

Abstract
Methamphetamine (Meth) is a widely abused psychostimulant that causes long-term dopamine (DA) and serotonin (5-HT) depletions. Stress and Meth abuse are comorbid events in society and stress exacerbates Meth-induced monoaminergic terminal damage. Stress is also known to produce neuroinflammation. This study examined the role of the neuroinflammatory mediator, cyclooxygenase (COX), in the depletions of monoamines caused by serial exposure to chronic unpredictable stress (CUS) and Meth. CUS produced an increase in COX-2 protein expression and enhanced Meth-induced monoaminergic depletions in the striatum and hippocampus. The enhanced DA and 5-HT depletions in the striatum, but not the hippocampus, were prevented by pretreatment with COX inhibitor, ketoprofen, during stress or during Meth; however, ketoprofen did not attenuate the monoaminergic damage caused by Meth alone. The COX-dependent enhancement by stress of Meth-induced monoaminergic depletions was independent of hyperthermia, as ketoprofen did not attenuate Meth-induced hyperthermia. In addition, the EP1 receptor antagonist, SC-51089, did not attenuate DA or 5-HT depletions caused by stress and Meth. These findings illustrate that COX activity, but not activation of the EP1 receptor, is responsible for the potentiation of Meth-induced damage to striatal monoamine terminals by stress and suggests the use of anti-inflammatory drugs for mitigating the neurotoxic effects associated with the combination of stress and Meth.
Keywords: methamphetamine, stress, cyclooxygenase, neurodegeneration, neuroinflammation
1. INTRODUCTION
The widely abused psychostimulant, methamphetamine (Meth) produces long-term monoaminergic terminal damage that persists for months and up to years in rodents and non-human primates (De Vito and Wagner, 1989; Preston et al., 1985; Ricaurte et al., 1982; Wagner et al., 1980; Woolverton et al., 1989). The mechanisms that have been attributed to the damage to dopamine (DA) and 5-HT terminals include oxidative stress and excitotoxicity as evidenced by increases in free radicals and spectrin proteolysis (Giovanni et al., 1995; Staszewski and Yamamoto, 2006; Yamamoto and Zhu, 1998), and the ability of antioxidants and NMDA receptor antagonists to attenuate the Meth-induced monoaminergic depletions (Battaglia et al., 2002; De Vito and Wagner, 1989; Fukami et al., 2004; Mark et al., 2004; Sonsalla et al., 1991; Stephans and Yamamoto, 1994; Wagner et al., 1985).
More recently, neuroinflammation has also been implicated in the neurotoxic effects of Meth (LaVoie et al., 2004; Thomas et al., 2004a; Thomas and Kuhn, 2005b; Thomas et al., 2004b). Meth produces neuroinflammation marked by microglial activation and increases in proinflammatory cytokines and chemokines (Loftis et al., 2011; Thomas et al., 2004a). Although the anti-inflammatory agents, indomethacin and minocycline, prevent Meth-induced glial activation, the association between inflammation and Meth-induced monoaminergic depletions remains to be established (Boger et al., 2009; Goncalves et al., 2010). No studies to date have established a role for inflammation in Meth-induced DA and 5-HT depletions or successfully prevented Meth-induced DA and 5-HT depletions using an anti-inflammatory drug.
Meth-induced monoaminergic depletions are enhanced by chronic stress (Matuszewich and Yamamoto, 2004b; Tata et al., 2007) and chronic stress is known to produce an inflammatory state in the brain (de Pablos et al., 2006; Garcia-Bueno et al., 2008a; Garcia-Bueno et al., 2008b; Nair and Bonneau, 2006; Sorrells and Sapolsky, 2007). Similarly, the induction of inflammation by exposure to LPS potentiates low dose Meth-induced decreases in striatal TH and DA (Jung et al., 2010). Although the mechanisms by which stress may potentiate Meth-induced damage to monoaminergic terminals are unknown, they could be related to the ability of stress to increase COX protein.
Acute stressors can increase COX-2 in the brain (Garcia-Bueno et al., 2008b; Madrigal et al., 2003; Yamaguchi et al., 2010). COX is an inflammatory mediator and produces prostaglandins. Prostaglandin E2 (PGE2) is the most characterized pro-inflammatory prostaglandin (Cimino et al., 2008) and can be neurotoxic through activation of the EP1 receptor and the enhancement of NMDA-induced dysregulation of neuronal calcium in vitro (Kawano et al., 2006). Conversely, antagonism of the EP1 receptor with SC-51089 reduces NMDA-induced neurotoxicity in mice (Kawano et al., 2006). Thus, activation of the EP1 receptor may be one way through which COX and stress produce neurotoxicity.
Although administration of neurotoxic doses of Meth increases expression of COX-2 (Kita et al., 2000; Thomas and Kuhn, 2005a), the role of COX-2 in Meth-induced monoaminergic terminal damage is unclear. COX-2 knock-out mice are refractory to Meth-induced striatal DA depletions (Thomas and Kuhn, 2005a); however, pharmacological inhibition of COX-2 activity does not afford protection against Meth-induced striatal DA depletions (Goncalves et al., 2010; Thomas and Kuhn, 2005a; Zhang et al., 2007). While COX activity does not appear to mediate Meth-induced damage to dopaminergic terminals, it may mediate the enhanced damage observed in response to serial exposure to chronic stress and Meth. Therefore, it is hypothesized that stress potentiates Meth-induced neurotoxicity through an increase in COX and inhibition of COX will attenuate the enhanced toxicity observed following serial exposure to stress and Meth.
2. METHODS
2.1 Animals
Male Sprague Dawley rats (180–275 g, Harlan Indianapolis, IN) were used in all experiments. Rats were housed 2 to 3 per cage, in clear plastic containers (45 × 24 × 20 cm), and allowed 4–5 days to acclimate to the animal colony before any experimentation. The environment in which the rats were housed was under a 12 hr light/dark cycle, temperature (23±1°C) and humidity (40±5%) controlled and rats had ad libitum access to food and water. All procedures were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the University of Toledo Institutional Animal Care and Use Committee. Efforts were made to minimize the number of animals used as well as to minimize the amount of suffering each animal might endure.
2.2 CUS paradigm
Rats were either handled daily or exposed to the chronic unpredictable stress (CUS) paradigm, which consisted of exposure to a variety of mild stressors at varying times during the day, for 10 days. The stress schedule was as follows: day 1, 50 minute cold room (10:00 h) and 30 minute cage agitation (14:00 h); day 2, 3 hrs lights off (9:00 h) and lights on overnight; day 3, 1 hr restraint (11:00 h) and food and water deprivation (17:00 h – 8:00 h); day 4, 1 hr cage agitation (10:00 h) and 50 minute cold room (15:00 h); day 5, 15 minute cold room isolation (14:00 h) and isolation overnight (17:15 h – 8:00 h); day 6, 1 hr restraint (9:00 h) and lights on overnight; day 7, 3 hrs lights off (10:00 h) and food and water deprivation (17:00 h – 8:00 h); day 8, 1 hr cage agitation (11:00 h) and isolation overnight (16:00 h – 8:00 h); day 9, 15 minute cold room isolation (9:00 h) and lights on overnight; day 10, 3 hrs lights off (10:00 h) and 20 minute cage agitation (15:00 h).
This CUS paradigm has been used previously to mimic the daily life stress humans encounter (Haile et al., 2001; Matuszewich and Yamamoto, 2004a; Stein-Behrens et al., 1994). This paradigm does not allow for adaptation, but rather produces an increase in basal corticosterone measured in plasma (Johnson and Yamamoto, 2009).
2.3 Drug Treatments
The day after the last stressor or daily handling, rats were treated with (+) methamphetamine-hydrochloride (Meth) (Sigma, St. Louis, MO, Cat. M-8750). Meth was dissolved in 0.9% NaCl (saline) and administered intraperitoneally (ip), at a dose of 7.5 mg/kg, for 4 injections, one injection every 2 hrs. Controls for Meth treatment were rats treated with 4 ip injections of saline (1 mL/kg), one injection every 2 hrs. This dosing paradigm for Meth was chosen based on previous studies that have shown the combination of 10 days of CUS and Meth (7.5 mg/kg × 4, q 2hrs) results in damage to dopaminergic and serotonergic terminals in the rat brain (Matuszewich and Yamamoto, 2004a, b; Tata et al., 2007).
Rectal temperatures were measured before and 1 hr after each Meth or saline injection.
2.4 Pharmacological Treatments
In order to investigate the role of CUS-induced COX-activity in Stress+Meth-induced monoaminergic depletions, some rats were pretreated with the non-specific COX inhibitor, ketoprofen, during the CUS paradigm. These rats received ketoprofen at a dose of 5 mg/kg, sc, 1 hr before each stressor, resulting in 2 injections of 5 mg/kg ketoprofen each day for 10 days of CUS. Ketoprofen was dissolved in 25% transcutol, at a concentration of 2.5 mg/mL, and 25% transcutol, at a dose of 2 mL/kg, was used as the vehicle control for ketoprofen injections. All non-stressed controls received the ketoprofen or transcutol injection at the same time as the stressed rats, but were not exposed to the stressors.
In order to investigate the role of Meth-induced COX-activity in Stress+Meth-induced monoaminergic depletions, a second group of rats was pretreated with ketoprofen during drug treatment. These rats received ketoprofen at a dose of 5 mg/kg, or transcutol, sc, 1 hr before each Meth or saline injection, for a total of 4 injections.
A third group of rats received ketoprofen pretreatments before each stressor and before each Meth or saline injection, in order to investigate the contribution of both stress and Meth-induced COX activity to monoaminergic depletions.
The dose and treatment paradigm of 5 mg/kg of ketoprofen was chosen based on a previous study that observed an attenuation of Meth-induced decreases in DAT immunoreactivity (Asanuma et al., 2003).
A fourth group of rats received the EP1R antagonist, SC-51089, in order to investigate the role of the EP1R in Stress+Meth-induced monoaminergic depletions. Rats received SC-51089 at a dose of 5, 10 or 20 μg/kg ip, 1 hr before each Meth or saline injection, for 4 injections of SC-51089, totaling 20, 40 or 80 μg/kg SC-51089 per rat in one day, respectively. SC-51089 was dissolved in 0.02% DMSO and 0.02% DMSO was used as the vehicle control for these experiments. These doses of SC-51089 were chosen based on previous studies which have identified the protective effects of SC-51089, at doses of 5–30 μg/kg, in ischemia and epilepsy models (Kawano et al., 2006; Abe et al., 2009; Fischborn et al., 2010; Fukumoto et al., 2010).
2.5 Western Blot for COX-2
The day after the last stressor, rats were killed via live decapitation and striatal and hippocampal tissue was dissected and rapidly frozen on dry ice. Tissue was stored at -80°C until use. Striatal or hippocampal tissue was homogenized in RIPA buffer (0.1 M PBS, 1% Igepal, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) with 1x Halt protease inhibitor cocktail (Thermo Scientific, Rockford, IL, Cat. 78430). The Bradford assay (BioRad, Hercules, CA) was used to measure total protein in samples, samples were diluted 1:4 with Novex 4x LDS sample buffer (Invitrogen, Carlsbad, CA) and heated to 85°C for 5 min.
Equal amounts of protein (25 μg) were loaded per well in a 4–12% Bis-Tris NuPAGE gel. After electrophoresis, proteins were transferred to a PVDF membrane. Membranes were blocked for 2 hrs at room temperature, with Tris-buffered saline (TBS) (10 mM Tris, 150 mM NaCl), containing 0.5% Tween-20 and 5% non-fat powdered milk. Membranes were then incubated with primary anti-COX-2 primary antibody (1:500, Santa Cruz, sc-1746) or the monoclonal mouse anti-α tubulin antibody (1:3000; Sigma, T6074) in blocking buffer for approximately 18 hours at 4°C. Following 3, 5 min, washes with TBS containing 0.5% Tween-20 (TBS-T), membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (rabbit anti-goat IgG, 1:2500, (Santa Cruz) or goat anti-mouse IgG, 1:2500, (Santa Cruz)) in blocking buffer for 1 hr at room temperature. Membranes were then washed 3 times, each for 5 min, with TBS-T.
HyGLO enhanced chemiluminescence (ECL) (Denville Scientific Inc., Metuchen, NJ) was used for antibody detection. A Fuji LAS-4000 mini system (FujiFilm Corp. Life Science Division, Tokyo, Japan) was used to image chemiluminescence and optical density was quantified using Multi Gauge software (FujiFilm Corp. Life Science Division, Tokyo, Japan). The optical density of COX-2 was normalized to the optical density of the internal loading control, α tubulin. Results were calculated and expressed as a percent of the non-stressed control group.
2.6 HPLC Detection of 5-HT and DA
Whole striatum and hippocampus were collected from rats killed via live decapitation, 7 days after exposure to Meth or saline, and rapidly frozen on dry ice. Tissue was stored at −80°C until use. Tissue was sonicated in 0.25 N perchloric acid and centrifuged at 14,000 x g for 20 min at 4°C to pellet insoluble proteins. Supernatants were used for detection of DA and 5-HT and pellets were used for protein quantification.
Twenty μL of each sample was injected onto a Varian Microsorb-MV 100, 5 μm C-18 column, 250 × 4.6 mm (Varian Inc., Lake Forest, CA) by an ESA model 542 autosampler (ESA, Chelmsford, MA). The mobile phase used for detection of monoamines consisted of 11 mM citric acid, 0.81 mM 1-octanesulfonic acid sodium salt, 75 mM sodium phosphate dibasic and 11% methanol, at pH 4.4. Electrochemical detection of DA and 5-HT was performed with an LC-4C amperometric detector (Bioanalytical Systems, Inc., Lafayette, IN) using a 6 mm glassy working electrode maintained at a potential of 0.7 V relative to an Ag-AgCl reference electrode. Data was collected and analyzed using EZ Chrome software (Scientific Software, Pleasanton, CA).
Pellets were resuspended in 1 N NaOH to solubilize proteins and proteins were quantified using the Bradford Assay (BioRad, Hercules, CA). DA and 5-HT HPLC values were normalized to protein levels and tissue content was presented as pg of DA or 5-HT per μg of protein.
2.7 Statistical Analysis
The effects of CUS on COX-2 immunoreactivity were determined by a t-test. Body temperatures were analyzed with a two-way analysis of variance (ANOVA) with repeated measures, followed by Tukey’s post hoc analyses. A three-way ANOVA was used to compare the effects of Meth or Saline, Stress or No Stress and ketoprofen or transcutol on measures of DA and 5-HT tissue content. When a significant main effect of Meth within a three-way ANOVA was evident, post hoc two-way ANOVAs with Tukey’s post hoc analysis, within Meth treated groups were used to identify effects of stress and ketoprofen for DA and 5-HT tissue content. In order to compare the effects of SC-51089 pretreatment on Stress+Meth-induced DA and 5-HT depletions, two-way ANOVAs were used. For all experiments, statistical significance was set at p<0.05.
3. RESULTS
3.1 Effects of CUS on striatal and hippocampal COX-2 protein expression
The effects of 10 days of CUS on COX-2 protein expression are illustrated in Figure 1. Western blot detection of COX-2 produced a dense immunoreactive doublet band at 75 kDa, as illustrated in Figure 1C & 1D. Stress significantly increased COX-2 immunoreactivity by 77±30% and 32±13% in the hippocampus (p<0.05) (Fig 1A) and striatum (p<0.05) (Fig 1B), respectively.
Figure 1.

Effects of CUS on COX-2 protein expression. Rats were exposed to 10 days of CUS or daily handling. On the day after the last stressor, A) hippocampal COX-2 and B) striatal COX-2 immunoreactivity were quantified via Western Blot. CUS significantly increased COX-2 protein expression in the A) hippocampus (*, p<0.05) and B) striatum (*, p<0.05), compared to No Stress, as indicated by a t-test (n=5–6 for each group). Representative Western Blot images of C) hippocampal and D) striatal COX-2 (~72 kDa) and the α tubulin (50 kDa) loading control.
3.2 Effects of ketoprofen during CUS on stress and Meth-induced hyperthermia and monoamine depletions
Figure 2 illustrates the effects of stress, Meth and ketoprofen pretreatments during stress on body temperatures recorded during drug treatment. No Stress+Meth, No Stress+Ketoprofen+Meth, Stress+Meth and the Stress+Ketoprofen+Meth treatments resulted in a significant increase in temperature over time. More specifically, at the last time point, No Stress+Meth treatment and No Stress+Ketoprofen+Meth treatment produced significant 1°C and 1.5°C increases in body temperature, respectively, compared to Saline treated controls. Stress+Meth and Stress+Ketoprofen+Meth treatment produced significant increases in body temperatures by 3°C and 2.75°C, respectively, compared to saline treated controls. The body temperatures of Stress+Meth treated rats were significantly greater, by approximately 1.5°C, than that of No Stress+Meth treated rats. Furthermore, ketoprofen pretreatment during stress did not attenuate Meth or Stress+Meth-induced hyperthermia. These effects were indicated by a two-way repeated measures ANOVA, which revealed a significant main effect of treatment (F7,299 = 30.268, p<0.001) and time (F4,299 = 35.306, p<0.001) and a significant treatment by time interaction (F28,299 = 7.466, p<0.001) followed by Tukey’s post hoc tests for individual comparisons.
Figure 2.

Effects of ketoprofen pretreatment during stress on stress and Meth-induced hyperthermia. Meth (7.5 mg/kg q 2hrs, ×4 ip) or saline (1 mL/kg q 2hrs, ×4 ip) was administered to previously stressed or control rats. Some rats received ketoprofen (5 mg/kg, sc) 1 hr before each stressor or handling. Body temperatures were measured before and every hr after each Meth or saline injection (indicated by the arrows on the x-axis). Meth significantly increased body temperature over time (*, p<0.001) and prior exposure to 10 days of CUS significantly enhanced Meth-induced hyperthermia (#, p<0.001), as revealed by a two-way repeated measures ANOVA. Ketoprofen had no significant effect on stress or Meth-induced hyperthermia. (n=6–9 for each group)
The effects of ketoprofen pretreatment during stress on stress and Meth-induced hippocampal 5-HT depletions are illustrated in Figure 3A. Stress+Meth resulted in a significant 34±9% depletion in hippocampal 5-HT, compared to No Stress+Meth treatment. Stress+Ketoprofen+Meth treatment also resulted in a significant 23±8% 5-HT depletion, compared to No Stress+Meth treatment. However, there was no difference between Stress+Ketoprofen+Meth and Stress+Meth treatments. These effects were indicated by a three-way ANOVA with a significant main effect of Meth (F1,36 = 11.600, p<0.01) and main effect of stress (F1,36 = 5.124, p<0.05). To determine the effects of stress and ketoprofen within the Meth treated rats, a two-way ANOVA, within the Meth treated groups, indicated a significant main effect of stress (F1,17 = 9.544, p<0.01), but no interaction between stress and ketoprofen.
Figure 3.

Effect of ketoprofen pretreatment during stress on stress and Meth-induced hippocampal and striatal monoamine depletions, 7 days after treatment. Meth (7.5 mg/kg q 2hrs, ×4 ip) or saline (1 mL/kg q 2hrs, ×4 ip) was administered to previously stressed or control rats. Some rats received ketoprofen (5 mg/kg, sc) 1 hr before each stressor or handling. A) Hippocampal 5-HT: Stress+Meth produced a significant hippocampal 5-HT depletion (#, p<0.05, Tukey post hoc test) compared to No Stress+Meth treatments. Ketoprofen pretreatment did not attenuate Stress+Meth-induced hippocampal 5-HT depletions. Striatal B) DA & C) 5-HT: Three-way ANOVA revealed a significant effect of Meth on DA and 5-HT content compared to No Stress+Saline groups (*, p<0.001). Stress+Meth produced a significant DA and 5-HT depletion (#, DA: p<0.001; 5-HT: p<0.001, 2-way ANOVA and Tukey’s post hoc test) compared to No Stress+Meth. Ketoprofen pretreatment attenuated Stress+Meth-induced striatal DA and 5-HT depletions (&, DA: p<0.01; 5-HT: p<0.01, Tukey’s post hoc test). (n=6–12 for each group)
Figure 3 also illustrates the effects of ketoprofen pretreatment during stress on stress and Meth-induced striatal B) DA and C) 5-HT depletions. Stress+Meth resulted in a significant 49±7% depletion in striatal DA tissue content, compared to No Stress+Meth treatment. Furthermore, Stress+Ketoprofen+Meth resulted in DA content that was significantly different from Stress+Meth treatment, but not No Stress+Meth. In addition, Stress+Meth resulted in a significant 35±5% depletion in striatal 5-HT, compared to No Stress+Meth treatment. Stress+Ketoprofen+Meth 5-HT content was significantly different from Stress+Meth treatment, but not No Stress+Meth. These effects were indicated by a three-way ANOVA with a significant main effect of Meth (F1,53 = 49.664, p <0.001) on striatal DA content (Fig 3B). In addition, a two-way ANOVA within the Meth treated groups revealed a significant main effect of stress (F1,38 = 16.534, p<0.001) and a significant interaction between stress and ketoprofen (F1,38 = 9.841, p<0.01). Tukey’s post hoc analysis revealed a significant difference between Stress+Meth and Stress+Ketoprofen+Meth (p<0.01) on striatal DA content.
For striatal 5-HT measurements (Fig 3C), a three-way ANOVA revealed a significant main effect of Meth (F1,53 = 21.300, p <0.001). A two-way ANOVA comparing the effects of stress and ketoprofen within the Meth treated groups, indicated a significant main effect of stress (F1,38 = 9.841, p<0.01) and ketoprofen (F1,38 = 9.841, p<0.01) and a significant interaction between stress and ketoprofen (F1,38 = 6.344, p<0.05). Tukey’s post hoc analyses revealed a significant difference between Stress+Ketoprofen+Meth and Stress+Meth treatment (p<0.01).
3.3 Effects of ketoprofen during Meth on stress and Meth-induced hyperthermia and monoamine depletions
Figure 4 illustrates the effects of ketoprofen pretreatments during drug treatment on stress and Meth-induced hyperthermia. No Stress+Meth, NoStress+Meth+Ketoprofen, Stress+Meth and the Stress+Meth+Ketoprofen treatments resulted in an increased body temperature over time. More specifically, No Stress+Meth treatment and No Stress+Meth+Ketoprofen treatment produced a 2°C increase in body temperature, compared to Saline treated controls. Stress+Meth and Stress+Meth+Ketoprofen significantly increased body temperatures by an additional 1°C, compared to No Stress+Meth treatments. Furthermore, ketoprofen pretreatment did not attenuate stress or Meth-induced hyperthermia. These effects were revealed by a two-way repeated measures ANOVA, which indicated a significant main effect of treatment (F7,284 = 76.628, p<0.001) and time (F4,284 = 25.835, p<0.001) and a significant treatment by time interaction (F28,284 = 12.927, p<0.001). Tukey’s post-hoc analyses indicated significant differences between the individual treatment groups that were mentioned previously.
Figure 4.

Effects of ketoprofen pretreatment during drug treatment on stress and Meth-induced hyperthermia. Meth (7.5 mg/kg q 2hrs, ×4 ip) or saline (1 mL/kg q 2hrs, ×4 ip) was administered to previously stressed or control rats. Some rats received ketoprofen (5 mg/kg, sc) 1 hr before each Meth or saline injection. Body temperatures were measured before and every hr after a Meth or saline injection. Larger arrows on the x-axis indicate the time of Meth or saline injections, while the smaller arrows indicate ketoprofen or vehicle injections. Meth significantly increased body temperature over time (*, p<0.001, two-way RM ANOVA) and prior exposure to 10 days of CUS significantly enhanced Meth-induced hyperthermia (#, p<0.001, Tukey’s post hoc test). Ketoprofen pretreatment had no significant effect on stress or Meth-induced hyperthermia. (n=6–10 for each group)
The effect of ketoprofen pretreatment administered during drug treatment on stress and Meth-induced hippocampal 5-HT depletions is illustrated in Figure 5A. Stress+Meth resulted in a significant 37±8% decrease and Stress+Meth+Ketoprofen resulted in a significant 40±13% decrease in hippocampal 5-HT, compared to No Stress+Meth treatments. Furthermore, ketoprofen had no effect on 5-HT in the hippocampus. These results were evaluated with a three-way ANOVA revealing a significant main effect of Meth (F1,40 = 15.299, p<0.001) and stress (F1,40 = 6.886, p<0.05) and a significant interaction of stress and Meth (F1,40 = 4.989, p<0.05). A two-way ANOVA comparing the effects of stress and ketoprofen pretreatment within all Meth treatments revealed a significant main effect of stress (F1,17 = 12.563, p<0.005).
Figure 5.

Effects of ketoprofen pretreatment during drug treatment on stress and Meth-induced hippocampal and striatal monoamine depletions, 7 days after treatment. Meth (7.5 mg/kg q 2hrs, ×4 ip) or saline (1 mL/kg q 2hrs, ×4 ip) was administered to previously stressed or control rats. Some rats received ketoprofen (5 mg/kg, sc) 1 hr before each Meth or saline injection. A) Hippocampal 5-HT: Stress+Meth produced a significant hippocampal 5-HT depletion (#, p<0.005, Tukey’s post hoc test) compared to No Stress+Meth treatments. Ketoprofen pretreatment did not significantly attenuate Stress+Meth-induced hippocampal 5-HT depletions. Striatal B) DA and C) 5-HT: Three-way ANOVA revealed a significant effect of Meth on DA and 5-HT content compared to No Stress+Saline groups (*, p<0.001). Stress+Meth produced a significant DA and 5-HT depletion (#, DA: p<0.01; 5-HT: p<0.05, two-way ANOVA and Tukey post hoc test) compared to No Stress+Meth. Ketoprofen pretreatment attenuated the Stress+Meth-induced DA and 5-HT depletions (&, DA: p<0.005; 5-HT: p<0.005, Tukey’s post hoc test). (n=6–10 for each group)
Figure 5 illustrates the effects of ketoprofen pretreatment during stress on stress and Meth-induced striatal B) DA and C) 5-HT depletions. Stress+Meth resulted in a significant 39±7% depletion in striatal DA tissue content (Fig 5B), compared to No Stress+Meth treatment. The Stress+Meth-induced decreases in striatal DA content were attenuated in the Stress+Meth+Ketoprofen treatment group. In addition, Stress+Meth resulted in a significant 25±5% decrease in striatal 5-HT content (Fig 5C), compared to No Stress+Meth treated groups. Stress+Meth+Ketoprofen treatment resulted in striatal 5-HT content that was significantly different from Stress+Meth, but not No Stress+Meth. For the striatal DA measures, a three-way ANOVA indicated a significant main effect of Meth (F1,72 = 64.055, p <0.001). A two-way ANOVA within the Meth treated groups revealed a significant interaction between stress and ketoprofen (F1,36 = 5.075, p<0.05). Tukey’s post hoc analysis revealed that the Stress+Meth+Ketoprofen treatment group was different than Stress+Meth (p<0.005).
For striatal 5-HT measures, the effects were tested by a three-way ANOVA that indicated a significant main effect of Meth (F1,72 = 32.698, p <0.001). A two-way ANOVA comparing the effects of stress and ketoprofen within the Meth treated groups showed a significant interaction of stress and ketoprofen (F1,36 = 4.495, p<0.05). Tukey’s post hoc analyses indicated that the significant difference between Stress+Meth and Stress+Meth+Ketoprofen (p<0.005).
3.4 Effects of ketoprofen during stress and during Meth on Stress+Meth-induced hyperthermia and monoamine depletions
Ketoprofen administered during stress and Meth did not afford any additional protection against Stress+Meth-induced 5-HT or DA depletions in the hippocampus or striatum (data not illustrated).
3.5 Effects of the EP1R antagonist, SC-51089, during Meth on stress+Meth-induced hyperthermia and monoamine depletions
Figure 6 illustrates the effects of the EP1 receptor antagonist, SC-51089, on stress and Meth-induced hyperthermia. Stress+Meth, Stress+Meth+5 μg/kgSC-51089, Stress+Meth+10 μg/kgSC-51089 and Stress+Meth+20 μg/kgSC-51089 treatments resulted in an increased body temperature over time, compared to the No Stress+Saline treated groups. Furthermore, SC-51089 pretreatment did not have any significant effect on body temperature in any treatment group. These effects were indicated by a two-way repeated measures ANOVA, which revealed a significant main effect of treatment (F7,224 = 52.860, p<0.001) and time (F4,224 = 37.486, p<0.001) and a significant interaction between treatment and time (F28,224 = 7.644, p<0.001). Tukey’s post-hoc analyses indicated the individual differences between Stress+Meth and the No Stress+Saline treated groups as well as the lack of effect of SC-51089 at each time point.
Figure 6.

Effects of the EP1 receptor antagonist, SC-51089, pretreatment during drug treatment on stress and Meth-induced hyperthermia. Meth (7.5 mg/kg q 2hrs, ×4 ip) or saline (1 mL/kg q 2hrs, ×4 ip) was administered to previously stressed or control rats. Some rats received SC-51089 (5, 10 or 20 μg/kg, ip) 1 hr before each Meth or saline injection. Body temperatures were measured before and every hr after a Meth or saline injection. Larger arrows on the x-axis indicate the time of Meth or saline injections, while the smaller arrows indicate SC-51089 or vehicle injections. Stress+Meth significantly increased body temperature over time (*, p<0.001, two-way RM ANOVA). Pretreatment with SC-51089, at any dose, did not alter No Stress+Saline or Stress+Meth-induced hyperthermia. (n=5–7 for each group)
Figure 7A illustrates the effects of the EP1R antagonist, SC-51089, on Stress+Meth-induced hippocampal 5-HT depletions. Stress+Meth treated groups exhibited a depletion of 5-HT compared to No Stress+Saline controls. In addition, 5-HT tissue content did not differ between the Stress+Meth and Stress+Meth+SC-51089 treatment groups. These effects were revealed by a two-way ANOVA, which indicated a significant effect of Stress+Meth (F1,43 = 40.666, p<0.001), but no significant interaction between Stress+Meth and SC-51089 treatments.
Figure 7.
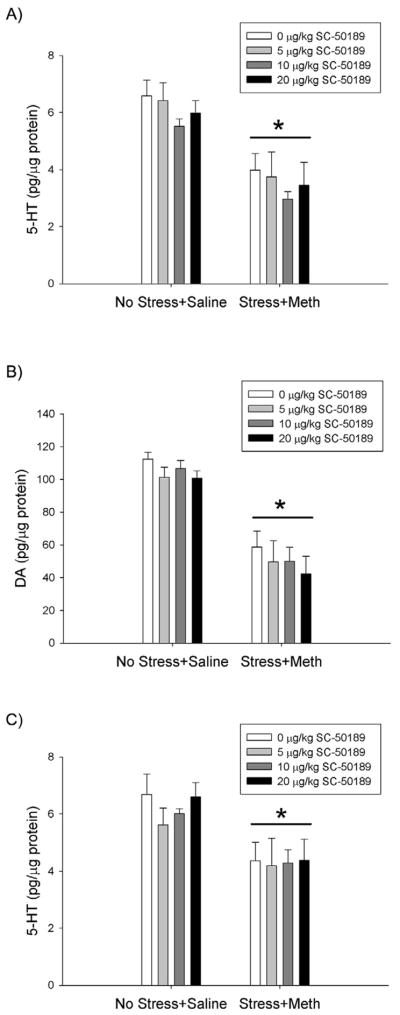
Effects of EP1R antagonist, SC-51089, on Stress+Meth-induced hippocampal and striatal monoamine depletions, 7 days after treatment. Meth (7.5 mg/kg q 2hrs, ×4 ip) or saline (1 mL/kg q 2hrs, ×4 ip) was administered to previously stressed or control rats. Some rats received the EP1R antagonist, SC-51089 (5, 10 or 20 μg/kg, ip) 1 hr before each Meth or saline injection. Stress+Meth treatment resulted in a significant depletion of A) hippocampal 5-HT, B) striatal DA and C) striatal 5-HT (*, p<0.001, two-way ANOVA) compared to No Stress+Saline treatments. SC-50189 treatments did not attenuate Stress+Meth-induced monoamine depletions. (n=5–7 for each group)
Figure 7 also illustrates the effects of the EP1R antagonist, SC-51089, on Stress+Meth-induced striatal B) DA and C) 5-HT depletions. Stress+Meth treated groups resulted in significant striatal DA and 5-HT depletions, compared to No Stress+Saline treated groups. Furthermore, SC-51089 pretreatment did not attenuate the effects of Stress+Meth on striatal DA or 5-HT. The effects of treatments on striatal DA and 5-HT tissue content were indicated by a two-way ANOVA comparing treatment (No Stress+Saline vs Stress+Meth) and SC-51089 pretreatment doses (0, 5, 10 and 20 μg/kg), which revealed a significant main effect of Stress+Meth treatment, compared to No Stress+Saline controls (DA: F1,43 = 89.295, p<0.001; 5-HT: F1,43 = 18.481, p<0.001), but no significant interaction between Stress+Meth treatment and SC-51089 pretreatments.
4. DISCUSSION
The role of COX activity in striatal and hippocampal monoaminergic depletions produced by serial exposure to stress and Meth was investigated. CUS exposure increased COX-2 protein in the hippocampus and striatum. In addition, COX activity mediated the enhanced striatal dopaminergic and serotonergic depletions, but not the enhanced hippocampal serotonergic depletions, resulting from serial exposure to stress and Meth, as ketoprofen pretreatment afforded protection in the striatum but not the hippocampus. Furthermore, COX-mediated damage was independent of hyperthermia and EP1 receptor activation.
Chronic stress increased COX-2 protein expression in the hippocampus and striatum (Fig 1). The upregulation of COX-2 protein is a likely consequence of stress-induced increases in oxidative stress (Madrigal et al., 2001) and pro-inflammatory cytokines (Glaser and Kiecolt-Glaser, 2005; Zhou et al., 1993), which in turn, can induce COX-2 transcription through activation of nuclear factor kappa B (NFκB) (Gloire et al., 2006; Laflamme et al., 1999; Nadjar et al., 2005). The new finding that the CUS paradigm increases COX-2 protein is consistent with other studies showing that acute restraint stress increases COX-2 protein expression in the brain (Garcia-Bueno et al., 2008b; Madrigal et al., 2003; Yamaguchi et al., 2010). In addition, our lab has shown that chronic unpredictable stress and Meth synergize to produce a long-term increase in COX-2 protein expression, observed 7 days after treatment (Northrop and Yamamoto, 2012). Furthermore, since COX plays a role in a variety of neurodegenerative disorders (Andreasson, 2010; Yang and Chen, 2008), it is plausible that stress-induced increases in COX-2 mediate the stress-induced enhancement of Meth-induced monoaminergic depletions in the hippocampus and striatum.
Inhibition of COX during stress prevented the stress-induced enhancement of dopaminergic and serotonergic depletions in the striatum, but not the hippocampus (Fig 3), after exposure to the combination of stress and Meth. Similarly, inhibition of COX during Meth prevented the stress-induced enhancement of dopaminergic and serotonergic depletions in the striatum, but not the hippocampus (Fig 5). Since hyperthermia plays a major role in the toxic effects of Meth (Bowyer et al., 1992; Xie et al., 2000), it is important to note that the protective effects of ketoprofen were independent of an attenuation of stress and Meth-induced hyperthermia. Ketoprofen pretreatment, regardless of the time of administration, did not attenuate Meth-induced hyperthermia or the enhancement of Meth-induced hyperthermia by stress (Fig 2 & Fig 4). However, while ketoprofen did not attenuate hyperthermia, there was a trend for ketoprofen to alter body temperature, especially in the NoStress+Meth treated groups. While it is surprising that ketoprofen did not attenuate Meth-induced hyperthermia, given that the products of COX activity, prostaglandins, are involved in temperature regulation (Mohaghegh et al., 1997), it is not surprising that ketoprofen treated rats may lack the ability to thermoregulate. Regardless, the results demonstrate that Meth-induced hyperthermia and the enhancement of Meth-induced hyperthermia by stress are independent of COX and prostaglandins. Furthermore, the enhancement of NoStress+Meth-induced hyperthermia by ketoprofen (Fig 2) without a concomitant increase in monoamine depletions in the NoStress+Ketoprofen+Meth, compared to the NoStress+Meth, treated groups, indicates that the mechanisms responsible for enhancement of Meth-induced hyperthermia are different than the mechanisms responsible for enhancement of Meth-induced monoamine depletions. Moreover, the protective effects of ketoprofen on striatal monoaminergic depletions can be attributed to its ability to inhibit COX activity rather than the attenuation of hyperthermia.
Interestingly, inhibition of COX during both stress and Meth did not afford any additional protection (data not shown), compared to ketoprofen when administered only during stress or only during Meth. In addition, regardless of whether ketoprofen was administered during stress or during Meth, ketoprofen pretreatment had no effect on the monoaminergic depletions observed in response to No Stress+Meth. These data suggest that COX activity mediates the enhancement of Meth-induced damage to striatal monoaminergic terminals by stress, but does not mediate the neurotoxic effects of Meth alone.
The lack of a role for COX activity in monoaminergic depletions produced by Meth alone is consistent with previous studies. Pretreatment with COX-2 specific inhibitors, rofecoxib or NS-398, the COX-1 specific inhibitor, SC-560, or the non-specific COX inhibitor, ketoprofen, did not afford protection against Meth (5 mg/kg ×4 ip, q 2hr) induced striatal DA depletions (Thomas and Kuhn, 2005a). Although non-specific COX inhibitors have successfully attenuated Meth-induced glial activation, decreases in beta III tubulin and decreases in DAT protein expression, the role of COX activity in Meth-induced DA depletions were not investigated (Asanuma et al., 2003; Goncalves et al., 2010). In contrast, Meth-induced striatal DA depletions are prevented in COX-2 knockout mice (Thomas and Kuhn, 2005a) but this protection could be attributed to either the slight hypothermia observed in the COX-2 knockout mice in response to Meth or an unknown compensation that occurred in the COX-2 knockout mice during their development. Regardless, the current results show that COX activity in normal rats is not involved in the monoaminergic damage to Meth alone but does in fact mediate the potentiation of Meth-induced striatal monoaminergic depletions.
COX activity may mediate the Stress+Meth-induced damage to dopaminergic or serotonergic terminals via its prostaglandin synthase activity. COX activity results in the production of prostaglandins and subsequent activation of prostaglandin receptors (Ricciotti and FitzGerald, 2011). The prostaglandin EP1R is associated with excitotoxic damage and activation of the EP1R potentiates NMDA-induced neurotoxicity, whereas antagonism of the receptor protects neurons against NMDA-induced cell death (Kawano et al., 2006). Because Meth increases extracellular concentrations of glutamate (Nash and Yamamoto, 1992; Northrop et al., 2011) and stress enhances Meth-induced glutamate release and excitotoxicity (Raudensky and Yamamoto, 2007; Tata and Yamamoto, 2008), the effects of the EP1R antagonist, SC-51089, on Stress+Meth-induced monoaminergic depletions were studied. Contrary to our expectations, antagonism of the EP1R during Meth treatment did not attenuate hippocampal or striatal monoaminergic depletions induced by the combination of stress and Meth (Fig 7). These data suggest that EP1R activation does not play a major role in the COX- or glutamate-dependent monoaminergic damage observed in response to the combination of stress and Meth.
Another mechanism by which COX may mediate Stress+Meth-induced monoaminergic damage is through its peroxidase activity. COX has peroxidase activity which functions to reduce PGG2 to PGH2 (Smith et al., 2000); however, one adverse effect of this peroxidase activity is the enzymatic oxidation of DA and formation of DA quinones (Hastings, 1995). DA quinones are highly reactive and can react with sulfhydryl groups of cysteinyl residues of proteins resulting in neuronal damage (Hastings and Zigmond, 1994; Tse et al., 1976). By extension, a peroxidase-mediated mechanism may be responsible for COX-dependent monoaminergic damage in response to the combination of stress and Meth. Furthermore, prior exposure to stress enhances Meth-induced DA release (Matuszewich and Yamamoto, 2004a), which provides more substrate for COX-related peroxidase activity and could explain the enhanced monoaminergic damage in response to Stress+Meth, compared to Meth alone.
It is known that DA quinones and protein cysteinyl-DA are observed as early as 3 days after Meth exposure (Miyazaki et al., 2006) and may contribute to Meth-induced monoaminergic damage (LaVoie and Hastings, 1999b); however, DA or DA quinone formation alone is not responsible for the monoaminergic depletions in response to Meth alone (LaVoie and Hastings, 1999a; Metzger et al., 2000; Yuan et al., 2010), since transient DA depletions produced by α-methyl-p-tyrosine just prior to Meth only attenuated the long-term DA depletions produced by Meth when hyperthermia was prevented (Metzger et al., 2000; Yuan et al., 2010). Thus, although COX may not mediate the monoaminergic depletions produced by Meth alone, the potentiation by stress of Meth-induced monoaminergic depletions in the striatum could be mediated by COX-dependent peroxidase activity and subsequent DA quinone formation.
The hypothesis that the peroxidase activity of COX mediates the Stress+Meth-induced DA and 5-HT depletions in the striatum is consistent with the selective protection by ketoprofen in the striatum but not in the hippocampus. Since the dopaminergic innervation of the hippocampus is sparse compared to the striatum (Scatton et al., 1980), the mechanism underlying hippocampal 5-HT terminal damage is not likely due to DA quinone formation and explains why ketoprofen did not protect against the damage to the hippocampus.
The mechanisms responsible for the enhancement of Meth-induced hippocampal serotonergic damage by stress remain undefined. One possibility is the enhanced Meth-induced hyperthermic response observed in animals with prior exposure to stress. For example, stress enhances Meth and MDMA-induced hyperthermia (Johnson and Yamamoto, 2010; Matuszewich and Yamamoto, 2004a) and inhibition of the enhanced hyperthermia prevents the enhanced hippocampal 5-HT depletions in response to stress and Meth or stress and MDMA (Doyle and Yamamoto, 2010; Johnson and Yamamoto, 2010). Alternatively, stress-induced CORT may mediate the enhanced hippocampal 5-HT depletions observed in response to the combination of stress and Meth as the enhanced damage observed in response to stress and MDMA is prevented by the CORT synthesis inhibitor, metyrapone (Johnson and Yamamoto, 2009). Regardless, while previous studies have observed higher levels of inflammatory mediators, specifically COX-2, in the hippocampus, compared to other brain regions under basal conditions (unpublished findings; Yamagata et al., 1993; Breder et al., 1995), COX activity does not appear to be involved in the enhanced hippocampal 5-HT terminal damage observed in response to the combination of stress and Meth.
In conclusion, COX activity mediates the enhanced striatal, but not hippocampal, monoaminergic damage caused by serial exposure to chronic stress and Meth in a manner that is independent of hyperthermia and EP1 receptor activation. These results support the use of anti-inflammatory drugs for mitigating the neurotoxic effects associated with the combination of stress and Meth and possibly other neurodegenerative disorders that involve DA and stress.
HIGHLIGHTS.
Chronic stress increases COX-2 in the striatum and hippocampus
COX mediates stress enhancement of Meth-induced monoamine depletions in striatum
COX effects on Meth toxicity are independent of hyperthermia and the EP1 receptor
Acknowledgments
This work was supported by the National Institute of Health Grant DA07606. We would like to thank Gattefossé Corporation for their generosity in supplying us with transcutol.
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Nicole A. Northrop, Email: nicole.fazo@utoledo.edu.
Bryan K. Yamamoto, Email: bryan.yamamoto@utoledo.edu.
References
- Abe T, Kunz A, Shimamura M, Zhou P, Anrather J, Iadecola C. The neuroprotective effect of prostaglandin E2 EP1 receptor inhibition has a wide therapeutic window, is sustained in time and is not sexually dimorphic. J Cereb Blood Flow Metab. 2009;29:66–72. doi: 10.1038/jcbfm.2008.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasson K. Emerging roles of PGE2 receptors in models of neurological disease. Prostaglandins Other Lipid Mediat. 2010;91:104–112. doi: 10.1016/j.prostaglandins.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanuma M, Tsuji T, Miyazaki I, Miyoshi K, Ogawa N. Methamphetamine-induced neurotoxicity in mouse brain is attenuated by ketoprofen, a non-steroidal anti-inflammatory drug. Neurosci Lett. 2003;352:13–16. doi: 10.1016/j.neulet.2003.08.015. [DOI] [PubMed] [Google Scholar]
- Battaglia G, Fornai F, Busceti CL, Aloisi G, Cerrito F, De Blasi A, Melchiorri D, Nicoletti F. Selective blockade of mGlu5 metabotropic glutamate receptors is protective against methamphetamine neurotoxicity. J Neurosci. 2002;22:2135–2141. doi: 10.1523/JNEUROSCI.22-06-02135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger HA, Middaugh LD, Granholm AC, McGinty JF. Minocycline restores striatal tyrosine hydroxylase in GDNF heterozygous mice but not in methamphetamine-treated mice. Neurobiol Dis. 2009;33:459–466. doi: 10.1016/j.nbd.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowyer JF, Tank AW, Newport GD, Slikker W, Jr, Ali SF, Holson RR. The influence of environmental temperature on the transient effects of methamphetamine on dopamine levels and dopamine release in rat striatum. J Pharmacol Exp Ther. 1992;260:817–824. [PubMed] [Google Scholar]
- Breder CD, Dewitt D, Kraig RP. Characterization of inducible cyclooxygenase in rat brain. J Comp Neurol. 1995;355:296–315. doi: 10.1002/cne.903550208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimino PJ, Keene CD, Breyer RM, Montine KS, Montine TJ. Therapeutic targets in prostaglandin E2 signaling for neurologic disease. Curr Med Chem. 2008;15:1863–1869. doi: 10.2174/092986708785132915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pablos RM, Villaran RF, Arguelles S, Herrera AJ, Venero JL, Ayala A, Cano J, Machado A. Stress increases vulnerability to inflammation in the rat prefrontal cortex. J Neurosci. 2006;26:5709–5719. doi: 10.1523/JNEUROSCI.0802-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vito MJ, Wagner GC. Methamphetamine-induced neuronal damage: a possible role for free radicals. Neuropharmacology. 1989;28:1145–1150. doi: 10.1016/0028-3908(89)90130-5. [DOI] [PubMed] [Google Scholar]
- Doyle JR, Yamamoto BK. Serotonin 2 receptor modulation of hyperthermia, corticosterone, and hippocampal serotonin depletions following serial exposure to chronic stress and methamphetamine. Psychoneuroendocrinology. 2010;35:629–633. doi: 10.1016/j.psyneuen.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischborn SV, Soerensen J, Potschka H. Targeting the prostaglandin E2 EP1 receptor and cyclooxygenase-2 in the amygdala kindling model in mice. Epilepsy Res. 2010;91:57–65. doi: 10.1016/j.eplepsyres.2010.06.012. [DOI] [PubMed] [Google Scholar]
- Fukami G, Hashimoto K, Koike K, Okamura N, Shimizu E, Iyo M. Effect of antioxidant N-acetyl-L-cysteine on behavioral changes and neurotoxicity in rats after administration of methamphetamine. Brain Res. 2004;1016:90–95. doi: 10.1016/j.brainres.2004.04.072. [DOI] [PubMed] [Google Scholar]
- Fukumoto K, Takagi N, Yamamoto R, Moriyama Y, Takeo S, Tanonaka K. Prostanoid EP1 receptor antagonist reduces blood-brain barrier leakage after cerebral ischemia. Eur J Pharmacol. 2010;640:82–86. doi: 10.1016/j.ejphar.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Garcia-Bueno B, Caso JR, Leza JC. Stress as a neuroinflammatory condition in brain: damaging and protective mechanisms. Neurosci Biobehav Rev. 2008a;32:1136–1151. doi: 10.1016/j.neubiorev.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Garcia-Bueno B, Madrigal JL, Perez-Nievas BG, Leza JC. Stress mediators regulate brain prostaglandin synthesis and peroxisome proliferator-activated receptor-gamma activation after stress in rats. Endocrinology. 2008b;149:1969–1978. doi: 10.1210/en.2007-0482. [DOI] [PubMed] [Google Scholar]
- Giovanni A, Liang LP, Hastings TG, Zigmond MJ. Estimating hydroxyl radical content in rat brain using systemic and intraventricular salicylate: impact of methamphetamine. J Neurochem. 1995;64:1819–1825. doi: 10.1046/j.1471-4159.1995.64041819.x. [DOI] [PubMed] [Google Scholar]
- Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: implications for health. Nat Rev Immunol. 2005;5:243–251. doi: 10.1038/nri1571. [DOI] [PubMed] [Google Scholar]
- Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72:1493–1505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Goncalves J, Baptista S, Martins T, Milhazes N, Borges F, Ribeiro CF, Malva JO, Silva AP. Methamphetamine-induced neuroinflammation and neuronal dysfunction in the mice hippocampus: preventive effect of indomethacin. Eur J Neurosci. 2010;31:315–326. doi: 10.1111/j.1460-9568.2009.07059.x. [DOI] [PubMed] [Google Scholar]
- Haile CN, GrandPre T, Kosten TA. Chronic unpredictable stress, but not chronic predictable stress, enhances the sensitivity to the behavioral effects of cocaine in rats. Psychopharmacology (Berl) 2001;154:213–220. doi: 10.1007/s002130000650. [DOI] [PubMed] [Google Scholar]
- Hastings TG. Enzymatic oxidation of dopamine: the role of prostaglandin H synthase. J Neurochem. 1995;64:919–924. doi: 10.1046/j.1471-4159.1995.64020919.x. [DOI] [PubMed] [Google Scholar]
- Hastings TG, Zigmond MJ. Identification of catechol-protein conjugates in neostriatal slices incubated with [3H]dopamine: impact of ascorbic acid and glutathione. J Neurochem. 1994;63:1126–1132. doi: 10.1046/j.1471-4159.1994.63031126.x. [DOI] [PubMed] [Google Scholar]
- Johnson BN, Yamamoto BK. Chronic unpredictable stress augments +3,4-methylenedioxymethamphetamine-induced monoamine depletions: the role of corticosterone. Neuroscience. 2009;159:1233–1243. doi: 10.1016/j.neuroscience.2009.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BN, Yamamoto BK. Chronic stress enhances the corticosterone response and neurotoxicity to +3,4-methylenedioxymethamphetamine (MDMA): the role of ambient temperature. J Pharmacol Exp Ther. 2010;335:180–189. doi: 10.1124/jpet.110.171322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung BD, Shin EJ, Nguyen XK, Jin CH, Bach JH, Park SJ, Nah SY, Wie MB, Bing G, Kim HC. Potentiation of methamphetamine neurotoxicity by intrastriatal lipopolysaccharide administration. Neurochem Int. 2010;56:229–244. doi: 10.1016/j.neuint.2009.10.005. [DOI] [PubMed] [Google Scholar]
- Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, Kunz A, Cho S, Orio M, Iadecola C. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- Kita T, Shimada K, Mastunari Y, Wagner GC, Kubo K, Nakashima T. Methamphetamine-induced striatal dopamine neurotoxicity and cyclooxygenase-2 protein expression in BALB/c mice. Neuropharmacology. 2000;39:399–406. doi: 10.1016/s0028-3908(99)00175-6. [DOI] [PubMed] [Google Scholar]
- Laflamme N, Lacroix S, Rivest S. An essential role of interleukin-1beta in mediating NF-kappaB activity and COX-2 transcription in cells of the blood-brain barrier in response to a systemic and localized inflammation but not during endotoxemia. J Neurosci. 1999;19:10923–10930. doi: 10.1523/JNEUROSCI.19-24-10923.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVoie MJ, Card JP, Hastings TG. Microglial activation precedes dopamine terminal pathology in methamphetamine-induced neurotoxicity. Exp Neurol. 2004;187:47–57. doi: 10.1016/j.expneurol.2004.01.010. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999a;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVoie MJ, Hastings TG. Peroxynitrite- and nitrite-induced oxidation of dopamine: implications for nitric oxide in dopaminergic cell loss. J Neurochem. 1999b;73:2546–2554. doi: 10.1046/j.1471-4159.1999.0732546.x. [DOI] [PubMed] [Google Scholar]
- Loftis JM, Choi D, Hoffman W, Huckans MS. Methamphetamine causes persistent immune dysregulation: a cross-species, translational report. Neurotox Res. 2011;20:59–68. doi: 10.1007/s12640-010-9223-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrigal JL, Moro MA, Lizasoain I, Lorenzo P, Fernandez AP, Rodrigo J, Bosca L, Leza JC. Induction of cyclooxygenase-2 accounts for restraint stress-induced oxidative status in rat brain. Neuropsychopharmacology. 2003;28:1579–1588. doi: 10.1038/sj.npp.1300187. [DOI] [PubMed] [Google Scholar]
- Madrigal JL, Olivenza R, Moro MA, Lizasoain I, Lorenzo P, Rodrigo J, Leza JC. Glutathione depletion, lipid peroxidation and mitochondrial dysfunction are induced by chronic stress in rat brain. Neuropsychopharmacology. 2001;24:420–429. doi: 10.1016/S0893-133X(00)00208-6. [DOI] [PubMed] [Google Scholar]
- Mark KA, Soghomonian JJ, Yamamoto BK. High-dose methamphetamine acutely activates the striatonigral pathway to increase striatal glutamate and mediate long-term dopamine toxicity. J Neurosci. 2004;24:11449–11456. doi: 10.1523/JNEUROSCI.3597-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matuszewich L, Yamamoto BK. Chronic stress augments the long-term and acute effects of methamphetamine. Neuroscience. 2004a;124:637–646. doi: 10.1016/j.neuroscience.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Matuszewich L, Yamamoto BK. Effects of chronic stress on methamphetamine-induced dopamine depletions in the striatum. Ann N Y Acad Sci. 2004b;1032:312–314. doi: 10.1196/annals.1314.049. [DOI] [PubMed] [Google Scholar]
- Metzger RR, Haughey HM, Wilkins DG, Gibb JW, Hanson GR, Fleckenstein AE. Methamphetamine-induced rapid decrease in dopamine transporter function: role of dopamine and hyperthermia. J Pharmacol Exp Ther. 2000;295:1077–1085. [PubMed] [Google Scholar]
- Miyazaki I, Asanuma M, Diaz-Corrales FJ, Fukuda M, Kitaichi K, Miyoshi K, Ogawa N. Methamphetamine-induced dopaminergic neurotoxicity is regulated by quinone-formation-related molecules. FASEB J. 2006;20:571–573. doi: 10.1096/fj.05-4996fje. [DOI] [PubMed] [Google Scholar]
- Mohaghegh RA, Soulsby ME, Skinner RD, Kennedy RH. The Interaction between the Central and Peripheral Nervous Systems in Mediating the Thermic Effect of Methamphetaminea. Annals of the New York Academy of Sciences. 1997;813:197–203. doi: 10.1111/j.1749-6632.1997.tb51693.x. [DOI] [PubMed] [Google Scholar]
- Nadjar A, Tridon V, May MJ, Ghosh S, Dantzer R, Amedee T, Parnet P. NFkappaB activates in vivo the synthesis of inducible Cox-2 in the brain. J Cereb Blood Flow Metab. 2005;25:1047–1059. doi: 10.1038/sj.jcbfm.9600106. [DOI] [PubMed] [Google Scholar]
- Nair A, Bonneau RH. Stress-induced elevation of glucocorticoids increases microglia proliferation through NMDA receptor activation. J Neuroimmunol. 2006;171:72–85. doi: 10.1016/j.jneuroim.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Nash JF, Yamamoto BK. Methamphetamine neurotoxicity and striatal glutamate release: comparison to 3,4-methylenedioxymethamphetamine. Brain Res. 1992;581:237–243. doi: 10.1016/0006-8993(92)90713-j. [DOI] [PubMed] [Google Scholar]
- Northrop NA, Smith LP, Yamamoto BK, Eyerman DJ. Regulation of glutamate release by alpha7 nicotinic receptors: differential role in methamphetamine-induced damage to dopaminergic and serotonergic terminals. J Pharmacol Exp Ther. 2011;336:900–907. doi: 10.1124/jpet.110.177287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northrop NA, Yamamoto BK. Persistent neuroinflammatory effects of serial exposure to stress and methamphetamine on the blood-brain barrier. J Neuroimmune Pharmacol. 2012;7:951–968. doi: 10.1007/s11481-012-9391-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston KL, Wagner GC, Schuster CR, Seiden LS. Long-term effects of repeated methylamphetamine administration on monoamine neurons in the rhesus monkey brain. Brain Res. 1985;338:243–248. doi: 10.1016/0006-8993(85)90153-2. [DOI] [PubMed] [Google Scholar]
- Raudensky J, Yamamoto BK. Effects of chronic unpredictable stress and methamphetamine on hippocampal glutamate function. Brain Res. 2007;1135:129–135. doi: 10.1016/j.brainres.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, Moore RY. Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res. 1982;235:93–103. doi: 10.1016/0006-8993(82)90198-6. [DOI] [PubMed] [Google Scholar]
- Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scatton B, Simon H, Le Moal M, Bischoff S. Origin of dopaminergic innervation of the rat hippocampal formation. Neurosci Lett. 1980;18:125–131. doi: 10.1016/0304-3940(80)90314-6. [DOI] [PubMed] [Google Scholar]
- Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- Sonsalla PK, Riordan DE, Heikkila RE. Competitive and noncompetitive antagonists at N-methyl-D-aspartate receptors protect against methamphetamine-induced dopaminergic damage in mice. J Pharmacol Exp Ther. 1991;256:506–512. [PubMed] [Google Scholar]
- Sorrells SF, Sapolsky RM. An inflammatory review of glucocorticoid actions in the CNS. Brain Behav Immun. 2007;21:259–272. doi: 10.1016/j.bbi.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staszewski RD, Yamamoto BK. Methamphetamine-induced spectrin proteolysis in the rat striatum. J Neurochem. 2006;96:1267–1276. doi: 10.1111/j.1471-4159.2005.03618.x. [DOI] [PubMed] [Google Scholar]
- Stein-Behrens BA, Lin WJ, Sapolsky RM. Physiological elevations of glucocorticoids potentiate glutamate accumulation in the hippocampus. J Neurochem. 1994;63:596–602. doi: 10.1046/j.1471-4159.1994.63020596.x. [DOI] [PubMed] [Google Scholar]
- Stephans SE, Yamamoto BK. Methamphetamine-induced neurotoxicity: roles for glutamate and dopamine efflux. Synapse. 1994;17:203–209. doi: 10.1002/syn.890170310. [DOI] [PubMed] [Google Scholar]
- Tata DA, Raudensky J, Yamamoto BK. Augmentation of methamphetamine-induced toxicity in the rat striatum by unpredictable stress: contribution of enhanced hyperthermia. Eur J Neurosci. 2007;26:739–748. doi: 10.1111/j.1460-9568.2007.05688.x. [DOI] [PubMed] [Google Scholar]
- Tata DA, Yamamoto BK. Chronic stress enhances methamphetamine-induced extracellular glutamate and excitotoxicity in the rat striatum. Synapse. 2008;62:325–336. doi: 10.1002/syn.20497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DM, Dowgiert J, Geddes TJ, Francescutti-Verbeem D, Liu X, Kuhn DM. Microglial activation is a pharmacologically specific marker for the neurotoxic amphetamines. Neurosci Lett. 2004a;367:349–354. doi: 10.1016/j.neulet.2004.06.065. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Kuhn DM. Cyclooxygenase-2 is an obligatory factor in methamphetamine-induced neurotoxicity. J Pharmacol Exp Ther. 2005a;313:870–876. doi: 10.1124/jpet.104.080242. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Kuhn DM. MK-801 and dextromethorphan block microglial activation and protect against methamphetamine-induced neurotoxicity. Brain Res. 2005b;1050:190–198. doi: 10.1016/j.brainres.2005.05.049. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Walker PD, Benjamins JA, Geddes TJ, Kuhn DM. Methamphetamine neurotoxicity in dopamine nerve endings of the striatum is associated with microglial activation. J Pharmacol Exp Ther. 2004b;311:1–7. doi: 10.1124/jpet.104.070961. [DOI] [PubMed] [Google Scholar]
- Tse DC, McCreery RL, Adams RN. Potential oxidative pathways of brain catecholamines. J Med Chem. 1976;19:37–40. doi: 10.1021/jm00223a008. [DOI] [PubMed] [Google Scholar]
- Wagner GC, Carelli RM, Jarvis MF. Pretreatment with ascorbic acid attenuates the neurotoxic effects of methamphetamine in rats. Res Commun Chem Pathol Pharmacol. 1985;47:221–228. [PubMed] [Google Scholar]
- Wagner GC, Ricaurte GA, Seiden LS, Schuster CR, Miller RJ, Westley J. Long-lasting depletions of striatal dopamine and loss of dopamine uptake sites following repeated administration of methamphetamine. Brain Res. 1980;181:151–160. doi: 10.1016/0006-8993(80)91265-2. [DOI] [PubMed] [Google Scholar]
- Woolverton WL, Ricaurte GA, Forno LS, Seiden LS. Long-term effects of chronic methamphetamine administration in rhesus monkeys. Brain Res. 1989;486:73–78. doi: 10.1016/0006-8993(89)91279-1. [DOI] [PubMed] [Google Scholar]
- Xie T, McCann UD, Kim S, Yuan J, Ricaurte GA. Effect of temperature on dopamine transporter function and intracellular accumulation of methamphetamine: implications for methamphetamine-induced dopaminergic neurotoxicity. J Neurosci. 2000;20:7838–7845. doi: 10.1523/JNEUROSCI.20-20-07838.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- Yamaguchi N, Ogawa S, Okada S. Cyclooxygenase and nitric oxide synthase in the presympathetic neurons in the paraventricular hypothalamic nucleus are involved in restraint stress-induced sympathetic activation in rats. Neuroscience. 2010;170:773–781. doi: 10.1016/j.neuroscience.2010.07.051. [DOI] [PubMed] [Google Scholar]
- Yamamoto BK, Zhu W. The effects of methamphetamine on the production of free radicals and oxidative stress. J Pharmacol Exp Ther. 1998;287:107–114. [PubMed] [Google Scholar]
- Yang H, Chen C. Cyclooxygenase-2 in synaptic signaling. Curr Pharm Des. 2008;14:1443–1451. doi: 10.2174/138161208784480144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Darvas M, Sotak B, Hatzidimitriou G, McCann UD, Palmiter RD, Ricaurte GA. Dopamine is not essential for the development of methamphetamine-induced neurotoxicity. J Neurochem. 2010;114:1135–1142. doi: 10.1111/j.1471-4159.2010.06839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Dong F, Mayer GE, Bruch DC, Ren J, Culver B. Selective inhibition of cyclooxygenase-2 exacerbates methamphetamine-induced dopamine depletion in the striatum in rats. Neuroscience. 2007;150:950–958. doi: 10.1016/j.neuroscience.2007.09.059. [DOI] [PubMed] [Google Scholar]
- Zhou D, Kusnecov AW, Shurin MR, DePaoli M, Rabin BS. Exposure to physical and psychological stressors elevates plasma interleukin 6: relationship to the activation of hypothalamic-pituitary-adrenal axis. Endocrinology. 1993;133:2523–2530. doi: 10.1210/endo.133.6.8243274. [DOI] [PubMed] [Google Scholar]