

Introduction
Valproic acid (VPA) is a branched short-chain fatty acid derived from naturally occurring valeric acid. VPA is used primarily in the treatment of epilepsy and seizures, but is also used in migraine, bipolar, mood, anxiety, and psychiatric disorders [1]. Recent work has explored its use as an adjuvant agent in cancer, HIV therapy, and neurodegenerative disease because of its action as histone deacetylase (HDAC) inhibitor [2].
VPA is widely used in pediatric epilepsy because of its multiple mechanisms of action and acceptable safety profile [3]. The dose requirements for VPA are highly variable (10-fold differences in mean dose in adults) [4] and interactions with other drugs are common (discussed below). Therapeutic drug monitoring is commonly used, although both the clinical and toxic effects of the drug are considered to be poorly correlated with total serum concentrations [3].
The drug label carries a black box warning for life-threatening adverse drug reactions (ADR) including hepatoxocity, teratogenicity, and pancreatitis [1]. Compared with adults, children appear to be at an increased risk for severe hepatotoxic reactions to VPA, in particular those younger than 2 years of age undergoing polytherapy, with existing developmental delays and coincident metabolic disorders [1]. Hyperammonemia is also a documented ADR of VPA treatment, although this is usually successfully resolved by cessation of VPA and treatment with carnitine [5]. The Food and Drug Administration has issued recommendations for patient testing and advises that VPA is contraindicated in patients with known urea cycle disorders (see Clinical PGx tab at http://www.pharmgkb.org/drug/PA451846). However, specific genetic tests for the determination of at-risk patients are not mentioned.
The aim of this study was to introduce candidate genes in the pharmacokinetics (PK) (Fig. 1), pharmacodynamics (PD) of VPA (Fig. 2), and discuss results from pharmacogenomic studies so far.
Fig. 1.
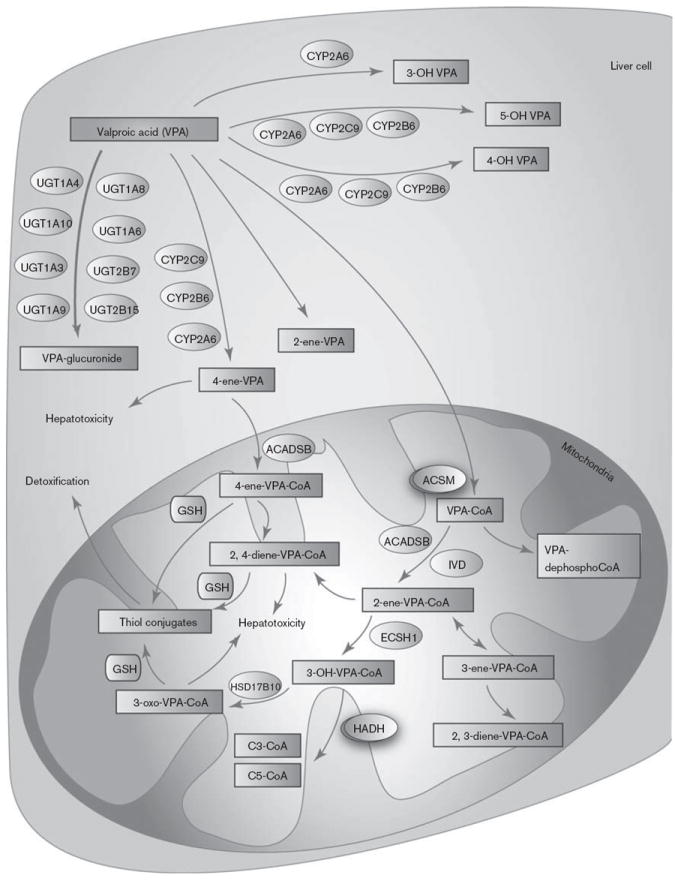
Graphic representation of the candidate genes involved in valproic acid (VPA) pharmacokinetics. A fully interactive version of this pathway is available online at PharmGKB at http://www.pharmgkb.org/pathway/PA165964265. CYP, cytochrome P450.
Fig. 2.
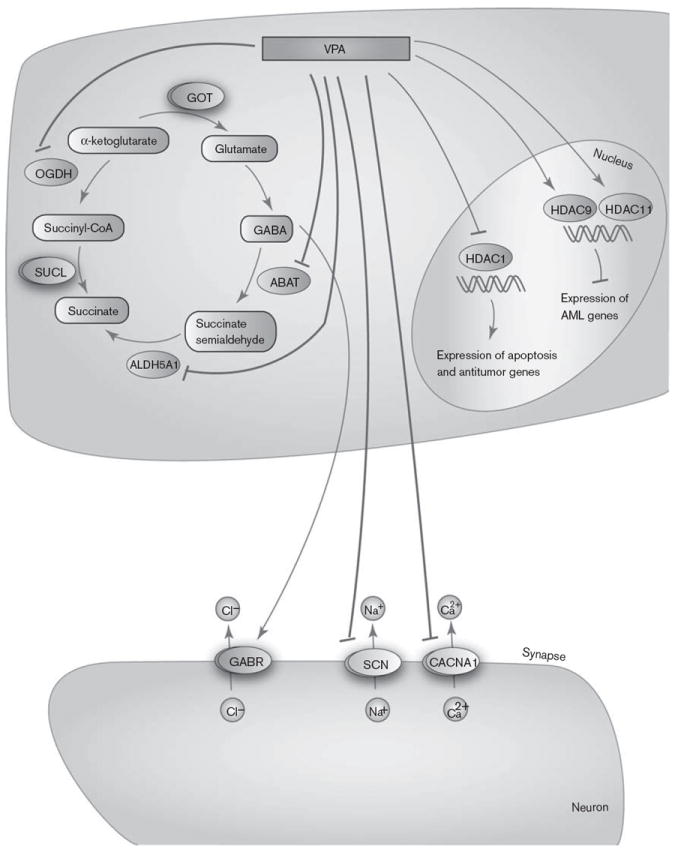
Graphic representation of the candidate genes involved in valproic acid (VPA) pharmacodynamics. A fully interactive version of this pathway is available online at PharmGKB at http://www.pharmgkb.org/pathway/PA165959313.
Pharmacokinetics
VPA is highly protein bound (87–95%) resulting in low clearance (6–20 ml/h/kg) [6]. There are at least three routes of VPA metabolism in humans: glucuronidation, β oxidation in the mitochondria (both considered major routes accounting for 50 and 40% of dose, respectively), and cytochrome P450 (CYP)-mediated oxidation (considered a minor route, ~10%) [7-9].
Valproate glucuronide is the major urinary metabolite of VPA (~30–50%) [8]. In-vitro studies of human liver microsomes and purified recombinant proteins have reported glucuronidation of VPA by UGT1A3, UGT1A4, UGT1A6, UGT1A8, UGT1A9, UGT1A10, UGT2B7, and UGT2B15 [8,10,11]. Other studies have disputed the role of UGT2B15, suggesting that VPA inhibits UGT2B15, but is not glucuronidated by it [12]. UGT1A1 does not have activity against VPA in vitro [8,12].
VPA is a fatty acid and can be metabolized through endogenous pathways in the mitochondria (Fig. 1). It has been observed that some of the mitochondrial metabolites of VPA generated by this pathway are hepatotoxic. The current understanding of VPA bioactivation involves the entry of 4-ene-VPA into the mitochondria, formation of a 4-ene-VPA-CoA ester with the help of ACADSB, and subsequent β-oxidation to form the reactive 2,4-diene-VPA-CoA ester [13,14]. Studies have demonstrated that the β-oxidation is blocked in fluorinated derivatives of 4-ene-VPA [15], and that the fluoro derivative of 4-ene-VPA cannot form a CoA ester [16], indicating a specific role of β oxidation of 4-ene-VPA in the formation of the 2,4-diene metabolite. This putative cytotoxic metabolite (2,4-diene-VPA-S-CoA) further gets conjugated with glutathione to form thiol conjugates. These chemically reactive metabolites generated from 4-ene-VPA have the potential to deplete mitochondrial glutathione pools [13] and form conjugates with CoA [17], in turn inhibiting enzymes in the β-oxidation pathway [18,19]. Identification of N-acetylcysteine conjugates of (E)-2,4-diene-VPA in human urine demonstrated that the reactive thiol conjugates of VPA arise primarily from the biotransformation of (E)-2,4-diene VPA in humans [20].
The key CYP-mediated branch of the VPA pathway is the generation of the metabolite 4-ene-VPA by CYP2C9, CYP2A6, and to a lesser extent by CYP2B6 [21,22]. In addition, these metabolizing enzymes also mediate the metabolism of VPA to the inactive 4-OH-VPA and 5-OH-VPA [23]. CYP2A6 also contributes partially to the formation of 3-OH-VPA [22].
Pharmacodynamics
VPA exhibits its PD effects in different ways: it acts on γ amino butyric acid (GABA) levels in the brain, blocks voltage-gated ion channels, and also acts as an HDAC inhibitor.
Impairment of GABAergic inhibitory activity can lead to convulsions, making the control of this pathway a target for antiepileptic drugs. GABA is formed from α-ketoglutarate through the tricarboxylic acid cycle and metabolized to succinate semialdehyde by GABA transa-minase (ABAT) and then to succinate by succinate semialdehyde dehydrogenase (ALDH5A1). α-Ketoglutarate can also be converted to succinyl CoA through the action of α-ketoglutarate dehydrogenase (OGDH), shunting it away from the formation of GABA. Ex-vivo and in-vitro studies have shown that VPA inhibits ABAT and ALDH5A1, both of which are involved in the GABA degradation pathway [24]. One in-vitro study also showed that OGDH was inhibited by high concentrations of VPA [24].
Besides increasing GABA levels, VPA may also have antiepileptic activity by reducing the high-frequency firing of neurons by blocking voltage-gated sodium, potassium, and calcium channels (including those coded for by CACNA1C, CACNA1D, CACNA1N, and CACNA1F and the SCN gene family) [24,25]. However, whether VPA increases or decreases the conductance of potassium channels is still controversial [1].
Recently, VPA was demonstrated to be an inhibitor of HDAC1 as well as other HDACs [26,27], which potentially increases the expression of genes involved in apoptosis and antitumor action. Therefore, VPA has been proposed to be a potential antitumor agent. VPA is an activator of HDAC9 and HDAC11 in cancer cell lines. HDAC inhibitor-induced activation/overexpression of specific deacetylases in tumor cells can increase the effectiveness of antineoplastic therapies by promoting selective killing of tumor cells [28].
Pharmacogenomics
There have been relatively few pharmacogenomic studies of VPA compared with other antiepileptic drugs carbamazepine and phenytoin. Most studies that focused on well-known polymorphisms in the UGT and CYP candidate genes included small numbers of individuals and were not informative about the risk for toxicity. However, studies involving some of the more promising candidate genes, which are yet to be replicated, involve mitochondrial genes that may be directly or indirectly affected by toxic VPA metabolites.
UGT variants
A study of recombinant UGT1A6 proteins showed that the *2 haplotype [which comprises rs6759892 T > G (Ser7Ala), rs2070959 A > G (Thr181Ala), and rs1105879 A > C (Arg184Ser)] was associated with increased glucuronidation of VPA compared with the *1 haplotype [10]. However, further work in this series with serotonin, another substrate of UGT1A6, found that the results with recombinant *2 did not correlate well with those seen with human liver microsomes with the *2/*2 genotype.
A recent study on 162 epileptic patients under maintenance with VPA monotherapy and stable seizure control demonstrated that carriers of the variant UGT1A6 19 T > G (rs6759892), 541 A > G (rs2070959), and 552 A > C (rs1105879) haplotypes required higher VPA dosages and had lower log-transformed concentration-to-dose ratios than noncarriers. This may be suggestive of higher activity of the UGT1A6 enzymes in patients with the variant genotypes, indicating a need for higher VPA maintenance dosages as compared with patients without variant alleles [29]. Despite the small sample size and lack of consensus on UGT1A6*2 effect, further study is warranted to determine if UGT1A6 haplotype is useful in guiding starting doses of VPA. Although no studies have shown a relevance of UGT1A6 to risk of toxicity in adult epilepsy, this enzyme is known to be developmentally regulated, and is not expressed at adult levels until sometime after 10 years of age [30]. This could, therefore, be a factor in the risk for VPA toxicity in infants.
Cytochrome P450 variants
In a PK study of 179 Asian patients with epilepsy, individuals with nonfunctional CYP2A6, CYP2D6, or CYP2C9 alleles had higher mean plasma VPA concentrations compared with those without [9]. Variant alleles in the CYP2A6, CYP2B6, and CYP2C9 genes may explain ~6–14% of interindividual variability in VPA PK, and individuals with the CYP2A6*4, CYP2B6*6, or CYP2C9*3 alleles in particular may have enhanced exposure to VPA; the effects of these allelic variations on toxicity has not been assessed to date. In-vitro studies of recombinant CYP2C9*2 and CYP2C9*3 proteins and human liver microsomes showed reduced formation of 4-ene-VPA, 4-OH-VPA, and 5-OH-VPA metabolites of VPA [23]. However, a small study of CYP2C9 variants on production of 4-ene-VPA in vivo showed no significant effects [31]. Although variants in the CYPs do not appear to play a significant role in the hepatotoxicity of VPA through generation of toxic metabolites, polytherapy has been shown to significantly increase the concentration of 4-ene-VPA [31]; thus, although a minor part of the PK pathway, flux in this pathway is important for toxicity and may be more important in patients with impaired UGTs.
Additional variants
As mentioned above, VPA is contraindicated in individuals with urea cycle disorders. Two urea cycle disorder genes have direct relationships with VPA: (a) carbamoyl phosphate synthetase 1 (CPS1) encodes a mitochondrial enzyme that catalyzes the conversion of ammonia to urea in the liver. CPS1 gene expression can be altered by epigenetic mechanisms [32]. Therefore, it is conceivable that VPA may influence CPS1 transcription and thus, ammonia metabolism. Alternatively, CPS1 may be impacted by general mitochondrial dysfunction caused by toxic VPA metabolites. In a small study (n = 79), the rs1047891A (4217C > A) polymorphism of CPS1 was associated with an increased risk of hyperammonemia when receiving combined treatment of VPA with two or more other antiepileptics [33]. This SNP is also associated with lower CPS1 activity [34]. (b) VPA has also been shown to interact with another gene in the urea cycle, N-acetylglutamate synthase, which is expressed in the mitochondrion. The metabolite valproyl-CoA inhibits N-acetylglutamate synthase in rat liver mitochondria [5]. There are three additional cytosolic genes that are known to be mutated in urea cycle disorders: arginase (ARG), argininosuccinase acid lyase (ASL), and argininosuccinic acid synthetase (ASS). Although no studies were found that examine these candidates and VPA, given the PD relationship of VPA to succinate, it is reasonable to suspect that there could be an indirect relationship through this part of the PD pathway.
Another mitochondrial gene, polymerase gamma gene (POLG) has been associated with toxicity to VPA. Mutations in POLG result in various disorders, including the neurometabolic disorder Alpers–Huttenlocher syndrome [35], which are associated with increased risk for VPA hepatotoxicity. Approximately one-third of Alpers–Huttenlocher patients, homozygous or heterozygous for POLG mutations, develop liver failure within 3 months of exposure to VPA [35]. It is unclear whether VPA interacts with POLG directly or whether this is the result of indirect mitochondrial dysfunction induced by toxic VPA metabolites. In addition to the rare variants in POLG, common yet functional polymorphisms of POLG may be present in 0.5% of the population, and are associated with migraine, epilepsy, and Parkinson’s disease, indications that might lead to the prescription of VPA [35]. Although replication of this association is necessary, these results suggest that screening for functional POLG polymorphisms might help to minimize the risk of liver failure in patients exposed to VPA.
Additional new pharmacogenomic candidates for VPA response include rs226957, a variant in the promoter of transcription factor XBP1. The G allele of rs226957 (− 116C > G) has reduced transcription compared with the C allele and is associated with increased VPA response in a small study of bipolar disorder patients (n = 51) [36,37]. Also, the rs1019385 G variant upstream of GRIN2B, a subunit of the NMDA receptor, is associated with decreased dose requirement for epilepsy patients (n = 162) [29]. The G allele is associated with decreased transcription compared with the T allele, and is overrepresented in patients with schizophrenia [38]. These results suggest that the GRIN2B polymorphism may cause NMDA receptor dysfunction, leading to glutamate-mediated neuronal excitation and in turn reducing the effective dose of VPA required to control epilepsies.
Conclusion
Although there is considerable evidence to link the candidate genes for both the PK and PD of VPA, very few studies have identified genomic variants that influence drug dosage, drug-induced toxicity, and treatment outcome. The major drawback of these studies is their small sample size, and conflicting results limit confidence about their pharmacogenomics relevance. There is a need for validating these associations in larger cohorts, and pediatric cohorts in particular. Glucuronidation is the primary route of VPA metabolism in adults, and expression of many UGTs is decreased in infants and young children because of developmental factors. Therefore, more VPA is available for mitochondrial and CYP-mediated biotransformation, particularly in infants. Induction of CYP metabolism by coadministered enzyme-inducing antiepileptic drugs theoretically could increase the relative formation of 4-ene-VPA, drive more substrate through the β-oxidation pathway, and increase the risk of mitochondrial stress and hepatotoxicity. However, the mechanisms underlying the increased risk of severe hepatotoxicity in infants and young children remains elusive.
Many drug–drug interactions have been reported for VPA and this list will likely increase as VPA is used in additional indications where polytherapy is common, such as HIV and cancer. A better understanding of VPA pathways should aid in predicting novel drug–drug interactions and avoiding ADRs from concomitant treatments. These may also aid in the design of large genomic studies that validate the effects of the genomic variants already reported and find new influential variants. Complementary metabolomics studies may also aid in profiling patients at risk for ADRs. Although severe urea cycle disorders are rare, there is preliminary evidence that other common variants may also influence risk for ADRs. More work is needed in larger cohorts to examine the role of newer candidate genes and variants in VPA PK and PD pathways.
Acknowledgments
The authors thank Fen Liu for assistance with the graphics. This work is supported by the NIH/NIGMS (R24 GM61374), NIH/NCI R01 CA132946, R21 CA155524, and R01 HD044239.
Footnotes
Conflicts of interest
There are no conflicts of interest.
References
- 1.Chateauvieux S, Morceau F, Dicato M, Diederich M. Molecular and therapeutic potential and toxicity of valproic acid. J Biomed Biotechnol. 2010 doi: 10.1155/2010/479364. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Terbach N, Williams RS. Structure–function studies for the panacea, valproic acid. Biochem Soc Trans. 2009;37(Pt 5):1126–1132. doi: 10.1042/BST0371126. [DOI] [PubMed] [Google Scholar]
- 3.Serrano BB, Garcia Sanchez MJ, Otero MJ, Buelga DS, Serrano J, Dominguez-Gil A. Valproate population pharmacokinetics in children. J Clin Pharm Ther. 1999;24:73–80. doi: 10.1046/j.1365-2710.1999.00202.x. [DOI] [PubMed] [Google Scholar]
- 4.Blanco-Serrano B, Otero MJ, Santos-Buelga D, Garcia-Sanchez MJ, Serrano J, Dominguez-Gil A. Population estimation of valproic acid clearance in adult patients using routine clinical pharmacokinetic data. Biopharm Drug Dispos. 1999;20:233–240. doi: 10.1002/(sici)1099-081x(199907)20:5<233::aid-bdd179>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 5.Aires CC, van Cruchten A, Ijlst L, de Almeida IT, Duran M, Wanders RJ, et al. New insights on the mechanisms of valproate-induced hyperammonemia: inhibition of hepatic N-acetylglutamate synthase activity by valproyl-CoA. J Hepatol. 2011;55:426–434. doi: 10.1016/j.jhep.2010.11.031. [DOI] [PubMed] [Google Scholar]
- 6.Leppik IE, Birnbaum AK. Epilepsy in the elderly. Ann N Y Acad Sci. 2010;1184:208–224. doi: 10.1111/j.1749-6632.2009.05113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ito M, Ikeda Y, Arnez JG, Finocchiaro G, Tanaka K. The enzymatic basis for the metabolism and inhibitory effects of valproic acid: dehydrogenation of valproyl-CoA by 2-methyl-branched-chain acyl-CoA dehydrogenase. Biochim Biophys Acta. 1990;1034:213–218. doi: 10.1016/0304-4165(90)90079-c. [DOI] [PubMed] [Google Scholar]
- 8.Argikar UA, Remmel RP. Effect of aging on glucuronidation of valproic acid in human liver microsomes and the role of UDP-glucuronosyltransferase UGT1A4, UGT1A8, and UGT1A10. Drug Metab Dispos. 2009;37:229–236. doi: 10.1124/dmd.108.022426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan L, Yu JT, Sun YP, Ou JR, Song JH, Yu Y. The influence of cytochrome oxidase CYP2A6, CYP2B6, and CYP2C9 polymorphisms on the plasma concentrations of valproic acid in epileptic patients. Clin Neurol Neurosurg. 2010;112:320–323. doi: 10.1016/j.clineuro.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 10.Krishnaswamy S, Hao Q, Al-Rohaimi A, Hesse LM, von Moltke LL, Greenblatt DJ, et al. UDP glucuronosyltransferase (UGT) 1A6 pharmacogenetics: II. Functional impact of the three most common nonsynonymous UGT1A6 polymorphisms (S7A, T181A, and R184S) J Pharmacol Exp Ther. 2005;313:1340–1346. doi: 10.1124/jpet.104.081968. [DOI] [PubMed] [Google Scholar]
- 11.Chung JY, Cho JY, Yu KS, Kim JR, Lim KS, Sohn DR, et al. Pharmacokinetic and pharmacodynamic interaction of lorazepam and valproic acid in relation to UGT2B7 genetic polymorphism in healthy subjects. Clin Pharmacol Ther. 2008;83:595–600. doi: 10.1038/sj.clpt.6100324. [DOI] [PubMed] [Google Scholar]
- 12.Ethell BT, Anderson GD, Burchell B. The effect of valproic acid on drug and steroid glucuronidation by expressed human UDP-glucuronosyltransferases. Biochem Pharmacol. 2003;65:1441–1449. doi: 10.1016/s0006-2952(03)00076-5. [DOI] [PubMed] [Google Scholar]
- 13.Kassahun K, Farrell K, Abbott F. Identification and characterization of the glutathione and N-acetylcysteine conjugates of (E)-2-propyl-2,4-pentadienoic acid, a toxic metabolite of valproic acid, in rats and humans. Drug Metab Dispos. 1991;19:525–535. [PubMed] [Google Scholar]
- 14.Kassahun K, Hu P, Grillo MP, Davis MR, Jin L, Baillie TA. Metabolic activation of unsaturated derivatives of valproic acid. Identification of novel glutathione adducts formed through coenzyme A-dependent and -independent processes. Chem Biol Interact. 1994;90:253–275. doi: 10.1016/0009-2797(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 15.Tang W, Borel AG, Fujimiya T, Abbott FS. Fluorinated analogues as mechanistic probes in valproic acid hepatotoxicity: hepatic microvesicular steatosis and glutathione status. Chem Res Toxicol. 1995;8:671–682. doi: 10.1021/tx00047a006. [DOI] [PubMed] [Google Scholar]
- 16.Grillo MP, Chiellini G, Tonelli M, Benet LZ. Effect of alpha-fluorination of valproic acid on valproyl-S-acyl-CoA formation in vivo in rats. Drug Metab Dispos. 2001;29:1210–1215. [PubMed] [Google Scholar]
- 17.Kassahun K, Abbott F. In vivo formation of the thiol conjugates of reactive metabolites of 4-ene VPA and its analog 4-pentenoic acid. Drug Metab Dispos. 1993;21:1098–1106. [PubMed] [Google Scholar]
- 18.Rettenmeier AW, Prickett KS, Gordon WP, Bjorge SM, Chang SL, Levy RH, et al. Studies on the biotransformation in the perfused rat liver of 2-N-propyl-4-pentenoic acid, a metabolite of the antiepileptic drug valproic acid. Evidence for the formation of chemically reactive intermediates. Drug Metab Dispos. 1985;13:81–96. [PubMed] [Google Scholar]
- 19.Baillie TA. Metabolic activation of valproic acid and drug-mediated hepatotoxicity. Role of the terminal olefin, 2-N-propyl-4-pentenoic acid. Chem Res Toxicol. 1988;1:195–199. doi: 10.1021/tx00004a001. [DOI] [PubMed] [Google Scholar]
- 20.Gopaul SV, Farrell K, Abbott FS. Identification and characterization of N-acetylcysteine conjugates of valproic acid in humans and animals. Drug Metab Dispos. 2000;28:823–832. [PubMed] [Google Scholar]
- 21.Sadeque AJ, Fisher MB, Korzekwa KR, Gonzalez FJ, Rettie AE. Human CYP2C9 and CYP2A6 mediate formation of the hepatotoxin 4-ene-valproic acid. J Pharmacol Exp Ther. 1997;283:698–703. [PubMed] [Google Scholar]
- 22.Kiang TK, Ho PC, Anari MR, Tong V, Abbott FS, Chang TK. Contribution of CYP2C9, CYP2A6, and CYP2B6 to valproic acid metabolism in hepatic microsomes from individuals with the CYP2C9*1/*1 genotype. Toxicol Sci. 2006;94:261–271. doi: 10.1093/toxsci/kfl096. [DOI] [PubMed] [Google Scholar]
- 23.Ho PC, Abbott FS, Zanger UM, Chang TK. Influence of CYP2C9 genotypes on the formation of a hepatotoxic metabolite of valproic acid in human liver microsomes. Pharmacogenomics J. 2003;3:335–342. doi: 10.1038/sj.tpj.6500210. [DOI] [PubMed] [Google Scholar]
- 24.Johannessen CU, Johannessen SI. Valproate: past, present, and future. CNS Drug Rev. 2003;9:199–216. doi: 10.1111/j.1527-3458.2003.tb00249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van den Berg RJ, Kok P, Voskuyl RA. Valproate and sodium currents in cultured hippocampal neurons. Exp Brain Res. 1993;93:279–287. doi: 10.1007/BF00228395. [DOI] [PubMed] [Google Scholar]
- 26.Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- 27.Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20:6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bradbury CA, Khanim FL, Hayden R, Bunce CM, White DA, Drayson MT, et al. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia. 2005;19:1751–1759. doi: 10.1038/sj.leu.2403910. [DOI] [PubMed] [Google Scholar]
- 29.Hung CC, Ho JL, Chang WL, Tai JJ, Hsieh TJ, Hsieh YW, et al. Association of genetic variants in six candidate genes with valproic acid therapy optimization. Pharmacogenomics. 2011;12:1107–1117. doi: 10.2217/pgs.11.64. [DOI] [PubMed] [Google Scholar]
- 30.McCarver DG, Hines RN. The ontogeny of human drug-metabolizing enzymes: phase II conjugation enzymes and regulatory mechanisms. J Pharmacol Exp Ther. 2002;300:361–366. doi: 10.1124/jpet.300.2.361. [DOI] [PubMed] [Google Scholar]
- 31.Amini-Shirazi N, Ghahremani MH, Ahmadkhaniha R, Mandegary A, Dadgar A, Abdollahi M, et al. Influence of CYP2C9 polymorphism on metabolism of valproate and its hepatotoxin metabolite in Iranian patients. Toxicol Mech Methods. 2010;20:452–457. doi: 10.3109/15376516.2010.497977. [DOI] [PubMed] [Google Scholar]
- 32.Liu H, Dong H, Robertson K, Liu C. DNA methylation suppresses expression of the urea cycle enzyme carbamoyl phosphate synthetase 1 (CPS1) in human hepatocellular carcinoma. Am J Pathol. 2011;178:652–661. doi: 10.1016/j.ajpath.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yagi M, Nakamura T, Okizuka Y, Oyazato Y, Kawasaki Y, Tsuneishi S, et al. Effect of CPS14217C > A genotype on valproic-acid-induced hyperammonemia. Pediatr Int. 2010;52:744–748. doi: 10.1111/j.1442-200X.2010.03157.x. [DOI] [PubMed] [Google Scholar]
- 34.Summar ML, Gainer JV, Pretorius M, Malave H, Harris S, Hall LD, et al. Relationship between carbamoyl-phosphate synthetase genotype and systemic vascular function. Hypertension. 2004;43:186–191. doi: 10.1161/01.HYP.0000112424.06921.52. [DOI] [PubMed] [Google Scholar]
- 35.Hudson G, Chinnery PF. Mitochondrial DNA polymerase-gamma and human disease. Hum Mol Genet. 2006;15(Spec No 2):R244–R252. doi: 10.1093/hmg/ddl233. [DOI] [PubMed] [Google Scholar]
- 36.Kakiuchi C, Iwamoto K, Ishiwata M, Bundo M, Kasahara T, Kusumi I, et al. Impaired feedback regulation of XBP1 as a genetic risk factor for bipolar disorder. Nat Genet. 2003;35:171–175. doi: 10.1038/ng1235. [DOI] [PubMed] [Google Scholar]
- 37.Kim B, Kim CY, Lee MJ, Joo YH. Preliminary evidence on the association between XBP1-116C/G polymorphism and response to prophylactic treatment with valproate in bipolar disorders. Psychiatry Res. 2009;168:209–212. doi: 10.1016/j.psychres.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 38.Miyatake R, Furukawa A, Suwaki H. Identification of a novel variant of the human NR2B gene promoter region and its possible association with schizophrenia. Mol Psychiatry. 2002;7:1101–1106. doi: 10.1038/sj.mp.4001152. [DOI] [PubMed] [Google Scholar]