

Abstract
Tauopathies are a group of neurological disorders characterized by the presence of intraneuronal hyperphosphorylated and filamentous tau. Mutations in the tau gene have been found in kindred with tauopathy. The expression of the human tau mutant in transgenic mice induced neurodegeneration, indicating that tau plays a central pathological role. However, the molecular mechanism leading to tau-mediated neurodegeneration is poorly understood. To gain insights into the role that tau plays in neurodegeneration, human tau proteins were immunoprecipitated from brain lysates of the tauopathy mouse model JNPL3, which develops neurodegeneration in age-dependent manner. In the present work, a novel EF-hand domain-containing protein was found associated with tau proteins in brain lysate of 12-month-old JNPL3 mice. The association between tau proteins and the novel identified protein appears to be induced by the neurodegeneration process as these two proteins were not found associated in young JNPL3 mice. Consistently, the novel protein co-purified with the pathological sarkosyl insoluble tau in terminally ill JNPL3 mice. Calcium-binding assays demonstrated that this protein binds calcium effectively. Finally, the association between tau and the novel calcium-binding protein is conserved in human and enriched in Alzheimer's disease brain. Taken together, the identification of a novel calcium-binding protein associated with tau protein in terminally ill tauopathy mouse model and its confirmation in human brain lysate suggests that this association may play an important physiological and/or pathological role.
Keywords: Alzheimer's disease, calcium, EF-hand domain, tau, tauopathy
Alzheimer's disease (AD) is the most known and studied member of a group of neurological disorders collectively referred to as tauopathies (Lee et al. 2001; Hernandez and Avila 2007). Tauopathies are characterized by the aggregation of hyperphosphorylated and filamentous tau proteins in an ultrastructure known as neurofibrillary tangles (Ballatore et al. 2007; Hernandez and Avila 2007). This family of neurological disorders also include frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), Pick's disease, progressive supranuclear palsy (PSP), and corticobasal degeneration. AD is also characterized by the neuropathological accumulation of senile plaques composed of β-amyloid (Aβ) peptides (Hardy 2006). However, senile plaques are not detected in other members of the tauopathy family (Ballatore et al. 2007). Recent studies demonstrated that the expression and aberrant modifications of tau proteins mediate the process of neurodegeneration, indicating that tau proteins play a central pathological role in tauopathies (Berger et al. 2007; Roberson et al. 2007).
The molecular events leading to tau-mediated neurodegeneration are still poorly understood. The dynamics of tau phosphorylation during the process of neurodegeneration has been intensively studied (Ballatore et al. 2007). Kinases, such as glycogen synthase kinase 3-β and cyclin-dependent kinase 5, promote the formation of tau aggregates (Hanger et al. 1992; Sengupta et al. 1997; Lee and Tsai 2003). Furthermore, deactivation of phosphatases led to accumulation of hyperphosphorylated tau proteins (Wang et al. 2007). Unfortunately, over-expression of these proteins in transgenic animals has not provided definite evidence as to their specific role in tau-mediated neurodegeneration (Ahlijanian et al. 2000; Lucas et al. 2001; Cruz et al. 2003). Other studies aimed to identify tau-associated proteins revealed the association of heat-shock protein (Hsp70) and Hsp90 with tau proteins. This association increases tau affinity for micro-tubules (Dou et al. 2003). However, the proteins heat shock cognate 70 (Hsc70), Hsp70, and Hsp90 have been also found associated with the E3 ubiquitin ligase C-terminal Hsp70 interacting protein (Petrucelli et al. 2004; Shimura et al. 2004; Dickey et al. 2007). These protein complexes bind hyperphosphorylated tau and mediate its ubiquitinylation and degradation, suggesting that these proteins play a neuroprotective role (Dickey et al. 2006). In contrast, a recent study demonstrated that tau induces the formation of actin bundles in vitro and in vivo (Fulga et al. 2007). Funga et al. suggested that the association between tau and actin could play an important role in neurodegeneration. The molecular mechanism as to how actin and tau association may mediate neuronal toxicity is still unknown. Nevertheless, the association of tau with different proteins suggests that the formation of these protein–protein complexes may be induced by physiological or pathological cues.
In the present work, we identified a novel tau-associated protein in the JNPL3 tauopathy mouse model (Lewis et al. 2000). This novel protein was first identified in CD8 lymphocytes and its sequence revealed the presence of two EF-hand calcium-binding domains (Nelson et al. 2002; Vuadens et al. 2004). This protein was named as swiprosin 1 or EF-hand domain protein 2 (EFHD2). In this report, we referred to this protein as EFHD2. The EFHD2 protein was found associated with human TauP301L in terminally ill JNPL3 mice and enriched in the sarkosyl insoluble fraction. The association between EFHD2 and tau was not found in young JNPL3 mice, suggesting that their association may be linked to the neurodegeneration process. Importantly, the association between EFHD2 and tau was validated in human and was found enriched in AD. The results established the novel calcium-binding protein EFHD2 as a tau-associated protein in terminally ill JNPL3 mice and human brains.
Materials and methods
Antibodies
A distinct EFHD2 peptide (from mouse protein sequence) was selected for polyclonal antibody production, using protein sequence and mass spectrometry (MS) analyses. The selected peptide [168SGLHVLARLSEIDVST184] sequence is conserved from mouse to human. Thus, the rabbit polyclonal antibody produced is expected to immunoreact with EFHD2 proteins from both organisms. The anti-EFHD2M (M for mouse) antibody was affinity purified using the recombinant glutathione-S-transferase fusion (GST)-EFHD2. Briefly, GST and GST-EFHD2 were conjugated to AffiGel10/15 as recommended by the manufacturer (BioRad, Hercules, CA, USA). The rabbit serum was incubated first with the GST-conjugated beads for 4 h at 4°C. The supernatant was transferred to a clean tube an incubated with GST-EFHD2 conjugated beads for 18 h at 4°C. The GST-EFHD2 beads were washed four times with 1 mL of 10 mM Tris base, pH 7.5. The antibodies were eluted with 1 mL 100 mM glycine, pH 2.5, and immediately neutralized with 250 μL 1 M Tris base, pH 8.0; 1 μg of anti-EFHD2M antibody was used for immunoprecipitation and 1 : 50 dilution for western blot analysis. A commercially available goat anti-human EFHD2 antibody was purchased (Novus Biological, Littleton, CO, USA). This antibody was raised against a synthetic peptide, representing the C-terminal part of the human EFHD2 protein (230–240 amino acid). For the competition experiment on Fig. 1d, the anti-human EFHD2 antibody (5 μg) was incubated with GST (10 μg) or GST-EFHD2 (10 μg) for 4 h. Then, western blot analysis was performed using the treated anti-human EHFD2 antibody.
Fig. 1.

Identification of a novel tau-associated protein. (a) Tau proteins were immunoprecipitated (IP) from post-nuclear brain extract of 12-month-old JNPL3 transgenic mice (JNPL3, lanes 3–4) and age-matched non-transgenic (NT, lanes 1–2) mice, using the human specific anti-Tau antibody Tau13 (lanes 2 and 4). Protein A beads (B) alone were used as negative control (lanes 1 and 3). The immunoprecipitated proteins were resolved on SDS–PAGE and visualized by silver staining. Those proteins enriched in Tau13 immunoprecipitates were identified by in-gel trypsin digestion and tandem mass spectrometry analysis. The identified proteins are pointed out. The asterisk points out to protein bands corresponding to cross-linked Tau13 antibody. (b) The EFHD2 cDNA was cloned in a bacterial expression vector and the protein was expressed as a GST-fusion protein. The immunoreactivity of the anti-EFHD2M antibody against a peptide of the mouse EFHD2 protein was tested by western blot analysis. Three different amounts of GST and GST-EFHD2 were resolved by SDS–PAGE [20 ng (lanes 1 and 4), 40 ng (lanes 2 and 5), and 80 ng (lanes 3 and 6)]. (c) The expression of EFHD2 protein in different organs of a 6-month-old NT mouse was determined by western blot analysis. The tissues used in the analysis were brain (lane 1), spinal cord (lane 2), liver (lane 3), lung (lane 4), heart (lane 5), kidney (lane 6), spleen (lane 7), and skeletal muscle (lane 8). As loading control, the nitrocellulose membrane was stained with Ponceau-S to detect differences in overall loading. (d) Brain lysate from NT mice was resolved on SDS–PAGE and western blot analysis was performed using anti-human EFHD2 antibody (anti-EFHD2). The anti-EFHD2 antibodies were pre-absorbed at 25°C with GST (lanes 1 and 2) or GST-EFHD2 (lanes 3 and 4).
Human specific anti-tau antibody Tau13 was commercially acquired (Convance, Madison, WI, USA). For immunoprecipitation, 5 μg of Tau13 were used and 1 : 100 000 for western blot analysis. Secondary antibodies used in western blot analysis were peroxidaseconjugated goat anti-rabbit (1 : 2000), goat anti-mouse (1 : 2000) antibodies, and rabbit anti-goat (1 : 2000) (Chemicon, Temecula, CA, USA).
Recombinant protein
The mouse EFHD2 cDNA was cloned into the bacterial-expression vector pGEX-KG (The RIKEN Genome Exploration Research Group Phase II Team and the FANTOM Consortium 2001; The RIKEN Genome Exploration Research Group I & II Team and the FANTOM Consortium 2002). The recombinant EFHD2 protein was expressed in Escherichia coli as GST-fusion protein. After Isopropyl β-D-1-thiogalactopyranoside-induction of the recombinant protein, bacteria cells were centrifuged at 15 000 g and resuspended in lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 2 mM EDTA, and 2 mM dithiothreitol). The bacterial cells were lysated by sonication. The cell slurry was incubated on ice for 4 h. After centrifugation at 18 000 g for 15 min at 4°C, the resulting bacterial protein extract was incubated overnight with glutathioneconjugated beads at 4°C. The beads were washed several times with lysis buffer and the purified proteins resolved in sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). The resolved proteins were visualized by coomassie staining. GST-EFHD2 was efficiently purified from the bacterial extract, using glutathione-conjugated beads and eluted from beads using lysis buffer containing 50 mM glutathione.
Calcium (45Ca) in vitro binding assay
One microgram of purified recombinant GST-EFHD2 (20 pmol) and GST (38 pmol) proteins were equilibrated in binding buffer (10 mM Tris–HCl, pH 7.5, and 100 mM KCl). To corroborate the amount of protein used in the assay, purified recombinant GST and GST-EFHD2 were resolved in a SDS–PAGE and visualized by coomassie blue staining. Beads containing the purified recombinant proteins were incubated in binding buffer containing 1 μCi 45CaCl2 with or without 10 mM EDTA and CaCl2 for 30 min at 25°C. The total reaction volume was 100 μL (10 μL beads and 90 μL binding buffer). The beads were allowed to settle down on the bottom of the tube and the radioactive supernatant was removed. The beads were washed five times with 500 μL of binding buffer. The washed beads were added to a vial containing scintillation liquid (10 mL) and the radioactivity associated with the bound proteins was determined using a Beckman Coulter scintillation counter (LS6500 Multi-Purpose Scintillation Counter, Fullerton, CA, USA). The assays were performed in triplicate on three independent experiments.
Brain protein lysate preparation
Transgenic JNPL3 and non-transgenic (NT) littermates were bred and genotyped as described previously (Lewis et al. 2000; Vega et al. 2005). This transgenic mouse model expresses the human tau isoform 0N4R with the mutation P301L (hTauP301L) commonly found in FTDP-17 (Lewis et al. 2000). To isolate sarkosyl-insoluble tau, JNPL3 brains were homogenized in Buffer A (20 mM Tris base, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM phenylmethylsulfonyl fluoride, 5 mM sodium pyrophosphate, 30 mM β-glycerol phosphate, and 30 mM sodium fluoride) and the protein extract centrifuged at high-speed (110 000 g) for 15 min. The supernatant, containing soluble non-pathological tau proteins, was removed. The proteins present in the pellet fraction were extracted in Buffer A containing high salt (800 mM NaCl) and 10% sucrose. After high speed centrifugation (110 000 g) for 15 min, the extracted proteins present in the supernatant were incubated with 1% sarkosyl at 37°C for 1 h. After centrifugation at high speed (110 000 g) for 30 min, sarkosyl-insoluble proteins were found in the pellet fraction. The isolated sarkosyl-insoluble tau protein band migrated at higher apparent molecular weight (~64 kDa) than those observed in the soluble fraction, consistent with the detection of hyperphosphorylated tau. For post-nuclear brain lysate preparation, JNPL3 (n = 6) and NT (n = 6) mice brains and human temporal cortex from normal aging (53 and 57 years of age), FTDP-17 (46 and 62 years of age), AD (57 and 62 years of age), and PSP (53 and 62 years of age) were homogenized in Buffer A. After homogenization, the protein extract was centrifuged at 16 000 g for 10 min. The supernatant was transferred to a clean tube and used for immunoprecipitation experiments and western blot analyses. The assays were repeated using brain tissue from different mice. Post-nuclear lysates from different organs of NT mice were also prepared using this procedure.
The brainstem, cerebellum, amygdala, striatum, hippocampus, cortex (includes posterior part of frontal lobe and parietal lobe), and prefrontal cortex (anterior part of the frontal lobe) were dissected from JNPL3 (n = 3) and NT (n = 3) mice brain. [The olfactory bulb was not taken into consideration in these studies.] Briefly, the brain was extracted and placed on a cold metal block. The brainstem and cerebellum were separated from the base of the cerebrum by performing a coronal section at the transverse cerebral fissure. The cerebellum and brainstem region were separated and placed on a pre-weighted centrifuge tube. A second coronal section was performed at the optic chiasm. From the two sections produced, the prefrontal cortex, striatum, amygdala, and hippocampus were dissected. The dissected brain regions were placed on a centrifuge tube and weighted. Then, these brain regions were homogenized in five volumes of Buffer A as explained above.
Immunoprecipitation and western blot analysis
The antibodies Tau13 and anti-EFHD2M were used to immunoprecipitate hTauP301L and EFHD2 proteins, respectively. Post-nuclear lysates (5 mg) from JNPL3 (n = 6), NT (n = 6), and human temporal cortex (n = 6) were incubated with Protein A-conjugated sepharose beads for 4 h at 4°C with constant agitation. The beads were allowed to settle at the bottom of the tube by gravity and the supernatant was transferred to a clean tube. Either Tau13 or anti-EFHD2M antibody was added to the pre-cleared post-nuclear lysate, followed by an overnight incubation at 4°C. Then, 40 μL of Protein A-conjugated sepharose beads were added and incubated for 4 h at 4°C. The beads were collected as described above and washed four times with Buffer A. The buffer was completely removed and 30 μL of sample buffer containing N-ethylmaleimide instead of reducing agent were added. The samples were not boiled. For western blot analysis, immunoprecipitates were resolved in either a 10% or 12% SDS–PAGE, and 12% SDS–PAGE was used to obtain a better resolution of the EFHD2 species. As a result of the difference in titter between Tau13 and anti-EFHD2M, 10 times more immunoprecipitate was loaded for western blot analysis using anti-EFHD2M antibodies than when Tau13 was used.
In-gel trypsin digestion and tandem mass spectrometry analysis
The in-gel trypsin digestion and tandem MS analysis were performed on protein bands detected by silver staining after immunoprecipitation with Tau13 antibodies. Post-nucelar brain lysate from JNPL3 (n = 3) and age-matched NT (n = 3) was used for these assays. Briefly, the protein bands detected by silver staining were sliced out from the gel, destained, and dehydrated in 100% acetonitrile (ACN). After removal of ACN by speed-vacuum, the gel slice was re-hydrated in 40 mM ammonium bicarbonate and 10% ACN. Trypsin (1 μg) was added to the samples and incubated overnight (18 h) at 37°C. The tryptic peptides were eluted from the gel slice by incubating it in a solution containing 50% ACN and 5% formic acid for 1 h at 25°C. The tryptic-peptides were loaded onto the Surveyor® HPLC system (Thermo Fisher, Waltham, MA, USA) and the peptides were eluted, using a gradient of ACN (0%–80%) in 0.2% formic acid/H2O, directly into the electro-spray ionization source. The eluted tryptic peptides were infused into a LTQ mass spectrometer (Thermo Fisher) for analysis and identification of the protein of interest. The peptides were identified by correlating MS–MS spectra with sequences from the NCBI (Bethesda, MA, USA) non-redundant protein and EST databases using SEQUEST (Thermo Fisher) database search algorithms. Only those identified peptides that pass selection filters imposed to the database search were taken into consideration for protein identification [XCorr higher than 1.5 (+1), 2.0 (+2), or 2.5 (+3); Delta Score > 0.1; 10 or more b and y ions; MS2 intensity of > 5 × 10−4, peptide probability >E × 10−2].
Results
Identification of a novel tau-associated protein in terminally ill JNPL3 mice
To uncover the identity of novel tau-associated proteins during the process of neurodegeneration, tau proteins were immunoprecipitated from brain extract of terminally ill JNPL3 mice, utilizing the human tau specific antibody Tau13 (Fig. 1a, JNPL3, lane 4). As control, brain extract from age-matched NT littermates were incubated with Tau13 antibodies (Fig. 1a, NT, lane 2). The immunoprecipitated proteins were resolved by SDS–PAGE and visualized by silver staining (Fig. 1a). In addition to the bands corresponding to the molecular weight of tau proteins (Fig. 1a, Tau), two distinct protein bands were observed enriched in tau immunoprecipitates from JNPL3 (Fig. 1a, lane 4). These protein bands correspond to a protein at ~70 kDa and another at 30 kDa (Fig. 1a, arrows). The enriched protein bands were excised from the gel, digested with trypsin, and the resulting peptides were analyzed using tandem MS (Table 1). As expected, the bands detected between 50 and 64 kDa corresponded to hTauP301L proteins (Fig. 1a, bracket Tau). The protein detected at 70 kDa was identified as Hsc70 (Fig. 1a, arrow Hsc70). Previous studies identified the chaperone protein Hsc70 as a tau-interacting protein (Shimura et al. 2004). The role of the association between Hsc70 and tau is still poorly understood. However, Shimura et al. (2004) suggested that Hsc70 and its interacting protein C-terminal Hsp70 interacting protein mediate tau ubiquitinylation and enhances cell survival.
Table 1.
Identified hTauP301L-associated proteins in terminally ill JNPL3 mice
| ID | Peptides | XCorr |
|---|---|---|
| Hsc70 | K.NQVAMNPTNTVFDAK | 4.028 |
| K.HWPFMVVNDAGRPK | 4.378 | |
| R.RFDDAVVQSDMK | 4.285 | |
| EFHD2 (NP_080270) | K.SMIQEVDEDFDSK | 5.043 |
| R.ADLNQGIGEPQSPSR | 4.107 | |
| R.FEEEIKAEQEER | 4.428 | |
| R.LSEIDVSTEGVK | 4.169 | |
| R.AAAGELQEDSGLHVLAR | 4.695 |
EFHD2, EF-hand domain protein 2.
Interestingly, however, the protein at ~30 kDa corresponds to a novel protein, which was first cloned from mouse and identified in human CD8 lymphocytes (Vuadens et al. 2004). The presence of this novel-protein in hTauP301L immunoprecipitates was confirmed in three independent experiments (data not shown). The novel protein was first named swiprosin 1 and afterward adopted the name of EFHD2. Sequence analysis of EFHD2 protein revealed the presence of two conserved EF-hand calcium-binding domains. The EF-hand domain is found in calcium-binding proteins such as calmodulin (Nelson et al. 2002). EFHD2 is a 240 amino acid long protein with a calculated molecular weight of 26 791 Da. The EFHD2 gene is found in chromosome 4 in mice, while in humans is found in chromosome 1. EFHD2 is highly conserved across species from Drosophila to human. The mouse EFHD2 protein is 94% identical to its human counterpart. A recent study indicated that over-expression of EFHD2 in immature murine B-cell induced apoptosis (Avramidou et al. 2007). However, the physiological role of this novel protein is still unknown.
EFHD2 proteins were widely expressed in all tissue and brain regions studied
The cDNA of mouse EFHD2 was cloned and expressed in bacteria as a GST fusion protein. The recombinant protein was used to test the antibodies produced against this novel protein (Fig. 1b). Polyclonal antibodies against mouse EFHD2 were generated by immunizing rabbits with a peptide corresponding to residues 168–184 of the EFHD2 protein. The anti-EFHD2M antibody cross-reacted with purified recombinant GST-EFHD2 but not GST control (Fig. 1b).
The anti-EFHD2M antibody was used to determine the protein expression pattern of EFHD2 protein in different organs from NT mice (Fig. 1c). Western blot analyses revealed two anti-EFHD2M antibody cross-reacting bands in brain, spinal cord, and spleen protein lysate (Fig. 1c, lanes 1–2 and 7). The fastest migrating protein band is closed to the theoretical molecular weight of EFHD2 protein (Fig. 1c, arrow). However, the slower migrating band has an estimated molecular weight of ~30 kDa. This protein band at 30 kDa represents the band that was excised and analyzed by tandem MS. The migration of EFHD2 as two protein species has also been reported in two independent studies using protein lysate from human and murine B lymphocytes (Vuadens et al. 2004; Avramidou et al. 2007). However, to further corroborate the identity of the two EFHD2 species, a commercially available anti-EFHD2 antibody was used (Fig. 1d). The anti-EFHD2 antibody recognized the same two proteins bands detected in the mouse brain lysate by anti-EFHD2M antibody (compare Fig. 1c, lane 1 and Fig. 1d, lanes 1–2). These two cross-reacting bands were efficiently reduced by pre-absorption with recombinant GST-EFHD2 (Fig. 1d, lanes 3–4) but not GST alone (Fig. 1d, lanes 1–2), suggesting that the two protein bands detected in brain lysates represent different species of the mouse EFHD2 protein. In contrast, the lung, heart, and kidney showed only the faster migrating band observed in brain and spinal cord. Furthermore, in the liver and skeletal muscle a smaller anti-EFHD2M antibody cross-reacting band was detected (Fig. 1c, lanes 3 and 8). The two EFHD2 species may be produced by selective splicing of the EFHD2 gene transcript or due to tissue specific post-translational modifications. Nevertheless, these results demonstrate that EFHD2 was widely expressed in all tissues that were studied.
EFHD2 is a calcium-binding protein
The presence of two EF-hand domains in EFHD2 suggests that this protein could bind calcium (Nelson et al. 2002). However, the calcium-binding activity of EFHD2 has not been determined. To determine the calcium-binding capacity of EFHD2 protein, the recombinant GST-EFHD2 was purified from bacterial extract (Fig.2a, lane 2). As negative control, purified recombinant GST protein was used (Fig. 2a, lane 1). Equal protein concentration of GST and GST-EFHD2 were incubated with radioactive calcium (45CaCl2). After extensive washing, the beads were resuspended in binding buffer and transferred to a scintillation vial. The 45Ca radiation associated with the beads was detected using a scintillation counter (Fig. 2b). The results showed that the beads containing GST-EFHD2 (Fig. 2b, bar 3) retained more than a 1000-fold more radioactivity than GST (Fig. 2b, bar 2) and beads alone (Fig. 2b, bar 1). Importantly, the radioactivity associated with beads containing GST-EFHD2 was reduced to background levels by CaCl2 and EGTA (Fig. 2b, bars 4 and 5). These results indicate that EFHD2 protein has calcium-binding activity.
Fig. 2.
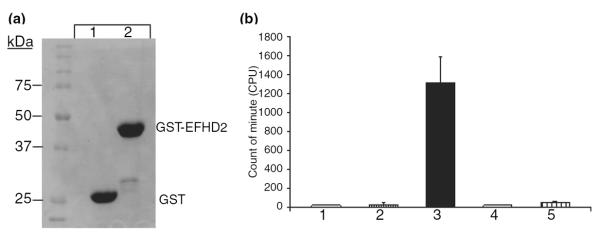
EFHD2 has calcium-binding activity. (a) The recombinant GST (lane 1) and GST-EFHD2 (lane 2) proteins were purified from bacterial extract, using glutathione-conjugated sepharose beads. The amount of protein used for the in vitro calcium-binding assay was verified by resolving the purified recombinant proteins on SDS–PAGE and visualized them by coomassie staining. (b) The same amount of recombinant proteins (1 μg) was used for the in vitro calcium-binding assay. The experiment was carried out as explained in the Materials and methods section. The radioactivity associated with the beads was determined by scintillation counter. Glutathione-conjugated sepharose beads (1) and GST bound to beads (2) were used as negative controls. GST-EFHD2 proteins bound to beads (3) showed the highest radioactivity after incubation with radioactive 45Ca and extensive washing of the beads. The associated radioactivity detected on GST-EFHD2 (3) was reduced by co-incubation with 10 mM CaCl2 (4) and EDTA (5). The bars represent the mean ± SD of three different experiments performed in triplicate.
EFHD2 preferentially co-immunoprecipitated with hTauP301L from terminally ill JNPL3 mice
To validate the results obtained from tandem MS analyses, hTauP301L proteins were immunoprecipitated from 12-month-old JNPL3 mice post-nuclear brain lysate, using the human specific anti-tau antibody Tau13 (Fig. 3a; lane 4, anti-Tau). As control, post-nuclear brain lysate from age-matched NT mice were used (Fig. 3a; lane 2, anti-Tau). Additionally, Protein A-conjugated beads alone were used as negative control to rule out non-specific protein precipitation (Fig. 3a; lanes 1, 3 and 5, anti-Tau). As expected, western blot against tau proteins demonstrated that hTauP301L proteins were efficiently immunoprecipitated from JNPL3 mice, but not NT mice post-nuclear brain lysate (Fig. 3a; compare lane 2 and 4, anti-Tau). Consistently with the tandem MS analysis, EFHD2 co-immunoprecipitated with hTauP301L proteins from old JNPL3 mice brain extracts (Fig. 3a; lane 4, anti-EFHD2M). EFHD2, however, was weakly detected in beads alone and NT immunoprecipitates (Fig. 3a; lanes 1–3). The amount of EFHD2 protein co-immunoprecipitated with tau proteins is substantially more than the one detected on beads and NT controls (Fig. 3a; compare lanes 1–3 and 4). The co-immunoprecipitated EFHD2 protein represents the higher molecular weight species present in brain extract (Fig. 1c, lane 1). The co-immunoprecipitated EFHD2 species migrates at ~30 kDa, which is the same molecular weight at which the silver stained protein band was detected (Fig. 1a). Therefore, this result validates the association between hTauP301L and endogenous EFHD2 proteins.
Fig. 3.
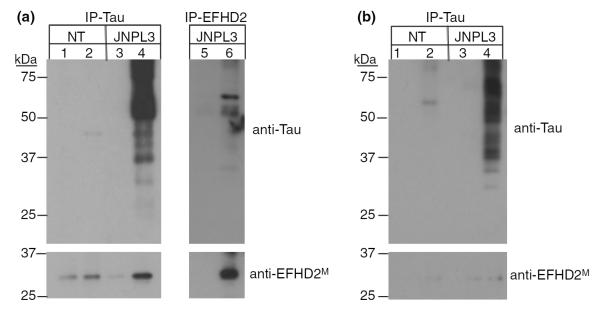
EFHD2 co-immunoprecipitated hyperphosphorylated tau. (a) The hTauP301L (IP-Tau, lane 4) and EFHD2 (IP-EFHD2, lane 6) proteins were immunoprecipitated from post-nuclear brain extract of 12-month-old JNPL3 mice. As control, post-nuclear brain extract from age-matched NT mice (lanes 1–2) and Protein A-conjugated beads alone (lanes 1, 3, and 5) were used. Additionally, EFHD2 proteins were immunoprecipitated from 12-month-old JNPL3 mice (lane 6), using anti-EFHD2M antibodies. The immunoprecipitated proteins were visualized by western blot, using the indicated antibodies. (b) hTauP301L was immunoprecipitated from 3-month-old JNPL3 mice (IP-Tau, lane 4). As control, post-nuclear brain extract from age-matched NT mice (lanes 1–2) and Protein A-conjugated beads alone (lanes 1 and 3) were also used. The immunoprecipitated proteins were visualized by western blot analysis, using the indicated antibodies.
To further validate the association between EFHD2 and hTauP301L, EFHD2 proteins were immunoprecipitated from terminally ill JNPL3 mice (Fig. 3a; lane 6). Brain lysate from terminally ill JNPL3 mice were incubated with anti-EFHD2M antibodies. The immunoprecipitated proteins were resolved on SDS–PAGE and western blot analysis was performed. The anti-EFHD2M antibody efficiently immunoprecipitated the 30 kDa EFHD2 protein species but not the lower molecular weight species (Fig. 4a; lane 6, anti-EFHD2M). This phenomenon could be explained by changes in conformation that prevent the antibody from binding to the lower molecular weight EFHD2 species presence in brain lysate. As control, beads alone were incubated with post-nuclear brain lysate from JNPL3 mice (Fig. 4a, lane 5, anti-EFHD2M). EFHD2 protein was not detected on beads alone (Fig. 4a, compare lanes 5 and 6, anti-EFHD2M). To confirm the identity of immunoprecipitated EFHD2 proteins, the anti-EFHD2M immunoprecipitated proteins were resolved by SDS–PAGE and visualized using silver staining. The band corresponding to the molecular weight of EFHD2 protein was excised and subjected to trypsin digestion and tandem MS analysis. After confronting the MS/MS spectra with the mouse protein database, the identity of EFHD2 was confirmed (data not shown).
Fig. 4.

EFHD2 co-purified with sarkosyl insoluble tau. (a) The expression of EFHD2 proteins in different brain regions of 12-month-old JNPL3 and age-matched NT mice was determined by western blot analysis, using the anti-EFHD2M antibody. The brain regions selected were brainstem (lanes 1 and 8), cerebellum (lanes 2 and 9), amygdala (lanes 3 and 10), striatum (lanes 4 and 11), hippocampus (lanes 5 and 12), cortex (lanes 6 and 13), and prefrontal cortex (lanes 7 and 14) from NT and JNPL3 mice. A non-specific cross-reacting band was used as loading control (LC). The arrows point out to the lower molecular weight EFHD2 species. (b) The intensity of the lower molecular weight EFHD2 band (arrow in a) was determined by densitometry. (c–d) Sarkosyl insoluble preparation of brain extracts from NT and JNPL3 mice at 12 months (c) and 3 months (d) of age. (c) The brain lysate from a 12-month-old JNPL3 and NT mice were fractionate. Lanes 1, 3, 5, and 7 represent the soluble fractions, while the sarkosyl insoluble fractions are in lanes 2, 4, 6, and 8. The graph represents the quantification of EFHD2 protein in the sarkosyl insoluble fraction in NT (white) and JNPL3 (black) brains. The bars represent mean ± SD of three different experiments. (d) The brain lysate from a 3-month-old JNPL3 and NT mice were fractionated. The lanes 1 and 3 represent the soluble fraction, while lanes 2 and 4 contain the sarksosyl insoluble fraction. hTauP301L and EFHD2 proteins were visualized by western blot analysis, using the indicated antibodies.
The hTauP301L proteins are heavily modified in terminally ill JNPL3 mice (Fig. 3a, lane 4). To determine the hTauP301L species associated with EFHD2 protein, the immunoprecipitated proteins with anti-EFHD2M antibody were subjected to western blot analysis using Tau13 antibodies. Consistently, hTauP301L co-immunoprecipitated with EFHD2 proteins from terminally ill JNPL3 mice post-nuclear brain lysate. The main co-immunoprecipitated hTauP301L species migrates at an apparent molecular weight of 64 kDa (Fig. 4a, lane 6, anti-Tau). This 64 kDa hTauP301L species represents the hyperphosphorylated form found in JNPL3 mice undergoing neurodegeneration and enriched in the sarkosyl insoluble fraction (Lewis et al. 2000; Sahara et al. 2002; Vega et al. 2005). In addition, hTauP301L proteins of lower molecular weight were also co-immunoprecipitated. These hTauP301L species could represent hyperphosphorylated forms or truncated fragments of the hyperphosphorylated form. Nevertheless, these results demonstrate that EFHD2 was associated with hTauP301L found in terminally ill JNPL3 mice.
The co-immunoprecipitation of EFHD2 proteins with the 64 kDa hTauP301L species in terminally ill JNPL3 mice suggest that the association between these proteins could be related to the neurodegeneration process. The 64 kDa hTauP301L species is not detected in 3-month old or younger JNPL3 mice. Therefore, EFHD2 may not be found associated with hTauP301L in young JNPL3 mice brain lysate. To corroborate this hypothesis, hTauP301L were immunoprecipitated from post-nuclear brain extracts of 3-month JNPL3 mice (Fig. 3b; lane 4, anti-Tau). EFHD2 proteins were not detected co-immunoprecipitating with hTauP301L from 3-month-old JNPL3 mice (Fig. 3b, lane 4, anti-EFHD2M). In contrast, Hsc70 co-immunoprecipitated with hTauP301L derived from both 12 and 3 months of age JNPL3 mice brain lysate, suggesting that the association between Hsc70 and hTauP301L is independent of the association between EFHD2 and hTauP301L (data not shown). These results suggest that the association between EFHD2 and hTauP301L may be induced in terminally ill JNPL3 mice.
EFHD2 co-purified with sarkosyl insoluble hTauP301L
In the tauopathy mouse model JNPL3, neurodegeneration takes place at specific brain regions (Lewis et al. 2000). Brain regions that undergo noticeable degeneration in the JNPL3 mice are brainstem, cerebellum, and hippocampus. The association of EFHD2 with hTauP301L in terminally ill JNPL3 mice might be because of differential expression of EFHD2 in brain areas affected by tau-mediated neurodegeneration. To determine the expression of EFHD2 protein, brain tissue from 12-month JNPL3 and age-matched NT mice were dissected. Post-nuclear brain lysates from each brain region were subjected to western blot analysis using anti-EFHD2M antibody. The higher molecular weight EFHD2 species was found equally expressed in all brain regions of JNPL3 and NT mice (Fig. 4a). However, the level of the lower molecular weight EFHD2 species was reduced in JNPL3 mice. In NT mice, however, both EFHD2 protein species were found in most brain regions, except hippocampus (Fig. 4a; NT, lane 5) and prefrontal cortex (Fig. 4a; NT, lane 7). Conversely, the lower molecular weight EFHD2 species was absent in most of the brain regions of the 12-month-old JNPL3 mice (Fig. 4a; lanes 8–14). The level of this lower molecular weight EFHD2 species, when added all brain regions, is almost four times more in NT than in JNPL3 mice (Fig. 4b). These results suggest that the higher molecular EFHD2 species associated with hTauP301L tends to be enriched in terminally ill JNPL3.
The hyperphosphorylated 64 kDa hTauP301L detected in post-nuclear brain lysate corresponds to the species found in the sarkosyl insoluble fraction (Lewis et al. 2000; Sahara et al. 2002; Vega et al. 2005). In humans, the detection of sarkosyl insoluble tau has been correlated with the severity of neurodegeneration (Lee et al. 2001). In JNPL3 mice, the amount of sarkosyl insoluble hTauP301L increases in an age-dependent manner and it correlates with the detection of intraneuronal hyperphosphorylated tau aggregates. The association between EFHD2 proteins and the 64 kDa hTauP301L species suggests that EFHD2 may co-purify with hTauP301L present in the sarkosyl insoluble fraction. Sarkosyl insoluble preparations were performed using brain tissue from 12-month-old JNPL3 mice and age-matched NT control (Vega et al. 2005). As previously reported, the 64 kDa hyperphosphorylated hTauP301L is enriched in the sarkosyl insoluble fraction in JNPL3 mice (Fig. 4c; lanes 6 and 8), but not in NT mice (Fig. 4c; lanes 2 and 4, anti-Tau) (Lewis et al. 2000; Sahara et al. 2002). Both EFHD2 species were found in the soluble fraction of NT mice (Fig. 4c, lanes 1 and 3). However, the lower molecular weight EFHD2 species was significantly reduced in JNPL3 mice (Fig. 4c, compare lanes 1 and 3 and 5 and 7). This result is consistent with the accumulation of the higher molecular weight EFHD2 species detected in the brain regions of 12-month-old JNPL3 mice (Fig. 4a). Consistently, the higher molecular weight EFHD2 species were enriched in the sarkosyl insoluble fraction derived from 12-month-old JNPL3 mice brain lysate (Fig. 4c; lanes 6 and 8, anti-EFHD2M) and not in age-matched NT mice (Fig. 4c; lanes 2 and 4, anti-EFHD2M). This result demonstrated that EFHD2 co-purified with sarkosyl insoluble hTauP301L in 12-month-old JNPL3 mice. Furthermore, this result indicates that EFHD2 associates with pathological-associated tau proteins, suggesting that EFHD2 may be involved in tau-mediated neurodegeneration.
To substantiate the association of EFHD2 with tau-mediated neurodegeneration and demonstrate that EFHD2 associates preferentially with hTauP301L from terminally ill JNPL3 mice, sarkosyl insoluble preparations were performed using brain tissue from 3-month-old JNPL3 (Fig. 4d; lanes 3–4) and age-matched NT control (Fig. 4d; lanes 1–2). As expected, hTauP301L proteins were not detected in the sarkosyl insoluble fraction of 3-month-old JNPL3 mice (Fig. 4d; lane 4, anti-Tau). Consistently, EFHD2 proteins were also not detected in the sarkosyl insoluble fraction (Fig. 4d; lane 4, anti-EFHD2M). These results further support the hypothesis that EFHD2 associates preferentially with hTauP301L during tau-mediated neurodegeneration.
EFHD2 and tau association in human tauopathies
As mentioned above, the mouse EFHD2 protein is 94% identical to its human counterpart. Furthermore, the mouse endogenous EFHD2 protein was found associated with the human TauP301L protein expressed in JNPL3 mice. Therefore, human EFHD2 and tau proteins should be associated in human brain lysate. The expression of human EFHD2 and its association with tau proteins was studied in human tauopathies. Protein lysate from the temporal cortex of normal aging, FTDP-17, AD, and PSP cases was analyzed by western blot, using anti-EFHD2M antibody (Fig. 5a). The anti-EFHD2M antibody detected two major protein bands in human post-nuclear protein lysate, similar to those detected in JNPL3 mouse brain lysate (Fig. 5a; compare lanes 1–4 with 5). Therefore, the result is consistent with what was observed in JNPL3 mice.
Fig. 5.

EFHD2 and tau are associated in human tauopathy. (a) The expression of EFHD2 proteins was determined in human post-nuclear brain lysate (lanes 1–4) from normal aging (N; lane 1), FTDP-17 (FTDP; lane 2), Alzheimer's disease (AD) (lane 3), and PSP (lane 4) diagnosed cases. Post-nuclear brain extract from JNPL3 mice was used as positive control (lane 5). EFHD2 was visualized by western blot analysis, using the indicated antibody. The arrow points out to the lower molecular weight EFHD2 species. (b) Human tau proteins were immunoprecipitated (IP-Tau) from normal aging (N, lane 2), FTDP-17 (FTDP, lane 4), and AD (lane 6) post-nuclear brain lysates. As negative control, Protein A-conjugated beads alone (lanes 1, 3, and 5) were used. The immunoprecipitated proteins were visualized by western blot analysis, using the indicated antibodies. (c) The presence of sarkosyl-insoluble tau from diagnosed human tauopathy cases was determined by western blot. Lanes 1, 3, and 5 represent the soluble fractions, while lanes 2, 4, and 6 represent sarkosyl-insoluble fraction. Temporal cortex from normal aging (N), FTDP-17 (FTDP), and AD diagnosed cases was used. Tau proteins were visualized by western blot, using the indicated antibody.
To validate the association of human EFHD2 with tau proteins, post-nuclear protein lysate from the temporal cortex of normal aging (Fig. 5b; lanes 1–2), FTDP-17 (Fig. 5b; lanes 3–4) and AD (Fig. 5b; lanes 5–6) were subjected to immunoprecipitation with human specific anti-Tau antibody Tau13. The immunoprecipitated proteins were resolved in SDS–PAGE and visualized by western blot analysis. As expected, human tau proteins were efficiently immunoprecipitated from all post-nuclear protein lysate (Fig. 5b; lanes 2, 4, and 6, anti-Tau). As negative control, Protein A-conjugated beads were also incubated with protein lysate (Fig. 5b; lanes 1, 3, and 5). A weak background signal corresponding to tau was detected only in AD protein lysate (Fig. 5b; lane 6, anti-Tau). Despite detecting a weak background in the beads alone control (Fig. 5b; lanes 1, 3, and 5, anti-EFHD2M), EFHD2 proteins were successfully co-immunoprecipitated with human tau proteins from all post-nuclear lysate (Fig. 5b; lanes 2, 4, and 6, anti-EFHD2M). Interestingly, the association between EFHD2 and tau was found slightly enriched in diagnosed AD cases. This AD case has a Braak and Braak stage of 6, while the FTDP-17 case corresponds to a Braak and Braak stage of 0.5. These data was confirmed by performing sarkosyl-insoluble preparation of the temporal cortex (Fig. 5c). As expected, sarkosyl insoluble tau was readily detected in the AD case and not in FTDP-17 or normal aging (Fig. 5c, compare lanes 2, 4, and 6). Therefore, the higher amount of EFHD2 co-immunoprecipitated from AD brain could be explained by the severity of neurodegeneration. A second AD case with similar Braak and Braak stage was examined and the same result was obtained (data not shown). These results validate the association between EFHD2 and human tau as observed in terminally ill JNPL3 mice. However, the relationship of the tau-EFHD2 association with the severity of neurodegeneration requires to be further investigated.
Discussion
The central role that tau proteins play in neurodegeneration is underscored by the identification of mutations found in FTDP-17 cases. Importantly, the expression of human tau mutants in transgenic mouse induces neurodegeneration. Recent studies, using human tau transgenic mice, have provided more evidence in support of the central pathological role of tau proteins in neurodegeneration. First, Le Corre et al. (2006) demonstrated that the treatment of JNPL3 mice with kinase inhibitor K252a reduces aggregated hyperphosphorylated tau. A subsequent study by Berger et al. (2007) demonstrated, in a tauopathy mouse model, that the accumulation of pathological tau species correlates with memory loss. Importantly, Roberson et al. (2007) demonstrated that reduction (or elimination) of the expression of endogenous tau ameliorates the neurological defects induced by the aggregation of Aβ peptides (plaques). Taken together, these results indicate that the process of neuronal death observed in tauopathy is induced by the aberrant expression and modification of the protein tau. However, the molecular mechanisms that mediate tau neurotoxicity are still unknown.
Several hypotheses have been generated to explain the pathobiology associated with the aggregation of tau proteins. The Amyloid-cascade hypothesis provides the basis for the current understanding of the etiology of AD. This hypothesis states that Aβ plaques are toxic to the cell (Hardy 2006; Hodges 2006). Several studies have provided convincing results in support of this hypothesis, demonstrating that Aβ plaques precede the intracellular aggregation of tau proteins (Oddo et al. 2004; Ballatore et al. 2007; Roberson et al. 2007). Immunotherapy using antibodies against Aβ peptides promoted the clearance of hyperphosphorylated tau proteins via the proteasome (Oddo et al. 2004). These results suggest that Aβ plaques could be seen as an inducer of the molecular mechanisms leading to tau-mediated neurodegeneration. However, the anti-Aβ-mediated clearance of tau pathology takes place only at early stages of the disease, suggesting that alteration or activation of other cellular processes may be involved in tau-mediated neurodegeneration. The absence of Aβ plaques in other tauopathies also supports the idea that unknown molecular processes may induce and/or mediate the neurotoxicity incited by aberrant expression or modification of tau proteins. Accordingly, research should be directed to understand the cellular and molecular changes induced during the process of tau-mediated neurodegeneration.
In this study, we showed the identification of a novel tau-associated protein. EFHD2 was found associated with hTauP301L and enriched in sarkosyl insoluble fractions derived from terminally ill JNPL3 mice. Co-immunoprecipitation of tau and EFHD2 proteins from human temporal cortex lysates validated the association. However, the association of EFHD2 with hTauP301L and its presence in sarkosyl insoluble fractions was not detected in 3-month-old JNPL3 mice. These results suggest that the association between EFHD2 and tau may be linked to the molecular mechanisms underlying tau-mediated neurodegeneration. Recently, Avramidou et al. (2007) demonstrated that EFHD2 regulates the lifespan of immature B lymphocytes. This study showed that over-expression of EFHD2 induced apoptosis through down-regulation of the antiapoptotic protein Bcl-xL. EFHD2 mediated the reduction of the Bcl-xL expression by interfering with B-cell receptor signaling and preventing the degradation of the nuclear factor-κB inhibitor. The EF-hand calcium-binding domain in EFHD2 suggests that its biological function may be regulated by calcium (Nelson et al. 2002). Here, we demonstrated that EFHD2 can bind calcium, but it is still unknown if calcium binding is required for its role in regulating apoptosis. Nevertheless, the correlation between increased intracellular calcium levels and AD suggests that EFHD2 and tau association may be induced by calcium (Huiping et al. 2006; Olivier et al. 2007). Taken together, the putative role of EFHD2 in regulating apoptosis and its association with tau proteins in terminally ill JNPL3 mice suggest that this protein may play a pathological role in neurodegeneration. Therefore, the characterization of the association between EFHD2 and tau in neurodegeneration requires further investigation.
Acknowledgements
This work was supported by NIH Grants (S06GM008102-35S1) and UPR intramural grant (8-80-109) to IEV. The authors are grateful to Dr Marla Gearing and the Emory Center for Neurodegenerative Disease Brain Bank for the brain tissue provided. The data showed here was presented as part of patent application and obtained a provisional patent on August 2007 (60/952,597) from USPTO.
Abbreviations used
- ACN
acetonitrile
- AD
Alzheimer's disease
- Aβ
amyloid-β peptide
- EFHD2
EF-hand domain protein 2
- FTDP-17
frontotemporal dementia with parkinsonism linked to chromosome 17
- GST
glutathione-S-transferase fusion
- Hsc70
heat shock cognate 70
- Hsp
heat-shock protein
- MS
mass spectrometry
- NT
non-transgenic
- PSP
progressive supranuclear palsy
- SDS-PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
References
- Ahlijanian MK, Barrezueta NX, Williams RD, et al. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc. Natl Acad. Sci. USA. 2000;97:2910–2915. doi: 10.1073/pnas.040577797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avramidou A, Kroczek C, Lang C, Schuh W, Jäck H-M, Mielenz D. The novel adaptor protein Swiprosin 1 enhances BCR signals and contributes to BCR-induced apoptosis. Cell Death Differ. 2007;14:1936–1947. doi: 10.1038/sj.cdd.4402206. [DOI] [PubMed] [Google Scholar]
- Ballatore C, Lee VM-Y, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Berger Z, Roder H, Hanna A, et al. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J. Neurosci. 2007;27:3650–3662. doi: 10.1523/JNEUROSCI.0587-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–483. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- Dickey C, Dunmore J, Lu B, et al. HSP induction mediates selective clearance of tau phosphorylated at praline-directed Ser/Thr sites but not KXGS (MARK) sites. FASEB J. 2006;20:753–755. doi: 10.1096/fj.05-5343fje. [DOI] [PubMed] [Google Scholar]
- Dickey CA, Kamal A, Lundgren K, et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Invest. 2007;117:648–658. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou F, Netzer WJ, Tanemura K, et al. Chaperones increase association of tau proteins with mircotubules. Proc. Natl Acad. Sci. USA. 2003;100:721–726. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulga TA, Elson-Schwab I, Khurana V, et al. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat. Cell Biol. 2007;9:139–148. doi: 10.1038/ncb1528. [DOI] [PubMed] [Google Scholar]
- Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH. Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: generation of paired-helical filament epitopes and neuronal localization of the kinase. Neurosci. Lett. 1992;147:58–62. doi: 10.1016/0304-3940(92)90774-2. [DOI] [PubMed] [Google Scholar]
- Hardy J. A hundred years of Alzheimer's disease research. Neuron. 2006;52:3–13. doi: 10.1016/j.neuron.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Hernandez F, Avila J. Tauopathies. Cell. Mol. Life Sci. 2007;64:2219–2233. doi: 10.1007/s00018-007-7220-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges JR. Alzheimer's centennial legacy: origins, landmarks and the current status of knowledge concerning cognitive aspects. Brain. 2006;129:2811–2822. doi: 10.1093/brain/awl275. [DOI] [PubMed] [Google Scholar]
- Huiping T, Nelson O, Bezprozvanny A, et al. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Corre S, Klafki HW, Plesnila N, et al. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc. Natl Acad. Sci. USA. 2006;103:9673–9678. doi: 10.1073/pnas.0602913103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Tsai LH. Cdk5: one of the links between senile plaques and neurofibrillary tangles? J. Alzheimers Dis. 2003;5:127–137. doi: 10.3233/jad-2003-5207. [DOI] [PubMed] [Google Scholar]
- Lee VM-Y, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Gen. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3 beta conditional transgenic mice. EMBO J. 2001;20:27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MR, Thulin E, Fagan PA, Forsen S, Chazin WJ. The EF-hand domain: a globally cooperative structural unit. Protein Sci. 2002;11:198–205. doi: 10.1110/ps.33302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Aβ immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Olivier T, Gant JC, Landfiled PW. Expansion of the calcium hypothesis of brain aging and Alzheimer's disease: minding the store. Aging Cell. 2007;6:307–317. doi: 10.1111/j.1474-9726.2007.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrucelli L, Dickson D, Kehoe K, Taylor J, et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 2004;13:703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, et al. Reducing endogenous tau ameliorates amyloid b-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Sahara N, Lewis J, DeTure M, McGowan E, Dickson DW, Hutton M, Yen SH. Assembly of tau in transgenic animals expressing P301L tau: alterations of phosphorylation and solubility. J. Neurochem. 2002;83:1498–1508. doi: 10.1046/j.1471-4159.2002.01241.x. [DOI] [PubMed] [Google Scholar]
- Sengupta A, Wu Q, Grundke-Iqbal I, Iqbal K, Singh TJ. Potentiation of GSK-3 catalyzed Alzheimer-like phosphorylation of human tau by cdk5. Mol. Cell. Biochem. 1997;167:99–105. doi: 10.1023/a:1006883924775. [DOI] [PubMed] [Google Scholar]
- Shimura H, Schwartz D, Gygi SP, Kosik KS. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J. Biol. Chem. 2004;279:4869–4876. doi: 10.1074/jbc.M305838200. [DOI] [PubMed] [Google Scholar]
- The RIKEN Genome Exploration Research Group I & II Team and the FANTOM Consortium Analysis of the mouse transcriptome base on functional annotation of 60,770 full-length cDNA. Nature. 2002;420:563–573. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- The RIKEN Genome Exploration Research Group Phase II Team and the FANTOM Consortium Functional annotation of 21,076 sequenced mouse cDNA prepared from full-length enriched libraries. Nature. 2001;409:685–690. [Google Scholar]
- Vega IE, Cui L, Props JA, et al. Increase in tau tyrosine phosphorylation correlates with the formation of tau aggregates. Mol. Brain Res. 2005;138:135–144. doi: 10.1016/j.molbrainres.2005.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuadens F, Rufer N, Kress A, Corthesy P, Schneider P, Tissot JD. Identification of swiprosin 1 in human lymphocytes. Proteomics. 2004;4:2216–2220. doi: 10.1002/pmic.200300779. [DOI] [PubMed] [Google Scholar]
- Wang JZ, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur. J. Neurosci. 2007;25:59–68. doi: 10.1111/j.1460-9568.2006.05226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]