilda Rosenbloom.
Footnotes
Author conflict of interest: none.
The editors and reviewers of this article have no relevant financial relationships to disclose per the JVS policy that requires reviewers to decline to review of any manuscript for which they may have a conflict of interest.
AUTHOR CONTRIBUTIONS Conception and design: NT, MK, MRK Analysis and interpretation: NT, MK, WF, CO, QJ, MRK Data collection: NT, MK, AV, WF, CO, QJ Writing the article: NT, MK, AV, WF, CO, QJ Critical revision of the article: NT, MRK Final approval of the article: NT, MK, AV, WF, CO, QJ, MRK Statistical analysis: NT, MK Obtained funding: NT, MRK Overall responsibility: MRK
REFERENCES
- 1.Shears LL, Kibbe MR, Murdock AD, Billiar TR, Lizonova A, Kovesdi I, et al. Efficient inhibition of intimal hyperplasia by adenovirus-mediated inducible nitric oxide synthase gene transfer to rats and pigs in vivo. J Am Coll Surg. 1998;187:295–306. doi: 10.1016/s1072-7515(98)00163-x. [DOI] [PubMed] [Google Scholar]
- 2.Pearce CG, Najjar SF, Kapadia MR, Murar J, Eng J, Lyle B, et al. Beneficial effect of a short-acting NO donor for the prevention of neointimal hyperplasia. Free Radic Biol Med. 2008;44:73–81. doi: 10.1016/j.freeradbiomed.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kapadia MR, Chow LW, Tsihlis ND, Ahanchi SS, Eng JW, Murar J, et al. Nitric oxide and nanotechnology: a novel approach to inhibit neointimal hyperplasia. J Vasc Surg. 2008;47:173–82. doi: 10.1016/j.jvs.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kapadia MR, Eng JW, Jiang Q, Stoyanovsky DA, Kibbe MR. Nitric oxide regulates the 26S proteasome in vascular smooth muscle cells. Nitric Oxide. 2009;20:279–88. doi: 10.1016/j.niox.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Amerik AY, Swaminathan S, Krantz BA, Wilkinson KD, Hochstrasser M. In vivo disassembly of free polyubiquitin chains by yeast Ubp14 modulates rates of protein degradation by the proteasome. EMBO J. 1997;16:4826–38. doi: 10.1093/emboj/16.16.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilkinson KD, Tashayev VL, O’Connor LB, Larsen CN, Kasperek E, Pickart CM. Metabolism of the polyubiquitin degradation signal: structure, mechanism, and role of isopeptidase T. Biochemistry. 1995;34:14535–46. doi: 10.1021/bi00044a032. [DOI] [PubMed] [Google Scholar]
- 7.Hadari T, Warms JV, Rose IA, Hershko A. A ubiquitin C-terminal isopeptidase that acts on polyubiquitin chains: role in protein degradation. J Biol Chem. 1992;267:719–27. [PubMed] [Google Scholar]
- 8.Dayal S, Sparks A, Jacob J, Allende-Vega N, Lane DP, Saville MK. Suppression of the deubiquitinating enzyme USP5 causes the accumulation of unanchored polyubiquitin and the activation of p53. J Biol Chem. 2009;284:5030–41. doi: 10.1074/jbc.M805871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lacombe T, Gabriel JM. Further characterization of the putative human isopeptidase T catalytic site. FEBS Lett. 2002;531:469–74. doi: 10.1016/s0014-5793(02)03586-x. [DOI] [PubMed] [Google Scholar]
- 10.Crews CM. Feeding the machine: mechanisms of proteasome-catalyzed degradation of ubiquitinated proteins. Curr Opin Chem Biol. 2003;7:534–9. doi: 10.1016/j.cbpa.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Gregori L, Poosch MS, Cousins G, Chau V. A uniform isopeptide-linked multiubiquitin chain is sufficient to target substrate for degradation in ubiquitin-mediated proteolysis. J Biol Chem. 1990;265:8354–7. [PubMed] [Google Scholar]
- 12.Li W, Ye Y. Polyubiquitin chains: functions, structures, and mechanisms. Cell Mol Life Sci. 2008;65:2397–406. doi: 10.1007/s00018-008-8090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pickart CM, Fushman D. Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol. 2004;8:610–6. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 14.Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–83. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 15.Finley D, Sadis S, Monia BP, Boucher P, Ecker DJ, Crooke ST, et al. Inhibition of proteolysis and cell cycle progression in a multiubiquitination-deficient yeast mutant. Mol Cell Biol. 1994;14:5501–9. doi: 10.1128/mcb.14.8.5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raasi S, Varadan R, Fushman D, Pickart CM. Diverse polyubiquitin interaction properties of ubiquitin-associated domains. Nat Struct Mol Biol. 2005;12:708–14. doi: 10.1038/nsmb962. [DOI] [PubMed] [Google Scholar]
- 17.Kibbe MR, Li J, Nie S, Watkins SC, Lizonova A, Kovesdi I, et al. Inducible nitric oxide synthase (iNOS) expression upregulates p21 and inhibits vascular smooth muscle cell proliferation through p42/44 mitogen-activated protein kinase activation and independent of p53 and cyclic guanosine monophosphate. J Vasc Surg. 2000;31:1214–28. doi: 10.1067/mva.2000.105006. [DOI] [PubMed] [Google Scholar]
- 18.Yu SM, Hung LM, Lin CC. cGMP-elevating agents suppress proliferation of vascular smooth muscle cells by inhibiting the activation of epidermal growth factor signaling pathway. Circulation. 1997;95:1269–77. doi: 10.1161/01.cir.95.5.1269. [DOI] [PubMed] [Google Scholar]
- 19.Tsihlis ND, Kapadia MR, Vavra AK, Jiang Q, Fu B, Martinez J, et al. Nitric oxide decreases activity and levels of the 11S proteasome activator PA28 in the vasculature. Nitric Oxide. 2012;27:50–8. doi: 10.1016/j.niox.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 20.Timms KM, Ansari-Lari MA, Morris W, Brown SN, Gibbs RA. The genomic organization of isopeptidase T-3 (ISOT-3), a new member of the ubiquitin specific protease family UBP. Gene. 1998;217:101–6. doi: 10.1016/s0378-1119(98)00341-2. [DOI] [PubMed] [Google Scholar]
- 21.Ansari-Lari MA, Muzny DM, Lu J, Lu F, Lilley CE, Spanos S, et al. A gene-rich cluster between the CD4 and triosephosphate isomerase genes at human chromosome 12p13. Genome Res. 1996;6:314–26. doi: 10.1101/gr.6.4.314. [DOI] [PubMed] [Google Scholar]
- 22.Kostin S, Pool L, Elsasser A, Hein S, Drexler HC, Arnon E, et al. Myocytes die by multiple mechanisms in failing human hearts. Circ Res. 2003;92:715–24. doi: 10.1161/01.RES.0000067471.95890.5C. [DOI] [PubMed] [Google Scholar]
- 23.Herrmann J, Saguner AM, Versari D, Peterson TE, Chade A, Olson M, et al. Chronic proteasome inhibition contributes to coronary atherosclerosis. Circ Res. 2007;101:865–74. doi: 10.1161/CIRCRESAHA.107.152959. [DOI] [PubMed] [Google Scholar]
- 24.Meiners S, Laule M, Rother W, Guenther C, Prauka I, Muschick P, et al. Ubiquitin-proteasome pathway as a new target for the prevention of restenosis. Circulation. 2002;105:483–9. doi: 10.1161/hc0402.102951. [DOI] [PubMed] [Google Scholar]
- 25.Faries PL, Rohan DI, Wyers MC, Marin ML, Hollier LH, Quist WC, et al. Relationship of the 20S proteasome and the proteasome activator PA28 to atherosclerosis and intimal hyperplasia in the human vascular system. Ann Vasc Surg. 2001;15:628–33. doi: 10.1007/s10016-001-0055-2. [DOI] [PubMed] [Google Scholar]
- 26.Stone DH, Sivamurthy N, Contreras MA, Fitzgerald L, LoGerfo FW, Quist WC. Altered ubiquitin/proteasome expression in anastomotic intimal hyperplasia. J Vasc Surg. 2001;34:1016–22. doi: 10.1067/mva.2001.119888. [DOI] [PubMed] [Google Scholar]
- 27.Lima B, Forrester MT, Hess DT, Stamler JS. S-Nitrosylation in cardiovascular signaling. Circ Res. 2010;106:633–46. doi: 10.1161/CIRCRESAHA.109.207381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahanchi SS, Varu VN, Tsihlis ND, Martinez J, Pearce CG, Kapadia MR, et al. Heightened efficacy of nitric oxide-based therapies in type II diabetes mellitus and metabolic syndrome. Am J Physiol Heart Circ Physiol. 2008;295:H2388–98. doi: 10.1152/ajpheart.00185.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kibbe MR, Li J, Nie S, Choi BM, Kovesdi I, Lizonova A, et al. Potentiation of nitric oxide-induced apoptosis in p53−/− vascular smooth muscle cells. Am J Physiol Cell Physiol. 2002;282:C625–34. doi: 10.1152/ajpcell.00119.2001. [DOI] [PubMed] [Google Scholar]