

Abstract
Colorectal cancer (CRC) is the third leading cause of cancer-related death in the United States, with the number of affected people increasing. There are many risk factors that increase CRC risk, including family or personal history of CRC, smoking, consumption of red meat, obesity, and alcohol consumption. Conversely, increased screening, maintaining healthy body weight, not smoking, and limiting intake of red meat are all associated with reduced CRC morbidity and mortality. Mouse models of CRC were first used in 1928 and have played an important role in understanding CRC biology and treatment and have long been instrumental in clarifying the pathobiology of CRC formation and inhibition. This review focuses on advancements in modeling CRC in mice.
Keywords: Colorectal cancer, human, mouse models, tumorigenesis, Apc, FAP, HNPCC, stem cells
Introduction
Colorectal cancer (CRC) is the third most common cancer worldwide, and the number one cause of nonsmoking cancer-related deaths in the world [1]. In the U.S., annual reported cases of CRC is approximately 142,000 and mortality 50,000 [2]. Clinically and histologically, colorectal cancer can be graded as 1 of 4 stages, with the highest grade and mortality associated with mainly liver or widespread metastasis [3]. CRC begins with specific molecular alterations in Wnt-β-catenin pathway. Additional loss of function or mutations in k-ras, DCC, DPC4 or JV18-1 or p53 contributes to CRC development [4,5]. Further, combinations of alterations in other pathways including the mitogen-activated protein kinase (MAPK) pathway, phosphatidylinositol 3-kinase (PI3K) pathway, nuclear factor-kappa B (NF-κB) pathway, and activator protein 1 (AP-1) pathway are additional contributors to stepwise CRC development [6-9]. As intestinal tumors develop, they quickly outgrow the local blood supply and must recruit new capillary blood vessels in order to sustain adequate blood supply for continued growth [9-11].
Numerous mouse models of CRC have been developed, providing insights into pathogenesis mechanisms, tools for discovery, validation of novel therapeutic targets, and a predictive platform in which to test new chemoprevention strategies. There are several excellent reviews in the literature on this subject, so in this review we provide an overview and update some of the latest genetic, chemical, and bacterial CRC studies employing animal models.
Genetic models of early events
Mouse model for FAP
Colorectal cancers begin with intestinal epithelial cells that lose the function of the Apc pathway (gatekeeper function), part of the Wnt signaling pathway [12]. Upon Wnt binding to the Frizzled receptor and receptor activation, Apc forms a complex in the cytoplasm that results in ß-catenin phosphorylation by glycogen synthase kinase-3 (GSK-3). β-catenin phosphorylation results in its proteolytic degradation [13]. However, loss of Apc function results in nuclear accumulation of β-catenin, where, in cooperation with the transcription factor Tcf-4, it modulates expression of a variety of Tcf-4 responsive target genes. Loss of Apc function has been shown to act through Tcf-4 to upregulate, c-Myc, Cdk4, and cyclin D1 proto-oncogene expression [14-16]. Therefore, Apc mutation affects the G1 to S transition of the cell cycle, causing cell growth dysregulation in intestinal epithelial cells, with resultant formation of intestinal polyps (Figure 1).
Figure 1.

Gross and microscopic images of the intestinal polyps. A: Multiple raised polyps are present within the small (top) and large (bottom) intestine. B: Ki67 staining of the microscopic section of the polyp showing prolifertive nuclear staining.
Patients with familial adenomatous polyposis (FAP) carry a germline mutation in one APC allele. They develop hundreds to thousands polyps within the large intestine and they are at high risk for developing CRC [17,18]. Mouse models have been useful for modeling FAP; the adenomas that arise in Apc mutant mice are similar to development in FAP patients, in that they are at least in part nonimmunogenic and arise in immunocompetent mice. On the other hand, Apc mutant mice develop large numbers of adenomas in their small intestine and fewer in the large intestine and rarely progress to invasive adenocarcinoma (perhaps due to a short lifespan), whereas FAP patients develop low numbers of adenomas in their small intestine and large numbers of adenomas in their large intestine which progress to invasive adenocarcinoma [19-21]. Homozygous knockout of the Apc gene in mice is embryonic lethal, but heterozygous mutant Apc mice (Apc -/+) develop between 3 and 300 intestinal adenomas/polyps in the intestine, with the overall number depending on the location of the truncating mutation, and other modifiers [22]. The first Apc mutant mouse model, multiple intestinal neoplasia (Min), was developed by Moser et al, with several other subsequent models showing multiple adenomas within the small and large intestine [19,22-27]. McCart et al reviewed these models and the application of the model in drug testing [28]. Studies have shown that NSAID’s inhibit adenoma formation in the Apc mutant mice [29-31]. This is in agreement with epidemiological studies suggesting that NSAID’s decrease colorectal cancer occurrence in humans [32,33]. While the Apc mutant mouse model is currently the best available model to study prevention strategies targeting early events in CRC development, one disadvantage of Apc mutant mice as a CRC model is that progression to malignant cancer and metastases occur late in the course of disease, so it is infrequently observed [19,20]. Robanus-Maandag and colleagues developed a new Apc mutant mouse with tumors developing mainly in the large intestine, similar to human FAP patients. This model, FabplCre; Apc 15lox/+, had an extended lifespan and developed a significant number of adenomas and adenocarcinomas in the large intestine, which should be useful to study the genetic alterations associated with the adenoma-carcinoma sequence in the mouse [34].
Mouse model for HNPCC
Hereditary Non-polyposis Colorectal Cancer (HNPCC), also known as Lynch Syndrome (LS), is the most common of the inherited CRC syndromes, and accounts for 3-5% of CRC cases [35,36]. HNPCC is caused by mutations in one of the DNA mismatch repair (MMR) genes MSH2, MLHJ, PMSJ, and PMS2. The disease is inherited in an autosomal dominant pattern and the mutations are associated with development of cancers [37,38]. There is also a limited, but increased incidence of hematological malignancies in patients with HNPCC [39]. Similarly, mouse models carrying disruptions of MMR genes develop lymphoma in addition to Figure 1. Gross and microscopic images of the intestinal polyps. A: Multiple raised polyps are present within the small (top) and large (bottom) intestine. B: Ki67 staining of the microscopic section of the polyp showing prolifertive nuclear staining. intestinal neoplasia [40-42]. A novel conditional knockout mouse was developed, in which the Msh2 is knocked down in villin-expressing tissues, mainly the small and large intestine, but normal MMR activity is preserved in the rest of the body. This model has similarities to HNPCC, as the mice do not develop lymphoma; however, they do develop intestinal adenomas and adenocarcinomas [43]. Lastly, mice homozygous for the Mlh1 gene are predisposed to developing tumors of the gastrointestinal tract. Introduction of the Apc gene (gatekeeper) in MMR homozygous mice enhanced Apc mediated intestinal tumorigenesis [44].
Modifiers that affect early events
Identification of mouse tumor susceptibility factors are an important strategy in finding second site modifier alleles that influence intestinal tumor development. The modifier of Min (Mom1) was identified in 1993 by introducing 35cM of distal AKR chromosome 4 from into C57BL/6 mouse. The results were that Mom1 is a semi-dominant modifier of intestinal adenoma size in Min mice [45]. The Mom2 locus, which is on chromosome 18, was introduced into Apc -/+ mice leading to greater reduction in adenoma multiplicity in both the small and large intestines than the Mom1 locus of Apc +/- mice [46]. Later, six recombinant lines presenting with limited intraline variation in adenoma multiplicity were established through selective breeding for homozygosity for distal chromosome 18 markers [47]. Mom5 was reported in 2009 to determine the impact of estrogen receptor β (ERβ) signaling on intestinal carcinogenesis in Apc -/+ mice. The results show 50% reduction in in adenoma formation [27]. Kwong et al. found that Mom7 on chromosome 18 regulates the loss of heterozygosity of distal elements and could be another pathway useful in chemoprevention [48]. The identification of Mom12 and Mom13 loci on chromosome 6 highlights the effects of residual donor DNA on tumorigenesis in Apc -/+ mice. Mom12, is linked to the D6Mit33 marker and results in increased tumorigenesis compared to Apc -/+ controls. Mom13 increases intestinal tumor multiplicity in the absence of the Mom12 [49].
Additional non-Apc gene considerations such as undefined genetic background effects and environmental factors can also act as modifiers. Genetic background affects adenoma multiplicity in Apc -/+ mice [24,45]. The average number of adenomas in a C57BL/6J background were around 29. However, the number of adenomas reduced to 6 when C57BL/6J was crossed to AKR mice [50]. Similarly, environmental factors such as chemical and bacterial agents have been shown to have implications for intestinal tumorigenesis. Exposure of MMR-deficient cells to mutagens and alkylating agents potentiate tumorigenesis and fail to induce apoptosis [51,52]. Dietary factors also play a role in CRC formation and inhibition; obese mice (ob/ob) are more susceptible to chemical- induced colon cancer. Tumor cell lines grew more rapidly in obese mice compared to lean mice [53]. Bacteria appear to be another important cofactor in CRC formation. Exposure of Apc mutant mice to enteriotoxigenic bacterial fragiles (ETBF) leads to enhanced high tumor load [54]. Further, Apc mutant mice have a high number of tumors upon infection with Citrobacter rodentium [55]. Smad3-/- mice develop colorectal adenocarcinoma after being inoculated with either Helicobacter bilis or Helicobacter hepaticus. Taken together, specific second site genetic modifiers, environmental factors, and genetic background can mediate dramatic differences in the dynamics of tumorigenesis in models of CRC.
Other elements in tumor multiplicity include the location of the mutation within the Apc gene and this has been reviewed in detail by McCart et al [28]. Additional factors such as posttranslational modifications affect formation of intestinal tumorigenesis. Laird et al found that DNA hypomethlyation suppresses intestinal neoplasia in Apc-/+ mice [56]. Similarly, hypermethylation of the APC promoter 1A has been described in sporadic CRC in humans with associated partial reduction in transcript levels [57].
Spontaneous and chemically induced intestinal tumorigenesis models
As the spontaneous incidence of colorectal cancer in mice is low (1%-4%), many chemicals have been used to induce CRC. These carcinogens include dimethyhydrazine (DMH) or its metabolites, azoxymethane (AOM), dextran sulfate sodium (DSS), 2-amino-1-methyl-6-phenylimidazol (4,5-b) pyridine (PhIP), N-methyl-N’-nitro-N-nitrosoguanidine (MNNG), MNU, 3,2’-dimethyl-4-aminobiphenyl (DMBA). The progression of these cancers depends on the duration and dosage of the chemical. Also, the background of the mice plays a significant role in the development of colorectal tumors [58-61]. AOM/DSS treatment in mice offers a powerful model in the initiation of aberrant crypt foci (early lesions) and is useful in evaluation of CRC chemopreventive strategies [58]. For example, it has been shown AOM/DSS treatment increases aberrant crypt foci in Nrf2 knockout mice (the transcription factor Nrf2 recognizes the antioxidant response element in the promoter of target genes) [62,63]. MNNG, DMBA, and PHIP have been used more frequently in rat models to date. MNNG does not require biochemical activation and can be injected directly in the rectum; therefore, it is considered a topical agent and is not an ideal model for humans due to the route of administration [59]. DMBA tumorigenic activity is less potent in inducing colorectal tumors and requires multiple doses [60]. In mice, PHIP has been used most widely in Apc -/+ mice and has been shown to increase the number and size of intestinal adenomas [61,64-66].
Mouse models of invasion and metastasis
Many models have been developed to monitor the invasiveness and metastasis of the implanted or injected tumors. Nude mice that lack T cell function or SCID mice that lack both B and T cell function have been useful for developing orthotopic tumor implantation models. Grafts from either human (xenografts) or murine (syngeneic autografts or allografts) tumors can be implanted into recipient mice. The tumor cells or tumor tissue can be implanted or injected at primary or metastatic tumor sites in immunodeficinet mice. In addition, spleen and kidney capsule can be useful for tumor cell implantation. The advantage of the model is that the starting material is from a parallel relevant site representing human cancer directly, as opposed to standard subcutaneous xenografts. In addition, intravascular and intrasplenic injection mimic vascular or portal spread of CRC [67-70]. However, the disadvantage of the xenograft implantation is that the tumor development is not exactly the same as human CRC development due to species differences. In addition, there are differences between native intestine and the subcutaneous microenvironment. Immunocompetent mice can be used as models as well. Mouse cell lines that escape immune detection have been used in these mice. Cell lines that lack major histocompatibility complex are able to grow without rejection in immunologically incompatible recipient mice [11,71]. These are useful in some cases where syngeneic cell lines are not available.
Orthotopic implantation has been used to produce a model more similar to human cancers than subcutaneous xenografts. In this model, the implant (colon cancer cell lines) is directly placed on the serosa of the intestine [11]. The advantage of orthotopic implantation is it’s relevance and that the metastatic site can be monitored by imaging. The disadvantage is that the orthotopic implantation is challenging procedure and can be associated with inflammation of the implanted site if stringent surgical technique is not followed.
Recently, a new colonoscopy system was developed for implanting human colorectal cancer into the mouse colonic submucosa. This promising model is non-invasive, fast, and was not associated with significant inflammation [72]. Magnetic resonance imaging and other related imaging modalities can effectively monitor internal tumor growth and invasiveness in vivo [73]. Similarly, many in vivo studies have successfully used a luciferase construct to monitor tumor growth, invasiveness, and metastasis [74,75].
Other models
In addition to the models mentioned above, Ramanathan et al found that a mutation in p53 gene increases progastrin-dependent colonic proliferation and subsequent formation of aberrant crypt foci [76]. Recently, a novel mouse model demonstrated the expression in the intestine of a dominant active form of the PI3K protein resulted in highly invasive mucinous adenocarcinomas [77]. p110, catalytic subunit of class Ib PI3-kinase, produces PIP3 in response to chemokines and other G protein-coupled receptor agonists [78]. Sasaki et al reported that p110 γ-/- mice developed spontaneous malignant CRC [79].
Another interesting model with relevance for age-dependent carcinogenesis relates to telomere maintenance. With each cell division and with aging, telomeres shorten and display degenerative defects. Both CAST/EiJ and mTR knockout mice can develop short telomeres and, in parallel, these mice have been shown to develop intestinal microadenomas [80]. In addition, a mouse model of obesity and colorectal cancer has been developed. Basically, (db/db) mouse-an animal model of type II diabetes-was bred to the Apc -/+ mouse. The double mutant mice, db/db- Apc -/+, developed larger numbers of adenomas when compared to Apc -/+ mice [81]. Similarly, a mouse model of alcohol consumption revealed that alcohol-fed Apc mutant mice exhibited an increase in number and sizes of adenomas in the intestine. Alcohol intake lead to increases in the number of mast cells and subsequent invasion of tumor cells [82].
Recent advances
Rapid advances have been made in identifying and understanding stem cell pathways affecting intestinal cell differentiation and proliferative capacity. These, in turn, have provided insights into gastrointestinal stem cell dynamics and CRC. Increasing evidence shows that stem cells are involved in development of CRC and other cancers [83-85]. The mucosal layer of the intestine is composed of epithelial cells, with villi at the luminal surface and crypts at the base of the villi [86]. The intestinal stem cells are located near the base of each crypt [87,88]. Each crypt contains about 30 stem cells with 4-6 lineage ancestors [89]. Stem cells, which develop and differentiate as they migrate from the crypt up to the villus, are the source of enterocytes, goblet cells, enteroendocrine, and paneth cells [90]. Similar to other cells, stem cells can go through apoptosis, which is believed to be a defensive mechanism against cancer development [91]. The signaling pathways which control stem cell propagation share some similarities to other non-stem cells. It is known that tumors arising from normal cells require many gene alterations [92]. Mutations in terminally differentiated cells, such as enterocytes, would in theory have little pathological significance for cancer in the intestine, since these cells are turned over constantly, in less than one week [90,93]. In contrast, mutations in stem cells, long term residents of the mucosa, can pass alterations to their progeny through self-renewal [94]. Consequently, the accumulation of mutations results in an opportunity for cells to go through a multistep carcinogenesis process and the eventual development of malignant cancer. Epithelial tumors may arise from adult stem cells and early daughter cells, because they are the only cells in the gut that persist long enough to accumulate multiple mutations.
Identifying intestinal cancer stem cells is a new important strategy for the identification of novel cancer biomarkers and developing more effective therapeutic interventions. The main intestinal stem cell biomarkers have been recently reviewed in detail [95]. Barker, et al identified that Lgr5-expressing crypt base columnar cells resist apoptosis, undergo self- renewal, give rise to terminally-differentiated cells, and have all the criteria of putative intestinal stem cells [96]. Another putative intestinal stem cell marker, DCAMKL-1 (doublecortin and CaM kinase-like-1), is predominantly observed in a unique quiescent cell population in the lower third of the intestinal crypt [97]. Furthermore, Prom1 (CD133 in humans), has been identified in colorectal, hepatocellular, and pancreatic cancer as a cancer stem cell marker, and has been used as a marker to predict colon cancer recurrence in humans [98-100]. It was recently found that Prom1 is a marker for stem cells and early progenitors in mouse small intestine (Figure 2) [101]. Similarly, Prom1-positive cells mark intestinal stem cells that are susceptible to neoplastic information [102]. In addition, Bmi1-positive cells are located at the bottom of crypts and have features of a stem cell marker [103]. Mouse telomerase reverse transcriptase (mTert) marks slowly cycling intestinal stem cells [104]. In addition, a sensitive model was recently developed to obtain a quantitative comprehensive in situ description of the location of stem-cell markers at the single-transcript level. In this model, co-expression of Lgr5, Bmi1, Dcamkl1, and mTert genes were detected at the crypt base [105].
Figure 2.
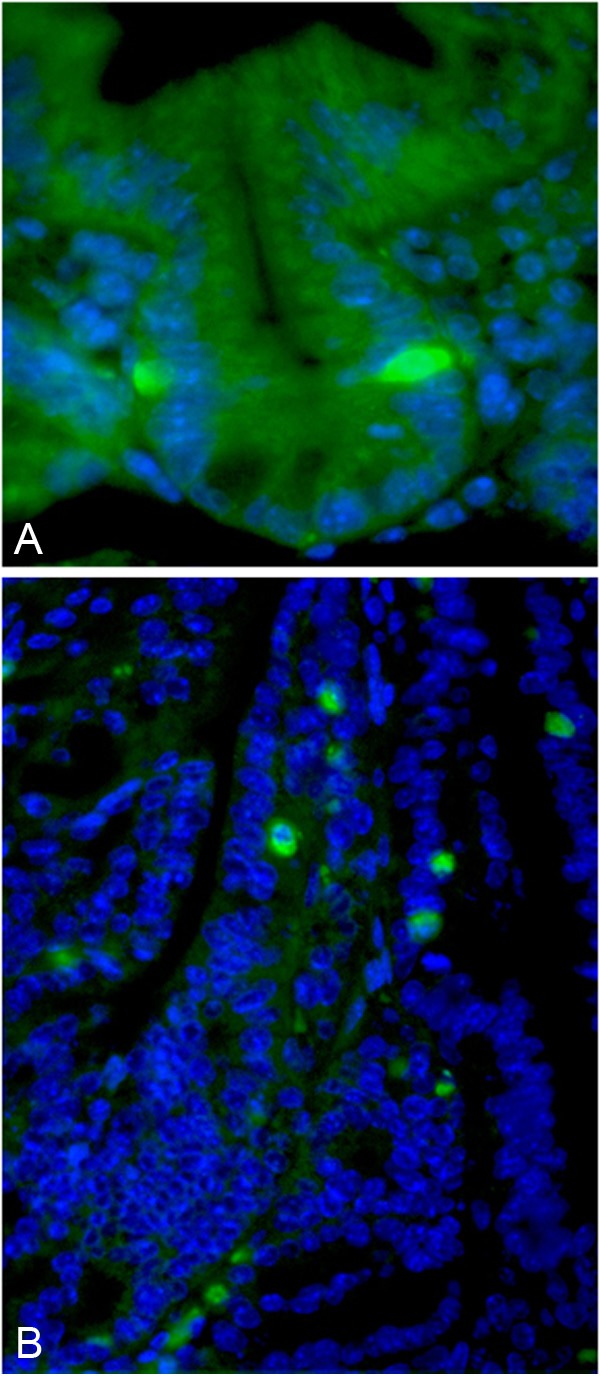
Prom1 immunofluroescsnese staining of normal crypt and polyp. A: Notice rare positive (green color) staining at the +4 crypt position. B: Notice expansion of the positive cells (green color) within the polyp.
To understand the role of stem cells in intestinal cancer stem cell initiation, an inducible Lgr5-EGFP-IRES-creERT2 cassette was used to delete the APC gene in crypt base columnar stem cells. After tamoxifen administration, β-catenin accumulation was observed in isolated Lgr5-EGFP+ stem cells and these transformed cells quickly became associated with clusters of β-catenin-expressing progeny migrating up the crypt [106].
Conclusion
In summary, genetically modified mouse models continue to play an important role in understanding the genome’s role in formation, progression, and inhibition of CRC. These models also offer robust methods to study naturally occurring and synthetic compounds for the inhibition or treatment of CRC. The spontaneous and chemically induced models are often used to study effect on the treatment or prevention of CRC formation. The mouse models for invasion and metastasis are useful for understanding the pathogenesis of progression and metastasis of CRC. Other models have been used to address specific questions like how aging, or alcohol consumption, or diabetes affect the risk of developing CRC. Finally, the recent advances in identifying roles for intestinal stem cells in CRC provide new insights for understanding the formation and inhibition of CRC.
With the advances in genomic sequencing of human CRC, the functional analysis of identified genomic alterations is necessary to distinguish driver gene alterations from passenger alterations in CRC [107,108]. Therefore developing mouse models and related methods to discover and validate candidate genomic CRC drivers that play an important role in human CRC is urgently needed for translation of CRC sequencing advances into new, safe and effective chemopreventives and treatments.
Acknowledgments
This research was supported by NIH CA134292 (DH) and a TEDCO grant from the state of Maryland (BK).
Disclosure of conflict of interest
All the authors do not have any conflict of interest.
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 3.Chirica M, Leconte M, Oberlin O, Dousset B. Surgical treatment of liver metastasis in patients with colorectal cancer. Presse Med. 2012;41:58–67. doi: 10.1016/j.lpm.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 4.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 5.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 7.Slattery ML, Lundgreen A, Wolff RK. MAP kinase genes and colon and rectal cancer. Carcinogenesis. 2012;33:2398–2408. doi: 10.1093/carcin/bgs305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anbalagan M, Huderson B, Murphy L, Rowan BG. Post-translational modifications of nuclear receptors and human disease. Nucl Recept Signal. 2012;10:e001. doi: 10.1621/nrs.10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakamoto K, Maeda S, Hikiba Y, Nakagawa H, Hayakawa Y, Shibata W, Yanai A, Ogura K, Omata M. Constitutive NF-kappaB activation in colorectal carcinoma plays a key role in angiogenesis, promoting tumor growth. Clin Cancer Res. 2009;15:2248–2258. doi: 10.1158/1078-0432.CCR-08-1383. [DOI] [PubMed] [Google Scholar]
- 10.Papetti M, Herman IM. Mechanisms of normal and tumor-derived angiogenesis. Am J Physiol Cell Physiol. 2002;282:C947–970. doi: 10.1152/ajpcell.00389.2001. [DOI] [PubMed] [Google Scholar]
- 11.Nanda A, Karim B, Peng Z, Liu G, Qiu W, Gan C, Vogelstein B, St Croix B, Kinzler KW, Huso DL. Tumor endothelial marker 1 (Tem1) functions in the growth and progression of abdominal tumors. Proc Natl Acad Sci U S A. 2006;103:3351–3356. doi: 10.1073/pnas.0511306103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakayama T, Morishita T, Kamiya T. Adenomatous polyposis coli gene as a gatekeeper. Rev Gastroenterol Peru. 2002;22:164–167. [PubMed] [Google Scholar]
- 13.Ikeda S, Kishida M, Matsuura Y, Usui H, Kikuchi A. GSK-3beta-dependent phosphorylation of adenomatous polyposis coli gene product can be modulated by beta-catenin and protein phosphatase 2A complexed with Axin. Oncogene. 2000;19:537–545. doi: 10.1038/sj.onc.1203359. [DOI] [PubMed] [Google Scholar]
- 14.Korinek V, Barker N, Morin PJ, Van Wichen D, De Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 15.Heinen CD, Goss KH, Cornelius JR, Babcock GF, Knudsen ES, Kowalik T, Groden J. The APC tumor suppressor controls entry into S-phase through its ability to regulate the cyclin D/RB pathway. Gastroenterology. 2002;123:751–763. doi: 10.1053/gast.2002.35382. [DOI] [PubMed] [Google Scholar]
- 16.Hermeking H, Rago C, Schuhmacher M, Li Q, Barrett JF, Obaya AJ, O’Connell BC, Mateyak MK, Tam W, Kohlhuber F, Dang CV, Sedivy JM, Eick D, Vogelstein B, Kinzler KW. Identification of CDK4 as a target of c-MYC. Proc Natl Acad Sci U S A. 2000;97:2229–2234. doi: 10.1073/pnas.050586197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M, Sargeant L, Krapcho K, Wolff E, Burt R, Hughes JP, Warrington J, McPherson J, Wasmuth J, Le Paslier D, Abderrahim H, Cohen DO, Leppert M, White R. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 18.Lynch HT, Lynch JF, Lynch PM, Attard T. Hereditary colorectal cancer syndromes: molecular genetics, genetic counseling, diagnosis and management. Fam Cancer. 2008;7:27–39. doi: 10.1007/s10689-007-9165-5. [DOI] [PubMed] [Google Scholar]
- 19.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 20.Halberg RB, Katzung DS, Hoff PD, Moser AR, Cole CE, Lubet RA, Donehower LA, Jacoby RF, Dove WF. Tumorigenesis in the multiple intestinal neoplasia mouse: redundancy of negative regulators and specificity of modifiers. Proc Natl Acad Sci U S A. 2000;97:3461–3466. doi: 10.1073/pnas.050585597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caldwell CM, Green RA, Kaplan KB. APC mutations lead to cytokinetic failures in vitro and tetraploid genotypes in Min mice. J Cell Biol. 2007;178:1109–1120. doi: 10.1083/jcb.200703186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moser AR, Luongo C, Gould KA, McNeley MK, Shoemaker AR, Dove WF. ApcMin: a mouse model for intestinal and mammary tumorigenesis. Eur J Cancer. 1995;31A:1061–1064. doi: 10.1016/0959-8049(95)00181-h. [DOI] [PubMed] [Google Scholar]
- 23.Nnadi SC, Watson R, Innocent J, Gonye GE, Buchberg AM, Siracusa LD. Identification of Five Novel Modifier Loci of ApcMin Harbored in theBXH14 Recombinant Inbred Strain. Carcinogenesis. 2012;33:1589–1597. doi: 10.1093/carcin/bgs185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moser AR, Dove WF, Roth KA, Gordon JI. The Min (multiple intestinal neoplasia) mutation: its effect on gut epithelial cell differentiation and interaction with a modifier system. J Cell Biol. 1992;116:1517–1526. doi: 10.1083/jcb.116.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koratkar R, Pequignot E, Hauck WW, Siracusa LD. The CAST/Ei strain confers significant protection against Apc(Min) intestinal polyps, independent of the resistant modifier of Min 1 (Mom1) locus. Cancer Res. 2002;62:5413–5417. [PubMed] [Google Scholar]
- 26.Koratkar R, Silverman KA, Pequignot E, Hauck WW, Buchberg AM, Siracusa LD. Analysis of reciprocal congenic lines reveals the C3H/HeJ genome to be highly resistant to ApcMin intestinal tumorigenesis. Genomics. 2004;84:844–852. doi: 10.1016/j.ygeno.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 27.Oikarinen SI, Cleveland AG, Cork KM, Bynote KK, Rafter JJ, Gustafsson JA, Mutanen M, Gould KA. Genetic mapping of Mom5, a novel modifier of Apc(Min)-induced intestinal tumorigenesis. Carcinogenesis. 2009;30:1591–1596. doi: 10.1093/carcin/bgp159. [DOI] [PubMed] [Google Scholar]
- 28.McCart AE, Vickaryous NK, Silver A. Apc mice: models, modifiers and mutants. Pathol Res Pract. 2008;204:479–490. doi: 10.1016/j.prp.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 29.Mohammed A, Janakiram NB, Li Q, Choi CI, Zhang Y, Steele VE, Rao CV. Chemoprevention of colon and small intestinal tumorigenesis in APC(Min/+) mice by licofelone, a novel dual 5-LOX/COX inhibitor. potential implications for human colon cancer prevention. Cancer Prev Res (Phila) 2011;4:2015–2026. doi: 10.1158/1940-6207.CAPR-11-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Torrance CJ, Jackson PE, Montgomery E, Kinzler KW, Vogelstein B, Wissner A, Nunes M, Frost P, Discafani CM. Combinatorial chemoprevention of intestinal neoplasia. Nat Med. 2000;6:1024–1028. doi: 10.1038/79534. [DOI] [PubMed] [Google Scholar]
- 31.Wang X, Kingsley PJ, Marnett LJ, Eling TE. The role of NAG-1/GDF15 in the inhibition of intestinal polyps in APC/Min mice by sulindac. Cancer Prev Res (Phila) 2011;4:150–160. doi: 10.1158/1940-6207.CAPR-10-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruder EH, Laiyemo AO, Graubard BI, Hollenbeck AR, Schatzkin A, Cross AJ. Non-steroidal anti-inflammatory drugs and colorectal cancer risk in a large, prospective cohort. Am J Gastroenterol. 2011;106:1340–1350. doi: 10.1038/ajg.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aalbers J. Substantial evidence for colorectal cancer reduction with daily low-dose aspirin. Cardiovasc J Afr. 2011;22:110. [PubMed] [Google Scholar]
- 34.Robanus-Maandag EC, Koelink PJ, Breukel C, Salvatori DC, Jagmohan-Changur SC, Bosch CA, Verspaget HW, Devilee P, Fodde R, Smits R. A new conditional Apc-mutant mouse model for colorectal cancer. Carcinogenesis. 2010;31:946–952. doi: 10.1093/carcin/bgq046. [DOI] [PubMed] [Google Scholar]
- 35.Leach FS, Nicolaides NC, Papadopoulos N, Liu B, Jen J, Parsons R, Peltomaki P, Sistonen P, Aaltonen LA, Nystrom-Lahti M, Guan XY, Zhang J, Meltzer PS, Yu JW, Kao FT, Chen DJ, Cerosaletti K, Fournier REK, Todd S, Lewis T, Leach RJ, Naylor SL, Weissenbach J, Mecklin JP, Järvinen H, Petersen GM, Hamilton SR, Green J, Jass J, Watson P, Lynch HT, Trent JM, Chapelle AAL, Kinzler KW, Vogelstein B. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–1225. doi: 10.1016/0092-8674(93)90330-s. [DOI] [PubMed] [Google Scholar]
- 36.Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM, Adams MD. Mutation of a mutL homolog in hereditary colon cancer. Science. 1994;263:1625–1629. doi: 10.1126/science.8128251. [DOI] [PubMed] [Google Scholar]
- 37.Montazer Haghighi M, Radpour R, Aghajani K, Zali N, Molaei M, Zali MR. Four novel germline mutations in the MLH1 and PMS2 mismatch repair genes in patients with hereditary nonpolyposis colorectal cancer. Int J Colorectal Dis. 2009;24:885–893. doi: 10.1007/s00384-009-0731-1. [DOI] [PubMed] [Google Scholar]
- 38.Liu B, Parsons R, Papadopoulos N, Nicolaides NC, Lynch HT, Watson P, Jass JR, Dunlop M, Wyllie A, Peltomaki P, de la Chapelle A, Hamilton SR, Vogelstein B, Kinzler KW. Analysis of mismatch repair genes in hereditary non-polyposis colorectal cancer patients. Nat Med. 1996;2:169–174. doi: 10.1038/nm0296-169. [DOI] [PubMed] [Google Scholar]
- 39.Pineda M, Castellsague E, Musulen E, Llort G, Frebourg T, Baert-Desurmont S, Gonzalez S, Capella G, Blanco I. Non-Hodgkin lymphoma related to hereditary nonpolyposis colorectal cancer in a patient with a novel heterozygous complex deletion in the MSH2 gene. Genes Chromosomes Cancer. 2008;47:326–332. doi: 10.1002/gcc.20536. [DOI] [PubMed] [Google Scholar]
- 40.Reiss C, Haneke T, Volker HU, Spahn M, Rosenwald A, Edelmann W, Kneitz B. Conditional inactivation of MLH1 in thymic and naive T-cells in mice leads to a limited incidence of lymphoblastic T-cell lymphomas. Leuk Lymphoma. 2010;51:1875–1886. doi: 10.3109/10428194.2010.510360. [DOI] [PubMed] [Google Scholar]
- 41.Carethers JM, Chauhan DP, Fink D, Nebel S, Bresalier RS, Howell SB, Boland CR. Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology. 1999;117:123–131. doi: 10.1016/s0016-5085(99)70558-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Wind N, Dekker M, Berns A, Radman M, te Riele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995;82:321–330. doi: 10.1016/0092-8674(95)90319-4. [DOI] [PubMed] [Google Scholar]
- 43.Kucherlapati MH, Lee K, Nguyen AA, Clark AB, Hou H Jr, Rosulek A, Li H, Yang K, Fan K, Lipkin M, Bronson RT, Jelicks L, Kunkel TA, Kucherlapati R, Edelmann W. An Msh2 conditional knockout mouse for studying intestinal cancer and testing anticancer agents. Gastroenterology. 2010;138:993–1002. e1. doi: 10.1053/j.gastro.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edelmann W, Yang K, Kuraguchi M, Heyer J, Lia M, Kneitz B, Fan K, Brown AM, Lipkin M, Kucherlapati R. Tumorigenesis in Mlh1 and Mlh1/Apc1638N mutant mice. Cancer Res. 1999;59:1301–1307. [PubMed] [Google Scholar]
- 45.Dietrich WF, Lander ES, Smith JS, Moser AR, Gould KA, Luongo C, Borenstein N, Dove W. Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell. 1993;75:631–639. doi: 10.1016/0092-8674(93)90484-8. [DOI] [PubMed] [Google Scholar]
- 46.Silverman KA, Koratkar R, Siracusa LD, Buchberg AM. Identification of the modifier of Min 2 (Mom2) locus, a new mutation that influences Apc-induced intestinal neoplasia. Genome Res. 2002;12:88–97. doi: 10.1101/gr.206002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haines J, Johnson V, Pack K, Suraweera N, Slijepcevic P, Cabuy E, Coster M, Ilyas M, Wilding J, Sieber O, Bodmer W, Tomlinson I, Silver A. Genetic basis of variation in adenoma multiplicity in ApcMin/+ Mom1S mice. Proc Natl Acad Sci U S A. 2005;102:2868–2873. doi: 10.1073/pnas.0500039102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kwong LN, Shedlovsky A, Biehl BS, Clipson L, Pasch CA, Dove WF. Identification of Mom7, a novel modifier of Apc(Min/+) on mouse chromosome 18. Genetics. 2007;176:1237–1244. doi: 10.1534/genetics.107.071217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crist RC, Roth JJ, Lisanti MP, Siracusa LD, Buchberg AM. Identification of Mom12 and Mom13, two novel modifier loci of Apc (Min)-mediated intestinal tumorigenesis. Cell Cycle. 2011;10:1092–1099. doi: 10.4161/cc.10.7.15089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuraguchi M, Yang K, Wong E, Avdievich E, Fan K, Kolodner RD, Lipkin M, Brown AM, Kucherlapati R, Edelmann W. The distinct spectra of tumor-associated Apc mutations in mismatch repair-deficient Apc1638N mice define the roles of MSH3 and MSH6 in DNA repair and intestinal tumorigenesis. Cancer Res. 2001;61:7934–7942. [PubMed] [Google Scholar]
- 51.Sansom OJ, Bishop SM, Court H, Dudley S, Liskay RM, Clarke AR. Apoptosis and mutation in the murine small intestine. loss of Mlh1- and Pms2-dependent apoptosis leads to increased mutation in vivo. DNA Repair (Amst) 2003;2:1029–1039. doi: 10.1016/s1568-7864(03)00111-3. [DOI] [PubMed] [Google Scholar]
- 52.Colussi C, Fiumicino S, Giuliani A, Rosini S, Musiani P, Macri C, Potten CS, Crescenzi M, Bignami M. 1,2-Dimethylhydrazine-induced colon carcinoma and lymphoma in msh2(-/-) mice. J Natl Cancer Inst. 2001;93:1534–1540. doi: 10.1093/jnci/93.20.1534. [DOI] [PubMed] [Google Scholar]
- 53.Flores MB, Rocha GZ, Damas-Souza DM, Osorio-Costa F, Dias MM, Ropelle ER, Camargo JA, de Carvalho RB, Carvalho HF, Saad MJ, Carvalheira JBC. Obesity-Induced Increase in Tumor Necrosis Factor-alpha Leads to Development of Colon Cancer in Mice. Gastroenterology. 2012;143:741–753. doi: 10.1053/j.gastro.2012.05.045. [DOI] [PubMed] [Google Scholar]
- 54.Housseau F, Sears CL. Enterotoxigenic Bacteroides fragilis (ETBF)-mediated colitis in Min (Apc+/-) mice: a human commensal-based murine model of colon carcinogenesis. Cell Cycle. 2010;9:3–5. doi: 10.4161/cc.9.1.10352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Newman JV, Kosaka T, Sheppard BJ, Fox JG, Schauer DB. Bacterial infection promotes colon tumorigenesis in Apc(Min/+) mice. J Infect Dis. 2001;184:227–230. doi: 10.1086/321998. [DOI] [PubMed] [Google Scholar]
- 56.Laird PW, Jackson-Grusby L, Fazeli A, Dickinson SL, Jung WE, Li E, Weinberg RA, Jaenisch R. Suppression of intestinal neoplasia by DNA hypomethylation. Cell. 1995;81:197–205. doi: 10.1016/0092-8674(95)90329-1. [DOI] [PubMed] [Google Scholar]
- 57.Esteller M, Sparks A, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Gonzalez S, Tarafa G, Sidransky D, Meltzer SJ, Baylin SB, Herman JG. Analysis of adenomatous polyposis coli promoter hypermethylation in human cancer. Cancer Res. 2000;60:4366–4371. [PubMed] [Google Scholar]
- 58.De Robertis M, Massi E, Poeta ML, Carotti S, Morini S, Cecchetelli L, Signori E, Fazio VM. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J Carcinog. 2011;10:9. doi: 10.4103/1477-3163.78279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Narisawa T, Magadia NE, Weisburger JH, Wynder EL. Promoting effect of bile acids on colon carcinogenesis after intrarectal instillation of N-methyl-N’-nitro-N-nitrosoguanidine in rats. J Natl Cancer Inst. 1974;53:1093–1097. doi: 10.1093/jnci/53.4.1093. [DOI] [PubMed] [Google Scholar]
- 60.Reddy BS, Ohmori T. Effect of intestinal microflora and dietary fat on 3,2’-dimethyl-4-aminobiphenyl-induced colon carcinogenesis in F344 rats. Cancer Res. 1981;41:1363–1367. [PubMed] [Google Scholar]
- 61.Andreassen A, Mollersen L, Vikse R, Steffensen IL, Mikalsen A, Paulsen JE, Alexander J. One dose of 2-amino-1-methyl-6-phenylimidazo[4,5-b] pyridine (PhIP) or 2-amino-3-methylimidazo[4,5-f] quinoline (IQ) induces tumours in Min/+ mice by truncation mutations or LOH in the Apc gene. Mutat Res. 2002;517:157–166. doi: 10.1016/s1383-5718(02)00065-7. [DOI] [PubMed] [Google Scholar]
- 62.Yu X, Kensler T. Nrf2 as a target for cancer chemoprevention. Mutat Res. 2005;591:93–102. doi: 10.1016/j.mrfmmm.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 63.Osburn WO, Karim B, Dolan PM, Liu G, Yamamoto M, Huso DL, Kensler TW. Increased colonic inflammatory injury and formation of aberrant crypt foci in Nrf2-deficient mice upon dextran sulfate treatment. Int J Cancer. 2007;121:1883–1891. doi: 10.1002/ijc.22943. [DOI] [PubMed] [Google Scholar]
- 64.Moser AR, Hegge LF, Cardiff RD. Genetic background affects susceptibility to mammary hyperplasias and carcinomas in Apc(min)/+ mice. Cancer Res. 2001;61:3480–3485. [PubMed] [Google Scholar]
- 65.Olstorn HB, Paulsen JE, Alexander J. Effects of perinatal exposure to acrylamide and glycidamide on intestinal tumorigenesis in Min/+ mice and their wild-type litter mates. Anticancer Res. 2007;27:3855–3864. [PubMed] [Google Scholar]
- 66.Svendsen C, Alexander J, Knutsen HK, Husoy T. The min mouse on FVB background: susceptibility to spontaneous and carcinogen-induced intestinal tumourigenesis. Anticancer Res. 2011;31:785–788. [PubMed] [Google Scholar]
- 67.Ager EI, Wen SW, Chan J, Chong WW, Neo JH, Christophi C. Altered efficacy of AT1R-targeted treatment after spontaneous cancer cell-AT1R upregulation. BMC Cancer. 2011;11:274. doi: 10.1186/1471-2407-11-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boll H, Nittka S, Doyon F, Neumaier M, Marx A, Kramer M, Groden C, Brockmann MA. Micro-CT based experimental liver imaging using a nanoparticulate contrast agent: a longitudinal study in mice. PLoS One. 2011;6:e25692. doi: 10.1371/journal.pone.0025692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Frampas E, Maurel C, Thedrez P, Remaud-Le Saec P, Faivre-Chauvet A, Barbet J. The intraportal injection model for liver metastasis: advantages of associated bioluminescence to assess tumor growth and influences on tumor uptake of radiolabeled anti-carcinoembryonic antigen antibody. Nucl Med Commun. 2011;32:147–154. doi: 10.1097/MNM.0b013e328341b268. [DOI] [PubMed] [Google Scholar]
- 70.Grimm M, Gasser M, Bueter M, Strehl J, Wang J, Nichiporuk E, Meyer D, Germer CT, Waaga-Gasser AM, Thalheimer A. Evaluation of immunological escape mechanisms in a mouse model of colorectal liver metastases. BMC Cancer. 2010;10:82. doi: 10.1186/1471-2407-10-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nakajima T, Masuda H, Okamoto T, Watanabe M, Yokoyama K, Yamada N, Tsukagoshi S, Taguchi T. [Antitumor effect of a benzoylphenylurea derivative HO-221] . Gan To Kagaku Ryoho. 1990;17:2353–2359. [PubMed] [Google Scholar]
- 72.Zigmond E, Halpern Z, Elinav E, Brazowski E, Jung S, Varol C. Utilization of murine colonoscopy for orthotopic implantation of colorectal cancer. PLoS One. 2011;6:e28858. doi: 10.1371/journal.pone.0028858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lavilla-Alonso S, Abo-Ramadan U, Halavaara J, Escutenaire S, Tatlisumak T, Saksela K, Kanerva A, Hemminki A, Pesonen S. Optimized mouse model for the imaging of tumor metastasis upon experimental therapy. PLoS One. 2011;6:e26810. doi: 10.1371/journal.pone.0026810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pichorner A, Sack U, Kobelt D, Kelch I, Arlt F, Smith J, Walther W, Schlag PM, Stein U. In vivo imaging of colorectal cancer growth and metastasis by targeting MACC1 with shRNA in xenografted mice. Clin Exp Metastasis. 2012;29:573–583. doi: 10.1007/s10585-012-9472-6. [DOI] [PubMed] [Google Scholar]
- 75.Sack U, Walther W, Scudiero D, Selby M, Kobelt D, Lemm M, Fichtner I, Schlag PM, Shoemaker RH, Stein U. Novel effect of antihelminthic Niclosamide on S100A4-mediated metastatic progression in colon cancer. J Natl Cancer Inst. 2011;103:1018–1036. doi: 10.1093/jnci/djr190. [DOI] [PubMed] [Google Scholar]
- 76.Ramanathan V, Jin G, Westphalen CB, Whelan A, Dubeykovskiy A, Takaishi S, Wang TC. P53 gene mutation increases progastrin dependent colonic proliferation and colon cancer formation in mice. Cancer Invest. 2012;30:275–286. doi: 10.3109/07357907.2012.657814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Leystra AA, Deming DA, Zahm CD, Farhoud M, Paul Olson TJ, Hadac JN, Nettekoven LA, Albrecht DM, Clipson L, Sullivan R, Washington MK, Torrealba JR, Weichert JP, Halberg RB. Mice Expressing Activated PI3K Develop Advanced Colon Cancer. Cancer Res. 2012;72:2931–2936. doi: 10.1158/0008-5472.CAN-11-4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 79.Sasaki T, Irie-Sasaki J, Horie Y, Bachmaier K, Fata JE, Li M, Suzuki A, Bouchard D, Ho A, Redston M, Gallinger S, Khokha R, Mak TW, Hawkinsk PT, Stephensk L, Scherer SW, Tsao M, Penninger JM. Colorectal carcinomas in mice lacking the catalytic subunit of PI(3)Kgamma. Nature. 2000;406:897–902. doi: 10.1038/35022585. [DOI] [PubMed] [Google Scholar]
- 80.Armanios M, Alder JK, Parry EM, Karim B, Strong MA, Greider CW. Short telomeres are sufficient to cause the degenerative defects associated with aging. Am J Hum Genet. 2009;85:823–832. doi: 10.1016/j.ajhg.2009.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hata K, Kubota M, Shimizu M, Moriwaki H, Kuno T, Tanaka T, Hara A, Hirose Y. C57BL/KsJ-db/db-Apc Mice Exhibit an Increased Incidence of Intestinal Neoplasms. Int J Mol Sci. 2011;12:8133–8145. doi: 10.3390/ijms12118133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wimberly AL, Forsyth CB, Khan MW, Pemberton A, Khazaie K, Keshavarzian A. Ethanol-Induced Mast Cell-Mediated Inflammation Leads to Increased Susceptibility of Intestinal Tumorigenesis in the APC(Delta468) Min Mouse Model of Colon Cancer. Alcohol Clin Exp Res. 2013;37:E199–208. doi: 10.1111/j.1530-0277.2012.01894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 84.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 85.Wang X, Kruithof-de Julio M, Economides KD, Walker D, Yu H, Halili MV, Hu YP, Price SM, Abate-Shen C, Shen MM. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009;461:495–500. doi: 10.1038/nature08361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hirano S, Kataoka K. Histogenesis of the mouse jejunal mucosa, with special reference to proliferative cells and absorptive cells. Arch Histol Jpn. 1986;49:333–348. doi: 10.1679/aohc.49.333. [DOI] [PubMed] [Google Scholar]
- 87.Kaur P, Potten CS. Cell migration velocities in the crypts of the small intestine after cytotoxic insult are not dependent on mitotic activity. Cell Tissue Kinet. 1986;19:601–610. doi: 10.1111/j.1365-2184.1986.tb00761.x. [DOI] [PubMed] [Google Scholar]
- 88.Qiu JM, Roberts SA, Potten CS. Cell migration in the small and large bowel shows a strong circadian rhythm. Epithelial Cell Biol. 1994;3:137–148. [PubMed] [Google Scholar]
- 89.Bach SP, Renehan AG, Potten CS. Stem cells: the intestinal stem cell as a paradigm. Carcinogenesis. 2000;21:469–476. doi: 10.1093/carcin/21.3.469. [DOI] [PubMed] [Google Scholar]
- 90.Potten CS, Loeffler M. Stem cells: attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt. Development. 1990;110:1001–1020. doi: 10.1242/dev.110.4.1001. [DOI] [PubMed] [Google Scholar]
- 91.Roberts SA, Hendry JH, Potten CS. Intestinal crypt clonogens. a new interpretation of radiation survival curve shape and clonogenic cell number. Cell Prolif. 2003;36:215–231. doi: 10.1046/j.1365-2184.2003.00279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 93.Kellett M, Potten CS, Rew DA. A comparison of in vivo cell proliferation measurements in the intestine of mouse and man. Epithelial Cell Biol. 1992;1:147–155. [PubMed] [Google Scholar]
- 94.Fearon ER, Hamilton SR, Vogelstein B. Clonal analysis of human colorectal tumors. Science. 1987;238:193–197. doi: 10.1126/science.2889267. [DOI] [PubMed] [Google Scholar]
- 95.Rizk P, Barker N. Gut stem cells in tissue renewal and disease: methods, markers, and myths. Wiley Interdiscip Rev Syst Biol Med. 2012;5:475–496. doi: 10.1002/wsbm.1176. [DOI] [PubMed] [Google Scholar]
- 96.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 97.May R, Sureban SM, Hoang N, Riehl TE, Lightfoot SA, Ramanujam R, Wyche JH, Anant S, Houchen CW. Doublecortin and CaM kinase-like-1 and leucine-rich-repeat-containing G-protein-coupled receptor mark quiescent and cycling intestinal stem cells, respectively. Stem Cells. 2009;27:2571–2579. doi: 10.1002/stem.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 99.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 100.Yasuda H, Tanaka K, Saigusa S, Toiyama Y, Koike Y, Okugawa Y, Yokoe T, Kawamoto A, Inoue Y, Miki C, Kusunoki M. Elevated CD133, but not VEGF or EGFR, as a predictive marker of distant recurrence after preoperative chemoradiotherapy in rectal cancer. Oncol Rep. 2009;22:709–717. doi: 10.3892/or_00000491. [DOI] [PubMed] [Google Scholar]
- 101.Snippert HJ, van Es JH, van den Born M, Begthel H, Stange DE, Barker N, Clevers H. Prominin-1/CD133 marks stem cells and early progenitors in mouse small intestine. Gastroenterology. 2009;136:2187–2194. doi: 10.1053/j.gastro.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 102.Zhu L, Gibson P, Currle DS, Tong Y, Richardson RJ, Bayazitov IT, Poppleton H, Zakharenko S, Ellison DW, Gilbertson RJ. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature. 2009;457:603–607. doi: 10.1038/nature07589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet. 2008;40:915–920. doi: 10.1038/ng.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Montgomery RK, Carlone DL, Richmond CA, Farilla L, Kranendonk ME, Henderson DE, Baffour-Awuah NY, Ambruzs DM, Fogli LK, Algra S, Breault DT. Mouse telomerase reverse transcriptase (mTert) expression marks slowly cycling intestinal stem cells. Proc Natl Acad Sci U S A. 2011;108:179–184. doi: 10.1073/pnas.1013004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Itzkovitz S, Lyubimova A, Blat IC, Maynard M, van Es J, Lees J, Jacks T, Clevers H. van Oudenaarden A, Single-molecule transcript counting of stem-cell markers in the mouse intestine. Nat Cell Biol. 2012;14:106–114. doi: 10.1038/ncb2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 107.Ji X, Tang J, Halberg R, Busam D, Ferriera S, Pena MM, Venkataramu C, Yeatman TJ, Zhao S. Distinguishing between cancer driver and passenger gene alteration candidates via cross-species comparison: a pilot study. BMC Cancer. 2010;10:426. doi: 10.1186/1471-2407-10-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bozic I, Antal T, Ohtsuki H, Carter H, Kim D, Chen S, Karchin R, Kinzler KW, Vogelstein B, Nowak MA. Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci U S A. 2010;107:18545–18550. doi: 10.1073/pnas.1010978107. [DOI] [PMC free article] [PubMed] [Google Scholar]