

Abstract
Nanotheranostics, the integration of diagnostic and therapeutic function in one system using the benefits of nanotechnology, is extremely attractive for personalized medicine. Because treating cancer is not a one-size-fits-all scenario, it requires therapy to be adapted to the patient’s specific biomolecules. Personalized and precision medicine (PM) does just that. It identifies biomarkers to gain an understanding of the diagnosis and in turn treating the specific disorder based on the precise diagnosis. By predominantly utilizing the unique properties of nanoparticles to achieve biomarker identification and drug delivery, nanotheranostics can be applied to noninvasively discover and target image biomarkers and further deliver treatment based on the biomarker distribution. This is a large and hopeful role theranostics must fill. However, as described in this expert opinion, current nanotechnology-based theranostics systems engineered for PM applications are not yet sufficient. PM is an ever-growing field that will be a driving force for future discoveries in biomedicine, especially cancer theranostics. In this article, the authors dissect the requirements for successful nanotheranostics-based PM.
Keywords: chemotherapy, drug delivery, molecular imaging, molecular profiling, nanotechnology, nanotheranostics, prodrugs
Personalized, or occasionally termed precision medicine (PM), is a new trend in medicine predominantly in cancer treatment that has promise in improving healthcare before, during and after disease. It has emerged because of the recognition that no single therapeutic agent has the same effect on a large number of patients with the same diagnosis. Instead of the most common prescription, PM would personalize a treatment best suited for the individual. PM relies on the molecular understanding of the disease and more importantly the tailoring of the treatment based on the patients’ genes, proteins and metabolites. In addition, with the help of genome sequencing, PM can also help with discerning a patient’s susceptibility to a disease, which in turn could provoke monitoring and disease-prevention regimens. Therefore, diagnostic testing of key molecules involved in a disease is central to the development of PM. As the efficiency of commercial drug research and development declines [1], the new direction of PM can redirect pharmaceutical success towards patients who are molecularly identified to respond successfully to the compound. Although there is no identifiable market in the private sector for PM yet, the NIH and the US FDA have designed various NIH-supported centers and public–private partnerships to move potential candidates toward the bench-to-bedside pipeline [2]. Significant advances in biomarker discovery and theranostics have been the driving force of PM.
Theranostics, the combined efforts of diagnostic imaging and therapy in one system, fits directly into PM [3,4]. By combining molecular imaging with molecular therapy, the theranostics field could be applied in many aspects of personalized treatment, such as early detection of disease, disease staging, therapy selection, treatment planning, recognizing adverse effects at early stages of the treatment and planning follow-up therapies. An ultimate PM theranostic system for cancer could first diagnose the type of cancer class, image the heterogeneity of the tumor, apply a tailored treatment based on the diagnostic and imaging results and finally monitor the treatment efficacy. A key player in theranostics is nanotechnology. Utilizing particles at the nanoscale level provides numerous advantages in diagnostics and treatment, leading to nanosensors and nanomedicine, respectively. For example, nanosensors can measure a large variety of biomarkers in a small sample volume [5–7], and nanomedicine can deliver drugs at higher doses with lower side effects by extravasating from the blood vessels into the tumor site or through receptor-mediated active targeting [8]. Because nanotechnology is an important tool in theranostics as well as a driving focus in our group [9,10], this review will focus on the use of nanotheranostics, and the application of nanoparticles in theranostics, in PM. Although such systems are just entering the clinical field, this review will first focus on the already developed nanosystems for diagnostics and therapy, separately. A list of highlighted studies is given in Table 1. Then, the authors will propose a Five-year view on how such systems will be combined specifically for PM.
Table 1.
Recent research and clinical studies of nanotheranostics.
| Molecular profiling and biomarkers | |||
|---|---|---|---|
| Biomarker | Disease | Drug | Ref. |
| UGT1A1 | Colorectal cancer | Camptosaw™ | [22] |
| Dihydropyrimidine dehydrogenase | Breakdown the chemotherapeutics, 5-FU | [22] | |
| DLBCL | Patients with activated B-like DLBCL | Cytarabine, daunorubicin | [25] |
| ERBB2 | Breast cancer | Herceptin™ | [31] |
| KRAS | Metastatic colorectal cancer | Erbitux™ | [34] |
| Targeted imaging and therapy | |||
| Name | Imaging modality | Therapy | Ref. |
| Zevalin™ | PET | Radiotherapy by chelating with 90Y | [38] |
| QD-RGD | Optical imaging | Chemotherapy | [51] |
| IONP-RGD | MRI | Chemotherapy | [44] |
| (18F)FPP(RGD)2 | PET | Chemotherapy | [47] |
| Activatable probes | |||
| Name | Nanocarrier | Purpose | Ref. |
| Apo-nanoparticle | Hyaluronic acid nanoparticle | Apoptosis imaging | [61] |
| Multiplex nanoquencher | Silica nanoparticle | Apoptosis imaging | [62] |
| GNR-DOX-cRGD | Gold nanorod | Imaging, therapy | [66] |
| MMP2P-GNR | Gold nanorod | Imaging, therapy | [67] |
| Activatable therapy | |||
| Target | Nanocarrier | Therapy | Ref. |
| MMP | PEGylated liposome | Gene therapy | [84] |
| RGD | Magnetic nanoparticle | siRNA therapy | [85] |
| Graphene | Magnetic nanoparticle | Photothermal therapy | [92] |
5-FU: 5-fluorouracil; (18F)FPP(RGD)2: 18F-labeled dimeric arginine–glycine–aspartic acid peptide tracer; cRGD: Cylic arginine–glycine–aspartic acid; DLBCL: Diffuse large B-cell lymphoma; DOX: Doxorubicin; GNR: Gold nanorod; IONP-RGD: Arginine–glycine–aspartic acid onto iron oxide nanoparticle; MMP: Matrix metalloprotease; MMP2P-GNR: Matrix metalloprotease 2-cleavable peptide sequence and photosensitizer-conjugated gold nanorod; QD-RGD: Cylic arginine–glycine–aspartic acid peptide-labeled quantum dots; RGD: Arginine–glycine–aspartic acid.
Nanotheranostics
Nanoparticles have intrinsic properties that offer unique imaging and functionalization utility. Due to their size, nanoparticles have advantages to localize towards disease sites in vivo, especially cancer. For example, depending on the surface functionalization, nanoparticles have increased circulation time in the blood in vivo over standard chemotherapeutics. The longer circulation half-life increases the chances of nanoparticles to extravasate from tumor blood vessels and into tumor tissues, through the leaky, underdeveloped tumor vasculature. Furthermore, nanoparticles have a high surface area to volume ratio, giving it high loading capacity for imaging probes, targeting ligands and therapeutic molecules. Furthermore, many nanoparticles have intrinsic imaging properties, which can further be functionalized to become nanotheranostics. Such a multifunctional system can greatly benefit PM to screen and diagnose the particular molecular make up of highly variable diseases such as cancer and optimize treatment strategies and delivery, and monitor treatment effects. In other words, nanotheranostics can greatly increase the quality of PM. In this section, the different platforms utilized for nanotheranostics are discussed.
Metallic nanoparticles
Inorganic nanoparticles have unique size and shape-dependent properties. Due to advanced synthesis techniques [11–14], nanoparticles can be fine-tuned for molecular imaging as well as increased surface functionalization efficacy with targeting ligands and therapeutic molecules. Of the numerous metallic nanoparticles being synthesized, gold nanoparticles have the highest potential for biomedical applications because of their low toxicity and unique surface characteristics.
Gold nanoparticles
Gold nanoparticles can be synthesized with a wide range of diameters, varying aspect ratios such as gold nanorods, and unique shapes such as gold nanoshells and nanocages. The changes in shape and size of gold nanoparticles cause specific shifts in the localized surface plasmon resonance (LSPR), which accounts for gold nanoparticle optical and thermal properties useful for theranostics. When excited with a laser energy tuned to the specific LSPR, gold-based nanomaterials exhibit strong vibrational energy, which causes high temperatures useful for tissue ablation. When the gold nanoparticles are targeted to a tumor site, photothermal therapy can be applied to ablate tumor tissue, specifically in difficult to operate regions. Furthermore, LSPR bands can be adjusted towards the near infrared region (NIR) to harness the optical window of biological tissue to penetrate deep into the tissue to harness the capabilities of LSPR in vivo. In addition to therapy, gold nanoparticles also have effective fluorescent-quenching capabilities because of the LSPR that can be utilized to detect specific molecular biomarkers, as discussed in the ‘Activatable probes’ section. In addition, gold nanoparticles can be utilized as computed tomography imaging agents or radiotherapy sensitizers, because of their high atomic number and x-ray absorption coefficient. Combined with well-established surface conjugation methods, gold nanoparticles can serve as effective nanotheranostics in PM [15].
Magnetic nanoparticles
Magnetic nanoparticles can serve as nanotheranostics because they can be noninvasively imaged by MRI, and with appropriate nanoformulation can also serve as platforms for drug delivery. MRI offers deep penetration into soft tissue with high spatio-temporal resolution, but may still require exogenous contrast agents to increase detection sensitivity (single/noise ratio). There are positive and negative contrast agents that shorten the T1 and T2 proton relaxation time in tissue, and therefore increase bright or dark contrast, respectively. Metal ions such as paramagnetic gadolinium (Gd3+) and manganese (Mn2+ and Mn3+) can increase T1 relaxation time and serve as T1 contrast agents, while super-paramagnetic iron oxide can serve as building blocks for T2 contrast agents. T1 Gd3+-based (e.g., Magnevist®, Bayer Schering Pharma, Berlin, Germany) and T2 iron oxide-based (e.g., Feridex® or GastroMARK®, AMAG Pharma, MA, USA) MRI contrast agents are FDA approved. However, most iron oxide MRI contrast agents, besides GastroMARK, have been discontinued and are not offered on the US market. Iron oxide nanoparticles (IONPs) are the most widely utilized magnetic nanoparticles that have intrinsic imaging properties for T2 contrast. In addition, IONPs have the advantage of a large surface area to volume ratio and loading capabilities for various drugs and serve as an attractive nanotheranostic. However, nanoparticle formulations such as micelles or liposomes that encompass such metallic particles are increasingly more popular to research as imaging agents and drug-delivery agents for theranostic capabilities.
Polymeric nanoparticles
The most clinically utilized nanoplatform is the nonmetallic nanoparticles, and, more specifically, polymer-based nanoparticles. Current nanomedicines, but not yet nanotheranostics, that are FDA approved and currently on the market include liposomal, lipid- or albumin-encapsulated or PEGylated drugs (Table 2). PEGylation has been applied to most nanotheranostic development to decrease immunogenicity of the nanoparticle and increase blood circulation half-life in vivo. Polymeric nanoparticles, usually made of amphiphilic polymers, provide inner cores for hydrophobic molecules such as chemotherapeutics or imaging agents and an external shell for ligand conjugation to target specific disease biomarkers. Applications of polymeric nanoparticles in nanotheranostics have been reviewed elsewhere [16,17,18]. Recently, we optimized a PEGylated tumor-homing polymeric nanoparticle system, P-HA-NPs, comprised of an FDA-approvable hyaluronic acid that can deliver hydrophobic compounds to the intracellular space of cancer cells with reduced toxicity. P-HA-NPs selectively target tumors through the following two distinct mechanisms: by passively accumulating in tumors through the enhanced permeability and retention effect followed by active targeting of CD44 and an antigen overexpressed on various tumors. For targeted therapy, the anticancer drug irinotecan was encapsulated into the hydrophobic cores of nanoparticles and for the diagnostic application an NIR dye, Cy5.5, was decorated on the surface of P-HA-NPs. Following the systemic injection of nanoparticles in colon tumor-bearing mouse models, P-HA-NPs clearly visualized tumors and also effectively suppressed tumor growth (Figure 1) [19].
Table 2.
US FDA-approved nanomedicines.
| Commercial name | Nanoplatform | Drug | Indication | Company (location) |
|---|---|---|---|---|
| Abraxane® | Nanoparticulate albumin | Paclitaxel | Metastatic NSCLC; metastatic breast cancer | Celgene Corporation (NJ, USA) |
| DaunoXome® | Lipid | Daunorubicin | Advanced HIV-associated Kaposi’s sarcoma | Galen Ltd. (Craigavon, UK) |
| Doxil® | PEGylated liposome | Doxorubicin hydrochloride | Ovarian cancer | Janssen Biotech, Inc. (PA, USA) |
| Oncaspar® | PEGylated | Asparaginase | Acute lymphoblastic leukemia | Enzon Pharmaceuticals (NJ, USA) |
NSCLC: Non-small-cell lung cancer. Data taken from [18].
Figure 1. The design of PEGylated tumor-homing polymeric nanoparticle system, P-HA-NP.
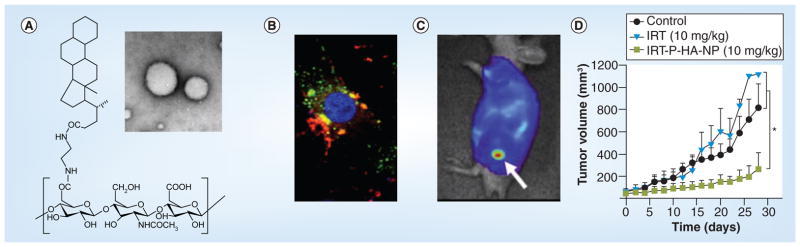
(A) Chemical structure of P-HA-NP. Inset: transmission electron microscopy image of P-HA-NP. (B) Selective tumor cell update and drug release of P-HA-NPs (green, P-HA-NP; red, drug; blue, nucleus). (C) Near-infrared optical imaging of tumor accumulation (arrow) of Cy5.5-labeled P-HA-NPs in a HT29 colon tumor-bearing mouse. (D) Therapeutic efficacy of irinotecan encapsulated polymeric nanoparticles, IRT-P-HA-NPs. *p < 0.05.
HA: Hyaluronic acid; IRT: Irinotecan; NP: Nanoparticles.
Reproduced with permission from [19].
Molecular profiling & biomarkers
Currently, theranostics has not been applied for true PM because there is a lack of knowledge regarding the type and number of biomarkers in individuals during early stages of cancer. Biomarkers are any type of biomolecule, such as proteins, lipids, genes and metabolites, which when present or altered signify important physiological conditions occurring in the body. Molecular profiling is the most common form of finding gene biomarkers, by DNA arrays, correlated with different forms of cancer using clustering algorithms [20]. In this way, specific signatures of cancer have been identified and classes of tumors can be assigned to individual patients. With the current knowledge of the human genome, detailed information about a patient’s genotype and clinical data can be used to select medication, therapy or preventative measures that are particularly suited to that patient at the time of treatment [21]. For example, doctors must perform genetic tests on patients before prescribing certain types of drugs to look for variations in specific genes that affect the patient’s metabolism and response to the drug, as in tamoxifen (Nolvadex™; AstraZeneca, London, UK) for cancer and warfarin (Coumadin™; Bristol-Myers Squibb, NY, USA) to manage blood coagulation [22]. The UGt1A1 TA repeat genotype test is used to determine variations in the UGT1A1 enzyme, which is important in the use of irinotecan (Camptosaw™; Pfizer, NY, USA), a chemotherapeutic for colorectal cancer. If the patient is deficient of this enzyme, the medication can cause severe side effects, because it will not be metabolized [22]. Another example is the genetic test for dihydropyrimdinedehydrogense deficiency to determine if the patient can break down the chemotherapeutic 5-fluorouracil. If a deficiency exists, the unmetabolized 5-fluorouracil can lead to severe or fatal buildup [22]. Such readouts for additional biomarkers could hopefully be available in the future to not only identify harmful regimens but also to guide effective ones.
In addition to identifying genes in a disease state, antibodies, peptides and aptamers can target specific protein biomarkers that have been identified and validated at the disease site. Predominantly, these biomarkers are ligands naturally expressed on many cells, but under a disease condition, they are overexpressed. Protein expression profiles of tumors using immunohistochemistry and flow cytometry and mRNA expression profiles are required for biomarker selection. Once a biomarker is validated, it can be targeted and blocked for inhibition, such as the case of numerous biologics in the clinic (e.g., Erbitux® [cetuximab]; Bristol-Myers Squibb) or targeted to deliver conjugated drugs or imaging probes to treat and monitor the specific disease site (described in more detail in the ‘Targeted imaging & therapy’ section). For example, antibody–drug conjugates (ADCs) are made up of cytotoxic drugs attached to the antibody specifically optimized with a high binding affinity to the target. When an ADC is intravenously injected, the antibody targets the biomarker that is aberrantly overexpressed and carries the conjugated drug into the cell by endocytosis. Once inside the cell, the linker is cleaved by the various proteases present in the vesicles released by the drug to perform its intended cytotoxic activity (described in more detail in the following ‘Activatable therapy’ section). Target selection for antibodies, aptamers and peptides are screened by high-throughput protein expression profiling, such as by using tissue microarrays and flow cytometry, as well as indirectly using mRNA expression profiling using microarrays, quantitative real-time PCR, cDNA hybridization and RNA in situ hybridizations [23,24]. A list of targeted biomarkers currently in clinical development for ADCs and their indications is listed in [24].
Generally, biomarkers can serve as prognostic, predictive or therapeutic response indicators. Prognostic biomarkers can predict the path of a specific class of cancer, which can help to choose appropriate therapies and doses. This is the most commonly used biomarker to date. One of the first studies using DNA arrays to find prognostic biomarkers was used to distinguish acute myeloid leukemia from acute lymphoblastic leukemia [25]. This study exemplified the role that molecular profiling can determine tumor pathology, and more importantly, the discovery of biomarkers at different tumor pathologies. For example, in another study, gene expression of diffuse large B-cell lymphoma (DLBCL) can be used to predict overall patient survival by distinguishing patients with germinal center B-like DLBCL, who have significantly better overall survival, from patients with activated B-like DLBCL [26]. This profiling revolution during the turn of the millennium led to the discovery of different genes correlated with clinical outcomes of different types of cancers such as breast [27], melanoma [28] and prostate [29]. Some of these genes could also serve as predictive biomarkers, which are used to determine the disease state that will or will not respond to certain types of treatments. When using a monoclonal antibody treatment [30], patients must be tested for their expression of the specifically targeted receptor, and the predictive biomarker, to determine therapy efficacy. Approximately 30% of breast cancer patients overexpress the ERBB2 gene, which in turn leads to overexpression of the protein human HER2 (also known as Neu and ErbB2) [31]. These specific patients can greatly benefit from trastuzumab antibody (Herceptin™; Genentech [CA, USA]/Roche [NJ, USA]) treatment, because it targets and interferes with HER2, while other patients remain unaffected [32]. Finally, therapeutic biomarkers signify the course the disease is taking after the treatment is initiated. This type of biomarker is extremely necessary in the clinic and during drug development to quickly identify whether the selected treatment course is having an effect on the tumor. Therefore, efficient assays to determine such biomarkers are required. To detect ERBB2, for example, a FISH (PathVysion™; Vysis–Abbott, IL, USA) and an immunohistochemistry (HercepTest™; Dako, Glostrup, Denmark) kit were FDA approved as in vitro diagnostics to determine whether trastuzumab treatment is beneficial to a specific patient [33]. Similarly, patients with a wild-type KRAS gene, which encodes for a protein in the EGFR pathway, are significantly more susceptible to treatment by cetuximab (Erbitux™; Eli Lilly, IN, USA), an FDA-approved antibody drug, over patients with a mutated form of the KRAS gene [34].
Most commonly, in vitro diagnostics, such as the ones mentioned earlier, require the collected tissue sample to be destroyed and mixed into one solution. In other words, in vitro diagnostics require invasive procedures while losing important morphological information of the tumor. Since most cancers are made up of numerous different cell types that can be benign, new methods in molecular profiling and diagnostics may be required. To collect important and quantitative correlations among different biomarkers and cancer phenotypes, preserved cancer tissue samples can be analyzed. However, many clinical specimens are formalin-fixed paraffin-embedded and require numerous complicated treatments to retrieve such biomarkers. Therefore, Xing and coworkers developed a quantum dot (QD) staining method to spatially localize inter- and intra-cellular biomarkers in archived tissue samples, surpassing the preservation disadvantage [35]. QDs are semiconductors that have confined electrons to a nanometer scale, which in turn increases the energy band gap, allowing the dot to have bright fluorescence after photon excitation [36,37]. The antibody-conjugated QDs, created by Xing and coworkers, could be tuned to different fluorescent emission wavelengths and were therefore utilized to probe multiple biomarkers by QD immunohistochemistry [35]. Above all, once the biomarkers are targeted by the QDs, the image is processed, and the biomarkers can be quantified. The large-scale quantification of many populations of cancer specimens can then be correlated with disease progression and aid in biomarker and therapy discovery. These correlations are vital to PM.
Another alternative to current in vitro diagnostics and biomarker discovery techniques is targeted molecular imaging, which can noninvasively localize different types of biomarkers in vivo. The imaging of multiple targeted biomarkers can aid in a deeper understanding of biomarker distribution and correlations, and eventually towards the development of effective PM. Greater insight into which biomarkers signify a disease state and which can be correlated with clinical outcomes are greatly needed. The addition of projects and policies related to identifying biomarkers in diseases in various population groups will aid in that understanding [2].
Targeted imaging & therapy
Targeted molecular imaging, a vital segment of a theranostic platform, can target, noninvasively image and track biomarkers involved in disease progression. This type of information can go beyond only identifying a genetic disorder, because it provides a whole-body scan of molecular events in real time. With targeted molecular imaging, clinicians can identify the degree of which certain biomarkers exist in the body, which is important in disease staging and treatment planning such as drug dosing and timing [38]. For effective molecular imaging, the identification of biomarkers in early stages of disease is required to specifically design imaging probes targeting genes, mRNA, DNA, metabolites, proteins and protein–protein interactions. One of the most effective techniques to study biomarkers is through nuclear medicine. Nuclear medicine can be easily applied to determine receptor binding and hence detect disease biomarkers by PET or single-photon emission computed tomography imaging [39]. Radiotherapy can then be conducted with the same molecules after changing the chelated imaging radionuclide to a therapeutic radionuclide [38]. In this way, nuclear medicine is a true theranostic PM system. Some radiotracers useful for nuclear medicine are reviewed in the study by Groves et al. [40]. For example, 16-α [18F] fluoroestradiol binds to receptors found in breast cancer that can be used to image receptor binding before and after treatment with an aromatase inhibitor, a therapy to reduce estrogen production. The FDA-approved drug for non-Hodgkin’s lymphoma, [90Y] ibritumomabtiuxetan (Zevalin™, Spectrum Pharmaceuticals, CA, USA), was initially chelated with 111In for imaging. The preliminary images are used to determine the dosimetry and biodistribution of the same compound before being used as radiotherapy by chelating with 90Y. In this way, a more personalized treatment regimen is applied to prevent harmful side effects [38].
Beyond antibodies, peptides can also be used to target specific integrins in vivo with ease of functionalization for different imaging techniques. For example, the RGD peptide targets the cell adhesion molecule, αvβ3 integrin, which is known to be overexpressed on activated endothelial and tumor cells, mainly metastatic cancer cells, but not in resting endothelial or healthy cells [41,42]. Integrin targeting in vivo has been greatly explored in our group and others by various imaging modalities such as optical imaging [43], MRI [44–46] and PET [47–50]. To study the αvβ3 integrin expression in vivo, Cai et al. labeled the RGD peptide onto fluorescent QDs that emitted at the NIR region at 705 nm, allowing in vivo optical imaging. The QD–RGD conjugate was intravenously administered into athymic nude mice bearing subcutaneous U87MG human glioblastoma tumors, the most common and lethal form of primary brain tumors [51]. The conjugated RGD targeted the tumor vasculature in vivo with high affinity to the integrin of active endothelial cells and by the QD fluorescence signal indicated αvβ3 integrin expression within the neovasculature of the tumor with high contrast. Another approach of the RGD peptide is to use it in treatment monitoring. By conjugating RGD onto iron oxide nanoparticles (IONP–RGD), one can image the tumor vasculature by MRI after an anticancer vascular-disrupting treatment [44]. IONPs are superparamagnetic nanoparticles that have been extensively used as contrast agents in T2 MRI [52–54]. T2 relaxation in MRI produces a signal reduction in the image – so-called negative contrast. To create the IONP–RGD, IONPs were firstly coated with a crosslinked poly(ethyleneglycol) (PEG) amphiphilic tri-block copolymer and then labeled with the RGD peptide [44]. Noninvasive treatment monitoring to VEGF121/rGEL therapy was accomplished using MRI based on the tumor vasculature-targeting ability of RGD and indicated successful and targeted disruption of the tumor vasculature after treatment. For PM, treatment monitoring for precise biomolecules will aid in early determination of the therapy success over standard anatomical approaches. In addition, improved quantitative measure of integrin targeting can help develop ideal targeting probe candidates, such as RGD in dimer form, for clinical use [55]. In fact, a clinical-grade 18F-labeled dimeric RGD-peptide tracer ([18F]FPP[RGD]2) has been optimized for noninvasive tumor imaging of αvβ3 expression for clinical trials using healthy human volunteers [47]. Targeted imaging and therapy continues to expand; however, proper diagnostics are required to understand which patients overexpress biomolecules and how specific patients will respond when those targets are affected. Using biopsies, molecular imaging and/or genetically identifying patients, targeted therapy will continue to become more precise for specific patients.
Activatable probes
More advanced disease targets could pursue molecular events/pathways such as proliferation, hypoxia, apoptosis, angiogenesis, inflammation and metastasis. Some optical probes developed by our group are based on targeting such molecular events. Fluorescence-activatable probes can activate a localized fluorescence signal upon cleavage of a specific substrate by the targeted biomarker peptide involved in the pathway [56–58]. Activatable probe designs are similar to molecular beacons. However, molecular beacons predominantly identify oligonucleotides only for in vitro applications, while nanoparticle-based activatable probes go beyond in vitro diagnostics and identify significant disease states in vivo [59]. One such strategy is using polymeric nanoparticles to load dye-labeled peptide substrates. For example, one of the initial probes was comprised of a NIR dye, Cy5.5, labeled on a caspase-3-cleavable peptide substrate – DEVDC conjugated to a self-assembled polymeric nanoparticle made up of a hydrophilic branched poly(ethylenimine) and an hydrophobic deoxycholic acid [60]. This causes many Cy5.5 dyes to be exposed on the surface of the nanoparticle in a self-quenched state. In the presence of caspase 3, an apoptotic marker, the substrate is hydrolyzed, the dye is released, and fluorescence is activated at approximately a tenfold increase. Yet, to increase the signal to noise ratio, a more advanced system for fluorescence quenching is required. Therefore, another polymeric nanoparticle platform was devised to boost fluorescence signal upon interaction with caspase-3 in vitro and in vivo [61]. The platform delivers dual-quenched caspase-3-sensitive fluorogenic peptides into cells, where they are then activated during apoptosis when caspase-3 is expressed. The probe consists of caspase-3-cleavable substrate GDEVEAPKGC with a NIR, Cy5.5, on one end and a dark quencher on the other end, black hole quencher-3. These probes are then conjugated on the surface of a biocompatible polymeric nanoparticle, hyaluronic acid–cholanic acid amphiphilic nanoparticle (HA–NP). The dye is quenched because of the quencher–dye interactions and dye–dye self-quenching mechanisms. This improved fluorescence quenching reduced the background fluorescence signal than previous designs, resulting in an overall clearly activated fluorescence signal in apoptotic cells. Importantly, this type of probe can be tuned to different peptide biomarkers when the nanoplatform is functionalized with a corresponding substrate. Recently, a more advanced system has been devised to image not just one caspase but multiple caspases (caspase 3, 8 and 9) to follow the complex protease signaling mechanism (Figure 2) [62]. The platform relies on a single nanoparticle that can encapsulate multiple quenchers (a nanoquencher) to produce multiplexed fluorescence signals in the presence of multiple proteases. Although the system was only tested in cells, the nanoquencher is cell penetrating, allowing it to be used in high-throughput screening of potential anticancer agents that induce apoptosis. More importantly, the particle has promise for in vivo applications to image a patient’s response to therapy in real time. Using these unique detection strategies, treatments can be tailored to patients that have a positive or negative response. Theranostic systems can be developed using similar principles or, more simply, in addition to such probes. As mentioned previously, targeted imaging is dependent on the discovery of additional biomarkers and molecular targets that provide clinically relevant information about the patient. Additional targets of interest are included in [63,64].
Figure 2. The design and function of an activatable nanoprobe that has broad-spectrum nanoquenching and multibiomarker sensing abilities.

(A) Nanoprobe design. First, the nanoquencher is engineered with a mixture of multiple quenchers and then the surface of the nanoparticle is labeled with multiple dye substrates, which are quenched. Finally, when the nanosensor comes into contact with its target proteases, fluorescence is recovered and the nanosensor is activated. (B) Multiplexed imaging of the caspase cascade in HCT116 cells at 2 h after apoptosis is initiated, as well as when apoptosis is inhibited (Inh. = with inhibitor). The colors indicate: blue (DAPI): nucleus; red (Cy5.5): caspase-3; green (FPG456): caspase-8; purple (FPG560): caspase-9.
Reproduced with permission from [62].
Another strategy is the use of inorganic nanoparticles as fluorescence activatable probes. These particles, such as iron oxide, silica, QDs and gold, have tunable imaging properties and multifunctionality, which have been extensively applied in multimodal molecular imaging probe development, such as IONPs as MRI contrast agents described in the ‘Targeted imaging and therapy’ section earlier [65]. When these metal-based activatable probes are combined with biomolecules, the in vivo functionality can also be extended towards activatable imaging and therapy. For the design of activatable probes, gold nano-particles (AuNPs) provide high quenching efficacy and therefore decrease nonspecific fluorescent background signals when fluorophores are near the AuNP surface based on the energy transfer between the fluorophore and the AuNP surface plasmon [66,67]. The deciding factors for nanometal surface energy-transfer and other energy transfer mechanisms between AuNPs and dyes are described in the study by Zhu et al. [58]. Lee et al. reported an AuNP-quenched activatable probe by conjugating a Cy5.5-labeled matrix metalloproteinase-2 (MMP-2) substrate, GPLGVRGC, onto the AuNP via a thiol–Au bond to detect MMP-2, a prognostic biomarker of cancer metastasis [68,69]. When the fluorophore-labeled substrate is bound to the AuNP, the fluorescence signal is quenched. Under in vivo conditions, the nanoparticles recover their fluorescence signal after reaching an area of MMP-2 overexpression. The fluorescence recovery was also linearly correlated to the MMP concentration, possibly providing a measurable level of cancer metastasis. Further discussion on proteases such as MMPs is included in the next section. The AuNP fluorescence activatable system allows a single nanoparticle to be used as a quencher for the dye. In addition, by changing the shape and size of AuNPs, various fluorescent dyes can be quenched to eventually signify and detect multiple proteases, similar to the nanoquencher. In terms of theranostics and PM, activatable probes are useful for in vivo imaging before or during treatment for therapy planning and monitoring. However, the markers used thus far are universally present in tumor growth, such as MMPs, or during apoptosis, such as caspases, and do not represent a precise molecule involved in the heterogeneity of the tumor. These current activatable probes can therefore be well-suited for in vivo drug screening.
Activatable therapy
Similar to activatable probes that can diagnose and detect disease states, current PM methods for treatment include ‘activated’ therapy, such as enzyme-cleavable prodrugs [62,70]. These drugs only become therapeutically active when they reach a specific biomarker in the cell. Upon transformation from an enzyme, or chemical or environmental stimuli, the parent drug is released at the area of the stimulus (Figure 3). This allows the drug to be active only at the site where the altered biomarker exists and importantly reduces toxic side effects in healthy areas of the body. Many deregulated proteolytic actions are involved in cancer [71]. Proteases are enzymes that hydrolyze peptide substrates to degrade extracellular matrices and are overexpressed during cancer progression, such as invasion and metastasis. Therefore, proteases are an important target for prodrug development [71]. MMPs are a popular target protease, because they are found abundantly in the tumor microenvironment in the extracellular and pericellular areas of the cell. MMPs are a versatile family, but one type of MMP, MMP-2, is strongly associated with the progression of various malignancies [69]. A prodrug with the MMP-2 cleavable sequence, GPLGIAGQ, and the anticancer drug, doxorubicin, is fourfold less toxic but with a higher anti-cancer effect than free doxorubicin at the same dose in vivo [72]. However, MMP targeting is not specific to one type of MMP. For example, the same cleavage site for MMP-2 is also active for MMP-9. Therefore, the addition of targeting moieties or stabilizing carriers can facilitate stability in the bloodstream and targeting specificity [73].
Figure 3. Prodrug constructs and hypothetical pathway of prodrug activation.

The development of a prodrug/activatable therapy (A–E) and the functional pathway (F) upon which the drug is activated inside the cell by the target protease. Reproduced with permission from [71].
Cathepsins are a popular target in prodrugs [74,75] because this type of protease is upregulated in several tumor tissues such as breast, liver, lung and thyroid cancer [76–78]. With a simple amino acid sequence, cathepsin overexpression can cleave the sequence intra- or extra-cellularly and release an anticancer drug conjugated to the end of the prodrug sequence. One notable example of an FDA-approved drug that targets cathepsin B is brentuximabvedotin (Adcetris®, Seattle Genetics, WA, USA) for Hodgkin’s lymphoma and systemic anaplastic large-cell lymphoma [79,101]. This is an ADC that uses an antibody to improve drug delivery and specificity in vivo, decreasing toxic side effects of the chemotherapeutic [80]. It is constructed from a chimeric anti-CD30 monoclonal antibody on one end, a valine–citrulline amino acid sequence making up the cathepsin B cleavable region, and the drug monomethylauristatin E on the opposite end. Because of its unique functionalities, Adcetris was the first FDA-approved drug for systemic anaplastic large-cell lymphoma. It has recently shown, however, cases of a serious side effect from brain injection [102].
Like their use in targeted molecular imaging, nanoparticles can serve as nanoplatforms to add multiple functionalities for prodrugs [81]. Hatakeyama et al. developed a gene delivery system for cancer therapy using an MMP-cleavable peptide comprised of a PEGylated liposome [82]. This nanoplatform showed a threefold higher gene transfection efficiency when compared with a noncleavable MMP peptide, even with an equivalent amount of PEG. The main reason cited for this effective delivery is the balance between the stability of the PEGylated liposome to increase circulation time in vivo and the increased intracellular delivery after the PEG molecule is removed by the extracellular MMPs at the tumor site. Plasmid DNA, the therapeutic molecule, was active the entire time but the activation of the delivery platform improved the efficacy of the gene therapy for cancer treatment.
A nanoplatform can also be utilized to not only carry prodrugs, additional targeting moieties and stabilizing molecules, but also imaging agents to make PM theranostic systems. For example, a magnetic nanoparticle, with the ability to be monitored by MRI, was constructed for targeted gene delivery [83]. The particle consists of 15-nm manganese-doped magnetism-engineered iron oxide (MnMEIO) particles for MRI, bovine serum albumin coating for nanoparticle stability, siRNA to silence green fluorescent protein-expressing genes inside cells (siGFP) to serve as a model therapeutic gene, cancer cell-specific targeting moiety, RGD peptide, to target the integrin αvβ3 overexpressed on specific metastatic tumor cells, and tumor endothelial cells and PEG (MW 3400) for increased in vivo blood circulation time, making MnMEIO–siGFP–Cy5/PEG–RGD. As a therapy, siRNA inhibits protein expression in the cell by suppressing the encoded gene via interference with post-transcriptional RNA. Importantly, for effective gene delivery, carriers are required to pass on the specific gene near the nuclear membrane of the cell so that it can interact with the RNA. In this system, the targeted αvβ3 ntegrin is overexpressed in metastatic cancer cells, allowing the particles to specifically enter the cancer cells. Targeting is an important aspect of PM in order to deliver treatment to metastatic cells in the heterogeneous tumor, which is made up of cancer, benign and stromal cells. Once targeted, the ‘all-in-one’ particle enters the cell through receptor-mediated endocytosis by RGD signaling and unloads siRNA in the endosome through chemical hydrolysis of a bond linking the siRNA to the nanoparticle like prodrugs described previously. Furthermore, the uptake and delivery of the model therapeutic gene can be imaged by MRI due to the T2 contrast provided by the IONP carrier and the fluorescence dye labeled on the siRNA. In this theranostics system, specific delivery of a therapy and treatment monitoring is achieved by specific targeting of the tumor via RGD to αvβ3 integrin interactions and MRI based on the unique imaging properties and functionality that inorganic nanoparticles provide.
Moreover, theranostics systems, especially nanoplatforms, can be activated by other environmental stimuli for a controlled drug release, such as by magnetic field, heat, light, sound, reactive oxygen species and pH [84]. For PM, mature theranostics systems can be developed to detect biomarkers, diagnose the disorder and finally deliver treatment based on a specific stimulus in vivo, like the presence of particular biomolecules or environmental and chemical condition, at a specific time. One example of an activatable drug-delivery system by a magnetic field used dextran-coated IONPs with MRI capability and conjugated fluorescein-labeled 18 base-pair oligonucleotide duplexes to the particle [85]. The conjugated particles were activated by a radiofrequency electromagnetic field at approximately 400 kHz, which does not heat water or tissue in the background and allows penetration into approximately 15 cm of the tissue [85]. Upon electromagnetic field activation, the duplex structure of the fluorescein-labeled oligo-nucleotides melted and released the fluorescein, which served as the model drug, into the tumor model. Using larger magnetic particles, such as microdiscs that possess a spin-vortex ground date, Kim et al. showed that a magnetic field can be used for controlled cancer cell destruction in vitro [86]. When an alternating low-frequency magnetic field is applied, the microdisc attached to the cell transmits mechanical oscillation to the cell, thereby compromising the cellular membrane and instigating the apoptosis pathway.
Hyperthermia is an established clinical treatment for cancer, where increased heat causes cell death [87,88]. Certain nanoparticles, with strong and tunable surface plasmon resonance absorption, can be activated by NIR light for imaging and therapy [89]. NIR light activation is required in order to penetrate through blood and tissue, known as the ‘biological optical window’. Different nanomaterials, such as gold- or carbon-based nanomaterials, can convert optical energy into heat. If targeted, these nanoparticles can cause hyperthermia and eventual targeted cell death. Furthermore, multifunctional nanoplatforms can be established for imaging, treatment monitoring and release of additional therapeutics for synergy with hyperthermia treatment. For example, Yang et al. developed a multimodal image-guided photothermal therapy by complexing reduced graphene oxide, iron oxide nanoparticles and PEG to make a reduced graphene oxide–IONP–PEG nanocomposite [90]. This combination allowed for strong NIR optical absorbance, superparamagnetic properties and physiological stability for in vivo applications. The composite was imaged by fluorescence, photoacoustic and MR imaging, and used for photothermal therapy using a low-power density of 0.5 W/cm2 with an 808-nm NIR laser in a tumor-bearing mouse model. A major advantage of this theranostic system is treatment monitoring by MRI. After therapy, T2-weighted MRIs were acquired to monitor the tumor size because of the pooling of the nanocomposite imaging agent.
Although theranostics systems are still far behind in the clinic and require deeper understanding between biomarkers and the heterogeneity of the tumor as well as among different biomarkers, there is an excellent potential for theranostics in PM. The type of targeting moiety, imaging modality, therapy and stimuli need to be well understood and balanced during the development of a theranostic system for PM. Specific nanoplatforms can be utilized for diagnosis, imaging and/or therapy. Sophisticated systems that integrate nanohybrids for multifunctionality and multiplexing further require rigourous study. Most importantly, pharmacokinetic tests, acute and long-term toxicity studies, and quality control during manufacturing are required steps on the preclinical scale for any future of PM theranostics. In fact, new discoveries and technology may be required for such steps. Some future directions are discussed in the ‘Five-year view’.
Expert commentary & five-year view
PM is currently one of the driving forces for new research and discoveries. In the next 5 years, PM is expected to be the main goal in biomedical research. With advanced development in biomarker discovery, diagnostics, drug-delivery systems and biologics, PM will only be further strengthened. Therapy will be precisely chosen based on the heterogeneity of the cancerous tumor and the biomarkers present as more options for therapy are made available. Nanotheranostics, in turn, will be developed in a broader sense so that therapy and diagnostics can work hand in hand. As seen during the development of Herceptin® described earlier, in vitro diagnostics may be paired with specific anticancer drugs to guide treatment decisions more effectively. Furthermore, diagnostic systems can be enhanced to include in vivo molecular imaging. Targeted molecular imaging can give more information on the heterogeneity of the disease rather than sampling a small subset of a diverse tissue. Activatable probes can increase spatial understanding of biomarkers as well as the distribution of targeted drug delivery. These diagnostic systems will greatly depend on the unique properties of nanotechnology to improve sensitivity and multiplexibility. Currently, biomarkers are being correlated with disease states, yet there is an understanding that a single biomarker cannot be the only indicator addressing the cancer subtype. The interplay of specific biomarkers can give more information of the disease state and, furthermore, the treatment response. Correlation among various biomarkers and the disease class will require expansion into more state-of-the-art molecular profiling techniques. Such biomarker discoveries will drive the design of diagnostic systems towards multiplexing, and furthermore into efficient treatment monitoring. Analysis of multiple biomarkers and their proportions with each other will greatly aid treatment discovery, planning and monitoring. For such advancements in biomarker discovery and correlation, significant policy changes may be required. Collecting genetic or other types of biomarker information from a patient would need to be properly compiled and interpreted to prevent a backlog of information. A classification system for genomic testing was established by Khoury et al. to prioritize tests ready for clinical studies and identify ones that require further research [91,92]. Yet, high volumes of information already exist in the proteomics and genomics fields, and the PM field will greatly expand both those databases. A database will be needed to find statistically significant associations among certain diseases and subclasses of diseases. Careful documentation and electronic recording will be essential for such collection. However, significant questions in regulation and privacy arise. How secure is the information? What negative effects can such a database have? Once treatments are developed based on PM, how will the industry make up for its limited population size in clinical trials?
In general, drug development for cancer therapy will move towards more stable and specific pharmaceuticals. By increasing the stability, the circulation time of the drug in the blood is increased and the probability of tumor uptake is increased by passive targeting. Targeting specific biomarkers will further improve cellular uptake of the drug and possibly reduce multidrug resistance. Both active and passive targeting strategies, as seen by various preclinical and clinical studies, decrease toxic side effects to the body that would otherwise deter efficient recovery. Targeted theranostics should also take into account the location and lifetime of its targets. Drugs exert an effective therapeutic effect when delivered to the intracellular space of the cancer cell. Many biomarkers are found extracellularly, leading to premature release of the therapy. Yet, activatable drug-delivery systems can balance active and passive targeting for efficient treatment – as seen in the PEGylated liposome example above [82], where the therapeutic had a higher activity intracellularly when it was activated extracellularly. In addition, some biomarkers, such as enzymes, are temporal. They only exist at specific stages of the molecular process and disease state. Thus, if molecular imaging indicates an overexpression of a certain biomarker at day 1, that biomarker may not be present for targeted therapy at day 4 and the treatment will not be effective. For example, the proteases and caspases 2 and 3, are markers of apoptosis, but caspase 3 is present only at late stages. Therefore, a huge advantage of targeted theranostics is the simultaneous detection of biomarker expression and treatment. With increased biomarker discovery, there will be more flexibility in the design of targeted imaging and therapy agents.
Key issues.
Personalized medicine requires theranostic nanoplatforms for simultaneous diagnosis and treatment in order to achieve its goal of identifying and treating patients based on their precise molecular make up.
For advancement, molecular profiling of patients is required as well as biomarker correlations with certain disease types and severities. Molecular profiling already exists to identify adverse effects with certain pharmaceuticals as well as to determine how effective targeted therapeutics will be.
Targeted imaging and therapy is an essential part of personalized medicine. Targeted molecular imaging can be used to noninvasively distinguish biomarkers spatially and temporally in the patient before or during treatment. Targets used in imaging can also be applied for targeted therapy, such as in nuclear medicine.
Activatable probes, such as dye-labeled protease substrates, can be used for noninvasive imaging of important biomarkers involved in different disease pathways, such as metastasis, or during treatment, such as apoptosis.
Activatable therapies involve the delivery of drugs or particles used in tumor eradication and then the initiation of that treatment only upon a stimulus.
Stimuli in activatable probes and therapies can be chemical, like the presence of reactive oxygen species; environmental, like a magnetic field or heat; and molecular, like protease overexpression.
The future of personalized medicine lies in the further development of theranostics systems, nanotechnology, biomarker discovery and healthcare policies.
Footnotes
For reprint orders, please contact reprints@expert-reviews.com
Financial & competing interests disclosure
This work was supported by the Intramural Research Program of the National Institute of Biomedical Imaging and Bioengineering (NIBIB), NIH and NIBIB award R00 EB013450. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- 1.Scannell JW, Blanckley A, Boldon H, Warrington B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discov. 2012;11(3):191–200. doi: 10.1038/nrd3681. [DOI] [PubMed] [Google Scholar]
- 2.Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med. 2010;363(4):301–304. doi: 10.1056/NEJMp1006304. [DOI] [PubMed] [Google Scholar]
- 3.Chen XS. Introducing Theranostics Journal – from the Editor-in-Chief. Theranostics. 2011;1:1–2. doi: 10.7150/thno/v01p0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen XS. One year after a successful start of theranostics. Theranostics. 2012;2(1):1–2. doi: 10.7150/thno.2.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swierczewska M, Liu G, Lee S, Chen X. High-sensitivity nanosensors for biomarker detection. Chem Soc Rev. 2012;41(7):2641–2655. doi: 10.1039/c1cs15238f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chikkaveeraiah BV, Bhirde AA, Morgan NY, Eden HS, Chen X. Electrochemical immunosensors for detection of cancer protein biomarkers. ACS Nano. 2012;6(8):6546–6561. doi: 10.1021/nn3023969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shao H, Min C, Issadore D, et al. Magnetic Nanoparticles and microNMR for diagnostic applications. Theranostics. 2012;2(1):55–65. doi: 10.7150/thno.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farokhzad OC, Langer R. Nanomedicine: developing smarter therapeutic and diagnostic modalities. Adv Drug Deliv Rev. 2006;58(14):1456–1459. doi: 10.1016/j.addr.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 9.Swierczewska M, Lee S, Chen X. Moving theranostics from bench to bedside in an interdisciplinary research team. Ther Deliv. 2011;2(2):165–170. doi: 10.4155/tde.10.106. [DOI] [PubMed] [Google Scholar]
- 10.Xie J, Lee S, Chen X. Nanoparticle-based theranostic agents. Adv Drug Deliv Rev. 2010;62(11):1064–1079. doi: 10.1016/j.addr.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hulteen JC, Treichel DA, Smith MT, Duval ML, Jensen TR, Van Duyne RP. Nanosphere lithography: size-tunable silver nanoparticle and surface cluster arrays. J Phy Chem B. 1999;103(19):3854–3863. [Google Scholar]
- 12.Haynes CL. Nanosphere lithography: a versatile nanofabrication tool for studies of size-dependent nanoparticle optics. J Phys Chem. 2001;105(24):5599–5611. [Google Scholar]
- 13.Grzelczak M, Pérez-Juste J, Mulvaney P, Liz-Marzán LM. Shape control in gold nanoparticle synthesis. Chem Soc Rev. 2008;37(9):1783–1791. doi: 10.1039/b711490g. [DOI] [PubMed] [Google Scholar]
- 14.Rozenberg BA, Tenne R. Polymer-assisted fabrication of nanoparticles and nano-composites. Progress Polymer Sci. 2008;33(1):40–112. [Google Scholar]
- 15.Akhter S, Ahmad MZ, Ahmad FJ, Storm G, Kok RJ. Gold nanoparticles in theranostic oncology: current state-of-the-art. Expert Opin Drug Deliv. 2012;9(10):1225–1243. doi: 10.1517/17425247.2012.716824. [DOI] [PubMed] [Google Scholar]
- 16.Mura S, Couvreur P. Nanotheranostics for personalized medicine. Adv Drug Deliv Rev. 2012;64(13):1394–1416. doi: 10.1016/j.addr.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 17.Wang LS, Chuang MC, Ho JA. Nano-theranostics – a review of recent publications. Int J Nanomedicine. 2012;7:4679–4695. doi: 10.2147/IJN.S33065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang R, Billone PS, Mullett WM. Nanomedicine in action: an overview of cancer nanomedicine on the market and in clinical trials. J Nanomat. 2013 doi: 10.1155/2013/629681. (Epub ahead of print) [DOI] [Google Scholar]
- 19.Choi KY, Jeon EJ, Yoon HY, et al. Theranostic nanoparticles based on PEGylated hyaluronic acid for the diagnosis, therapy and monitoring of colon cancer. Biomaterials. 2012;33(26):6186–6193. doi: 10.1016/j.biomaterials.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liotta L, Petricoin E. Molecular profiling of human cancer. Nat Rev Genet. 2000;1(1):48–56. doi: 10.1038/35049567. [DOI] [PubMed] [Google Scholar]
- 21.Sadée W, Dai Z. Pharmacogenetics/genomics and personalized medicine. Hum Mol Genet. 2005;14(Spec no 2):R207–R214. doi: 10.1093/hmg/ddi261. [DOI] [PubMed] [Google Scholar]
- 22.Vogenberg FR, Isaacson Barash C, Pursel M. Personalized medicine: part 1: evolution and development into theranostics. P T. 2010;35(10):560–576. [PMC free article] [PubMed] [Google Scholar]
- 23.Carter PJ, Senter PD. Antibody–drug conjugates for cancer therapy. Cancer J. 2008;14(3):154–169. doi: 10.1097/PPO.0b013e318172d704. [DOI] [PubMed] [Google Scholar]
- 24.Sievers EL, Senter PD. Antibody–drug conjugates in cancer therapy. Annu Rev Med. 2013;64:15–29. doi: 10.1146/annurev-med-050311-201823. [DOI] [PubMed] [Google Scholar]
- 25.Golub TR, Slonim DK, Tamayo P, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286(5439):531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 26.Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 27.Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 28.Bittner M, Meltzer P, Chen Y, et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406(6795):536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- 29.Dhanasekaran SM, Barrette TR, Ghosh D, et al. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412(6849):822–826. doi: 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- 30.Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23(9):1147–1157. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- 31.Harari D, Yarden Y. Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene. 2000;19(53):6102–6114. doi: 10.1038/sj.onc.1203973. [DOI] [PubMed] [Google Scholar]
- 32.Pegram MD, Lipton A, Hayes DF, et al. Phase II study of receptor-enhanced chemosensitivity using recombinant humanized anti-p185HER2/neu monoclonal antibody plus cisplatin in patients with HER2/neu-overexpressing metastatic breast cancer refractory to chemotherapy treatment. J Clin Oncol. 1998;16(8):2659–2671. doi: 10.1200/JCO.1998.16.8.2659. [DOI] [PubMed] [Google Scholar]
- 33.Dowsett M, Bartlett J, Ellis IO, et al. Correlation between immunohistochemistry (HercepTest) and fluorescence in situ hybridization (FISH) for HER-2 in 426 breast carcinomas from 37 centres. J Pathol. 2003;199(4):418–423. doi: 10.1002/path.1313. [DOI] [PubMed] [Google Scholar]
- 34.Van Cutsem E, Köhne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360(14):1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 35.Xing Y, Chaudry Q, Shen C, et al. Bioconjugated quantum dots for multiplexed and quantitative immunohistochemistry. Nat Protoc. 2007;2(5):1152–1165. doi: 10.1038/nprot.2007.107. [DOI] [PubMed] [Google Scholar]
- 36.Alivisatos AP. Semiconductor clusters, nanocrystals, and quantum dots. Science. 1996;271(5251):933–937. [Google Scholar]
- 37.Zhang Y, Wang TH. Quantum dot enabled molecular sensing and diagnostics. Theranostics. 2012;2(7):631–654. doi: 10.7150/thno.4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eckelman WC, Reba RC, Kelloff GJ. Targeted imaging: an important biomarker for understanding disease progression in the era of personalized medicine. Drug Discov Today. 2008;13(17–18):748–759. doi: 10.1016/j.drudis.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 39.Velikyan I. Molecular imaging and radiotherapy: theranostics for personalized patient management. Theranostics. 2012;2(5):424–426. doi: 10.7150/thno.4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Groves AM, Win T, Haim SB, Ell PJ. Non-[18F]FDG PET in clinical oncology. Lancet Oncol. 2007;8(9):822–830. doi: 10.1016/S1470-2045(07)70274-7. [DOI] [PubMed] [Google Scholar]
- 41.Cai W, Chen X. Anti-angiogenic cancer therapy based on integrin alphavbeta3 antagonism. Anticancer Agents Med Chem. 2006;6(5):407–428. doi: 10.2174/187152006778226530. [DOI] [PubMed] [Google Scholar]
- 42.Niu G, Chen X. Why integrin as a primary target for imaging and therapy. Theranostics. 2011;1:30–47. doi: 10.7150/thno/v01p0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ye Y, Chen X. Integrin targeting for tumor optical imaging. Theranostics. 2011;1:102–126. doi: 10.7150/thno/v01p0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang F, Huang X, Zhu L, et al. Noninvasive monitoring of orthotopic glioblastoma therapy response using RGD-conjugated iron oxide nanoparticles. Biomaterials. 2012;33(21):5414–5422. doi: 10.1016/j.biomaterials.2012.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie J, Chen K, Lee HY, et al. Ultrasmall c(RGDyK)-coated Fe3O4 nanoparticles and their specific targeting to integrin alpha(v)beta3-rich tumor cells. J Am Chem Soc. 2008;130(24):7542–7543. doi: 10.1021/ja802003h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tan M, Lu ZR. Integrin targeted MR imaging. Theranostics. 2011;1:83–101. doi: 10.7150/thno/v01p0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chin FT, Shen B, Liu S, et al. First experience with clinical-grade ([18F] FPP(RGD2): an automated multi-step radiosynthesis for clinical PET studies. Mol Imaging Biol. 2012;14(1):88–95. doi: 10.1007/s11307-011-0477-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao H, Lang L, Guo N, et al. PET imaging of angiogenesis after myocardial infarction/reperfusion using a one-step labeled integrin-targeted tracer 18F-AlF-NOTA-PRGD2. Eur J Nucl Med Mol Imaging. 2012;39(4):683–692. doi: 10.1007/s00259-011-2052-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jacobson O, Zhu L, Niu G, et al. MicroPET imaging of integrin avβ3 expressing tumors using 89Zr-RGD peptides. Mol Imaging Biol. 2011;13(6):1224–1233. doi: 10.1007/s11307-010-0458-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beer AJ, Kessler H, Wester HJ, Schwaiger M. PET imaging of integrin aVβ3 expression. Theranostics. 2011;1:48–57. doi: 10.7150/thno/v01p0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cai W, Shin DW, Chen K, et al. Peptide-labeled near-infrared quantum dots for imaging tumor vasculature in living subjects. Nano Lett. 2006;6(4):669–676. doi: 10.1021/nl052405t. [DOI] [PubMed] [Google Scholar]
- 52.Mornet S, Vasseur S, Grasset F, Duguet E. Magnetic nanoparticle design for medical diagnosis and therapy. J Mater Chem. 2004;14(14):2161–2175. [Google Scholar]
- 53.Bulte JW, Brooks RA, Moskowitz BM, Bryant LH, Jr, Frank JA. Relaxometry and magnetometry of the MR contrast agent MION-46L. Magn Reson Med. 1999;42(2):379–384. doi: 10.1002/(sici)1522-2594(199908)42:2<379::aid-mrm20>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 54.Xie J, Jon S. Magnetic nanoparticle-based theranostics. Theranostics. 2012;2:122–124. doi: 10.7150/thno.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo N, Lang L, Gao H, et al. Quantitative analysis and parametric imaging of (18) F-labeled monomeric and dimeric RGD peptides using compartment model. Mol Imaging Biol. 2012;14(6):743–752. doi: 10.1007/s11307-012-0541-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee S, Xie J, Chen X. Activatable molecular probes for cancer imaging. Curr Top Med Chem. 2010;10(11):1135–1144. doi: 10.2174/156802610791384270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee S, Xie J, Chen X. Peptide-based probes for targeted molecular imaging. Biochemistry. 2010;49(7):1364–1376. doi: 10.1021/bi901135x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu L, Xie J, Swierczewska M, et al. Real-time video imaging of protease expression in vivo. Theranostics. 2011;1:18–27. doi: 10.7150/thno/v01p0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tyagi S, Kramer FR. Molecular beacons: probes that fluoresce upon hybridization. Nat Biotechnol. 1996;14(3):303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 60.Kim K, Lee M, Park H, et al. Cell-permeable and biocompatible polymeric nanoparticles for apoptosis imaging. J Am Chem Soc. 2006;128(11):3490–3491. doi: 10.1021/ja057712f. [DOI] [PubMed] [Google Scholar]
- 61.Lee S, Choi KY, Chung H, et al. Real time, high resolution video imaging of apoptosis in single cells with a polymeric nanoprobe. Bioconjug Chem. 2011;22(2):125–131. doi: 10.1021/bc1004119. [DOI] [PubMed] [Google Scholar]
- 62.Huang X, Swierczewska M, Choi KY, et al. Multiplex imaging of an intracellular proteolytic cascade by using a broad-spectrum nanoquencher. Angew Chem Int Ed Engl. 2012;51(7):1625–1630. doi: 10.1002/anie.201107795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.El-Deiry WS, Sigman CC, Kelloff GJ. Imaging and oncologic drug development. J Clin Oncol. 2006;24(20):3261–3273. doi: 10.1200/JCO.2006.06.5623. [DOI] [PubMed] [Google Scholar]
- 64.Kelloff GJ, Krohn KA, Larson SM, et al. The progress and promise of molecular imaging probes in oncologic drug development. Clin Cancer Res. 2005;11(22):7967–7985. doi: 10.1158/1078-0432.CCR-05-1302. [DOI] [PubMed] [Google Scholar]
- 65.Swierczewska M, Lee S, Chen X. Inorganic nanoparticles for multimodal molecular imaging. Mol Imaging. 2011;10(1):3–16. [PMC free article] [PubMed] [Google Scholar]
- 66.Xiao Y, Hong H, Matson VZ, et al. Gold nanorods conjugated with doxorubicin and cRGD for combined anticancer drug delivery and PET imaging. Theranostics. 2012;2(8):757–768. doi: 10.7150/thno.4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jang B, Choi Y. Photosensitizer-conjugated gold nanorods for enzyme-activatable fluorescence imaging and photodynamic therapy. Theranostics. 2012;2(2):190–197. doi: 10.7150/thno.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee S, Cha EJ, Park K, et al. A near-infrared-fluorescence-quenched gold-nanoparticle imaging probe for in vivo drug screening and protease activity determination. Angew Chem Int Ed Engl. 2008;47(15):2804–2807. doi: 10.1002/anie.200705240. [DOI] [PubMed] [Google Scholar]
- 69.López-Otín C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nat Rev Cancer. 2007;7(10):800–808. doi: 10.1038/nrc2228. [DOI] [PubMed] [Google Scholar]
- 70.Rautio J, Kumpulainen H, Heimbach T, et al. Prodrugs: design and clinical applications. Nat Rev Drug Discov. 2008;7(3):255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 71.Choi KY, Swierczewska M, Lee S, Chen X. Protease-activated drug development. Theranostics. 2012;2(2):156–178. doi: 10.7150/thno.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mansour AM, Drevs J, Esser N, et al. A new approach for the treatment of malignant melanoma: enhanced antitumor efficacy of an albumin-binding doxorubicin prodrug that is cleaved by matrix metalloproteinase 2. Cancer Res. 2003;63(14):4062–4066. [PubMed] [Google Scholar]
- 73.Rautio J, Kumpulainen H, Heimbach T, et al. Prodrugs: design and clinical applications. Nat Rev Drug Discov. 2008;7(3):255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 74.Habibollahi P, Figueiredo JL, Heidari P, et al. Optical imaging with a cathepsin B activated probe for the enhanced detection of esophageal adenocarcinoma by dual channel fluorescent upper GI endoscopy. Theranostics. 2012;2(2):227–234. doi: 10.7150/thno.4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yhee JY, Kim SA, Koo H, et al. Optical imaging of cancer-related proteases using near-infrared fluorescence matrix metalloproteinase-sensitive and cathepsin B-sensitive probes. Theranostics. 2012;2(2):179–189. doi: 10.7150/thno.3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Leist M, Jäättelä M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol. 2001;2(8):589–598. doi: 10.1038/35085008. [DOI] [PubMed] [Google Scholar]
- 77.Sinha AA, Wilson MJ, Gleason DF, Reddy PK, Sameni M, Sloane BF. Immunohistochemical localization of cathepsin B in neoplastic human prostate. Prostate. 1995;26(4):171–178. doi: 10.1002/pros.2990260402. [DOI] [PubMed] [Google Scholar]
- 78.Roshy S, Sloane BF, Moin K. Pericellular cathepsin B and malignant progression. Cancer Metastasis Rev. 2003;22(2–3):271–286. doi: 10.1023/a:1023007717757. [DOI] [PubMed] [Google Scholar]
- 79.Doronina SO, Toki BE, Torgov MY, et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol. 2003;21(7):778–784. doi: 10.1038/nbt832. [DOI] [PubMed] [Google Scholar]
- 80.Senter PD. Potent antibody drug conjugates for cancer therapy. Curr Opin Chem Biol. 2009;13(3):235–244. doi: 10.1016/j.cbpa.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 81.Gullotti E, Yeo Y. Extracellularly activated nanocarriers: a new paradigm of tumor targeted drug delivery. Mol Pharm. 2009;6(4):1041–1051. doi: 10.1021/mp900090z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hatakeyama H, Akita H, Kogure K, et al. Development of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipid. Gene Ther. 2007;14(1):68–77. doi: 10.1038/sj.gt.3302843. [DOI] [PubMed] [Google Scholar]
- 83.Lee JH, Lee K, Moon SH, Lee Y, Park TG, Cheon J. All-in-one target-cell-specific magnetic nanoparticles for simultaneous molecular imaging and siRNA delivery. Angew Chem Int Ed Engl. 2009;48(23):4174–4179. doi: 10.1002/anie.200805998. [DOI] [PubMed] [Google Scholar]
- 84.Kim T, Huh Y-M, Haam S, Lee K. Activatable nanomaterials at the forefront of biomedical sciences. J Mater Chem. 2010;20(38):8194–8206. [Google Scholar]
- 85.Derfus AM, von Maltzahn G, Harris TJ, et al. Remotely triggered release from magnetic nanoparticles. Adv Mater. 2007;19(22):3932–3936. [Google Scholar]
- 86.Kim DH, Rozhkova EA, Ulasov IV, et al. Biofunctionalized magnetic-vortex microdiscs for targeted cancer-cell destruction. Nat Mater. 2010;9(2):165–171. doi: 10.1038/nmat2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhao Q, Wang L, Cheng R, et al. Magnetic nanoparticle-based hyperthermia for head and neck cancer in mouse models. Theranostics. 2012;2:113–121. doi: 10.7150/thno.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lim CK, Shin J, Lee YD, et al. Phthalocyanine-aggregated polymeric nanoparticles as tumor-homing near-infrared absorbers for photothermal therapy of cancer. Theranostics. 2012;2(9):871–879. doi: 10.7150/thno.4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Melancon MP, Zhou M, Li C. Cancer theranostics with near-infrared light-activatable multimodal nanoparticles. Acc Chem Res. 2011;44(10):947–956. doi: 10.1021/ar200022e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yang K, Hu L, Ma X, et al. Multimodal imaging guided photothermal therapy using functionalized graphene nanosheets anchored with magnetic nanoparticles. Adv Mater Weinheim. 2012;24(14):1868–1872. doi: 10.1002/adma.201104964. [DOI] [PubMed] [Google Scholar]
- 91.Chalise P, Batzler A, Abo R, Wang L, Fridley BL. Simultaneous analysis of multiple data types in pharmacogenomic studies using weighted sparse canonical correlation analysis. OMICS. 2012;16(7–8):363–373. doi: 10.1089/omi.2011.0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Korwar AM, Bhonsle HS, Ghole VS, Gawai KR, Koppikar CB, Kulkarni MJ. Proteomic profiling and interactome analysis of ER-positive/HER2/neu negative invasive ductal carcinoma of the breast: towards proteomics biomarkers. OMICS. 2013;17(1):27–40. doi: 10.1089/omi.2012.0054. [DOI] [PubMed] [Google Scholar]
Websites
- 101.US FDA approves Adcetris to treat two types of lympoma. 2011 www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm268781.htm.
- 102.US FDA. Drug safety communication: new boxed warning and contraindication for Adcetris (brentuximab vedotin) 2012 www.fda.gov/Drugs/DrugSafety/ucm287668.htm#data.