

Abstract
Association studies suggest that thyroid hormone receptor β (TRβ) could function as a tumor suppressor in breast cancer development, but unequivocal evidence is still lacking. To understand the role of TRβ in breast tumor development, we adopted the gain-of-function approach by stably expressing the THRB gene in a human breast cancer cell line, MCF-7 (MCF-7-TRβ). Parental MCF-7 cells express the estrogen receptor, but not TRs. MCF-7 cells, stably expressing only the selectable marker, the Neo gene, were also generated as control for comparison (MCF-7-Neo cells). Cell-based studies indicate that the estrogen (E2)-dependent growth of MCF-7 cells was inhibited by the expression of TRβ in the presence of the thyroid hormone (T3). In a xenograft mouse model, large tumors rapidly developed after inoculation of MCF-7-Neo cells in athymic mice. In contrast, markedly smaller tumors (98% smaller) were found when MCF-7-TRβ cells were inoculated in athymic mice, indicating that TRβ inhibited the E2-dependent tumor growth of MCF-7 cells. Further detailed molecular analysis showed that TRβ acted to activate apoptosis and decrease proliferation of tumor cells, resulting in inhibition of tumor growth. The TRβ-mediated inhibition of tumor growth was elucidated via down-regulation of the JAK-STAT-cyclin D pathways. This in vivo evidence shows that TRβ could act as a tumor suppressor in breast tumorigenesis. The present study provides new insights into the role of TR in breast cancer.
Keywords: Thyroid hormone receptor beta, tumor suppressor, tumorigenesis, STAT signaling, MCF-7 cells
Introduction
Thyroid hormone receptors (TRs) are ligand-dependent transcription factors that mediate the biological actions of the thyroid hormone (T3) in development, growth, differentiation, and maintenance of metabolic homeostasis [1,2]. Two human TR genes, THRA and THRB, located on different chromosomes, encode three major T3-binding TR isoforms. Although there has been significant progress in the understanding of molecular mechanisms by which TRs act to maintain normal physiological cellular functions, their role in human carcinogenesis is less well understood. Evidence suggesting TRs could be involved in human carcinogenesis came from the discoveries of mutated TRs in human hepatocellular carcinoma, renal clear cell carcinoma, breast cancer, pituitary tumor, and thyroid cancer [3-10]. The loss of normal expression of the THRB gene located on chromosome 3p due to truncation or deletion was also observed in many malignancies including lung, melanoma, breast, head and neck, renal cell, uterine cervical, ovarian, and testicular tumors [10-13]. Decreased THRB expression by promoter hypermethylation has been reported in human breast cancer, lung cancer, and thyroid carcinoma [14-16]. These findings raise the possibility that TRβ could act as a tumor suppressor in human cancers.
Although the function of most members of the nuclear receptor superfamily in breast tumor biology has been documented [17], much less is known about TRs. A low circulating thyroid hormone level (hypothyroidism) has been proposed to favor mammary hyperplasia in rodents and the development of breast tumors [18]. In addition, loss of TRß expression by gene deletion or silencing, or production of abnormal TRß proteins due to THRB mutations, has been reported in breast tumors. These correlative observations suggest that TRß could act as a tumor suppressor [6,19,20]. However, how TR can function as a suppressor in tumor development and/or progression of breast tumors is not clear.
Accordingly, in the present study, we adopted the gain-of-function approach by expression of the THRB gene in human MCF-7 cells. MCF-7 is a breast cancer cell line derived from a patient with invasive breast ductal carcinoma [21]. MCF-7 cells express the estrogen receptor, but lack TRs. Proliferation and tumorigenesis of MCF-7 cells are responsive to estrogen stimulation (E2). We therefore stably expressed the THRB gene in the MCF-7 cell line and evaluated the effects of the expression of TRβ on cell proliferation and tumor development in mouse xenograft models. We found that the xenograft tumor development was markedly inhibited in MCF-7 cells by the expression of TRβ. These inhibitory responses resulted from decreasing cell proliferation via activation of apoptotic activity and down-regulation of JAK-STAT signaling pathway. Thus, TRβ could act as a tumor suppressor in E2-mediated tumorigenesis of MCF-7 cells.
Materials and methods
Generation of MCF-7 cell lines stably expressing TRβ
MCF-7 cells were cultured in DMEM media containing 10% fetal bovine serum (FBS). Establishment of MCF-7 cells stably expressing either the human THRB gene or the control gene (Neo) was done much the same as described previously for HeLa cells [22]. Briefly, MCF-7 cells were transfected with the expression plasmid containing cDNA encoding Flag-Hemagglutinin-TRβ (FH-TRβ) or the empty vector containing only the cDNA for the selector marker, the Neo gene. After transfection, cells were selected with 400 μg/ml G418 (Invitrogen, Carlsbad, CA) for 2 weeks. G418-resistant colonies expressing FH-TRβ were expanded for subsequent experiments. The expression of FH-TRβ protein was verified by Western blot analysis using monoclonal anti-TRβ antibody (J53) [23].
Cell proliferation assay
Control (Neo) and TRβ stably expressing MCF-7 cells (MCF-7-TRβ) (5 x 104 cells per well) were plated in 6-well plates (in triplicates) and cultured for 3 days in the presence of T3 (100 nM) and/or E2 (10 nM) in phenol red free DMEM medium. Cell proliferation was measured after treatment with hormone for 72 hours using a cell and particle counter (Beckmann Coulter, Indianapolis, IN).
In vivo mouse xenograft study
The protocols for the use and care of the animals in the present studies were approved by the National Cancer Institute Animal Care and Use Committee. Six-week-old female athymic NCr-nu/nu mice were obtained from the NCI-Frederick animal facility. Administration of 17β-estradiol (E2, 10 μg/ml) in the drinking water was started for one week preceded the injection of cells [24]. Two clones each of the control MCF-7-Neo cells and TRβ-expressing cells (MCF-7-TRβ) (5 x 106 cells) in 200 μl suspension mixed with Matrigel basement membrane matrix (BD Biosciences, San Jose, CA) were inoculated subcutaneously into the right flank of mice. The tumor size was measured with calipers weekly until it reached ~2 cm in diameter. The mice were then sacrificed and the tumors dissected. The tumor volume was calculated as L x W x H x 0.5236.
Western blot analysis
Lysates from the tumors were prepared using a buffer containing 50 mM Tris, 250 mM NaCl, 5 mM EDTA, 0.5% NP-40, a cocktail of protease inhibitors (Roche Diagnostics cat. 11873580001), and cocktail of phosphatase inhibitors (Thermo Ltd: cat. 78420). After a 10-minute incubation on ice, cell lysates were centrifuged at 13,000 rpm for 10 minutes, and the supernatants were collected. Western blot analysis was carried out similarly as described previously [25]. The primary antibodies used were: antibodies against-TR [J53; 2 μg/ml [23], phospho-Jak2 (Tyr1007/1008) (1:500, Cell Signaling Technology cat. #3776), Jak2 (1:1000, Cell Signaling Technology cat. #3230), anti-phospho-STAT3 (Tyr705) (1:500, Cell Signaling Technology cat. #9131), STAT3 (1:1000, Cell Signaling Technology cat. #9132), phospho-STAT5 (Tyr694) (1:500, Cell Signaling Technology cat. #9359), STAT5 (1:1000, Cell Signaling Technology cat. #9363), phospho-Rb (Ser807/811) (1:500, Cell Signaling Technology cat. #9308), Rb (1:200, Santa Cruz Cat#. SC-50), GAPDH (1:1000, Cell Signaling Technology cat. #2118). Band intensities were quantified by using NIH ImageJ software version 1.44 (Wayne Rasband, National Institutes of Health, Bethesda, MD).
Immunohistochemical analysis
Immunohistochemistry was performed on formalin-fixed paraffin tumor sections, as previously described [26]. Primary antibodies used were anti-Ki-67 antibody (dilution 1:300; Thermo Scientific, Fremont, CA, USA; #RB-9043-P0) and anti-cleaved caspase-3 antibodies (1:300 dilution; Cell Signaling, Cat#: 9661). Staining was developed with 3,30 diaminobenzidine (DAB) using the DAB substrate kit for peroxidase (Vector Laboratories, Burlingame, CA, USA, SK-4100). For quantitative analysis Ki-67 or cleaved caspase-3 positive cells were counted by using NIH ImageJ software version 1.44 (Wayne Rasband, National Institutes of Health, Bethesda, MD).
Statistical analysis
All data are expressed as mean ± the standard error of the mean (SEM). Significant differences between groups were calculated using Student’s t-test with the use of GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA). p<0.05 is considered statistically significant.
Results
TRβ inhibits growth of MCF-7 cells
To elucidate the role of TRβ in tumorigenesis of MCF-7 cells, we generated MCF-7 cells stably expressing TRβ (TRβ#18 and TRβ#19) or only the neomycin as controls (Neo#1 and Neo#12) The expression of TRβ proteins in MCF-7 TRβ#18 and TRβ#19 was confirmed by Western blot analysis (Figure 1A, lanes 3 & 4), showing similar amounts in the two clones, whereas no TRβ was detected in control Neo cells (Figure 1A, lanes 1 & 2 for Neo#1 and 12, respectively). The lower band shows the loading control using glycerolaldehyde 3-phosphate dehydrogenase (GAPDH).
Figure 1.
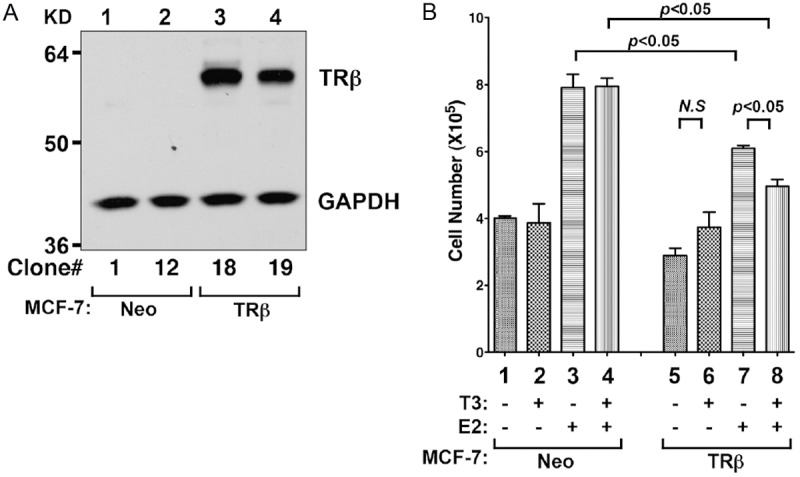
TRβ inhibits the proliferation of MCF-7 cells. A: TRβ was stably expressed in MCF-7 cell (lanes 3 and 4), but not in control MCF-7-Neo cells (lanes 1 and 2). The Western blot analysis was carried out as described in Materials and Methods. Two representative results are shown using two independent clones: MCF-7-Neo#1 (lane 1) and MCF-7-Neo#12 (lane 2) and MCF-7-TRβ#18 (lane 3) and MCF-7-TRβ#19 (lane 19). B: E2-dependent cell proliferation of MCF7 was inhibited by the expression of TRβ in the presence of T3. Cells with or without hormone (E2 and T3) treatment are as marked. Cell growth was analyzed as described in Materials and Methods. Data are expressed as mean ± standard error (SE) (n=3).
To ascertain whether the expressed TRβ in MCF-7 cells was functional, we evaluated the cell growth of MCF-7 cells with or without the expression of TRβ. Figure 1B shows that consistent with previous findings, the cell growth of MCF-7 cells (Neo control, no TRβ) was E2-dependent (compare bar 3 to 1). As expected, T3 had no significant effect on cell growth of MCF-7 cells with TRβ (compare bar 2 to 1; bar 4 to 3). However, while expression of TRβ had no significant effect on cell growth in the absence of E2 (compare bar 6 to 5), the expression of TRβ inhibited the E2-dependent cell growth in the presence of T3 and E2 (compare bar 8 to 7). The data shown in Figure 1 indicate that TRβ expressed in MCF-7 cells is functional and that TRβ-expressing cells are suitable model cell lines to ascertain the role of TRβ in tumorigenesis in vivo using xenograft models.
TRβ expression inhibits tumor growth of MCF-7 cells in mouse xenograft models
To ascertain the role of TRβ in E2-dependent tumorigenesis of MCF-7 cells, we injected Neo#1, Neo#12, TRβ#18 and TRβ#19 subcutaneously into the right flank of athymic nude mice. As shown in Figure 2A, the tumor volume induced by MCF-7-TRβ#18 and MCF-7-TRβ#19 was significantly less than that induced by control MCF-7 (Neo#1 and Neo#12) cells (p<0.05 and p<0.05, respectively). The quantitative comparison, illustrated in Figure 2B, showed that the expression of TRβ blocked the tumor growth induced by MCF-7 cell by 98%. These findings indicate that TRβ could act as a tumor suppressor in E2-dependent tumorigenesis of MCF-7 cells in vivo.
Figure 2.
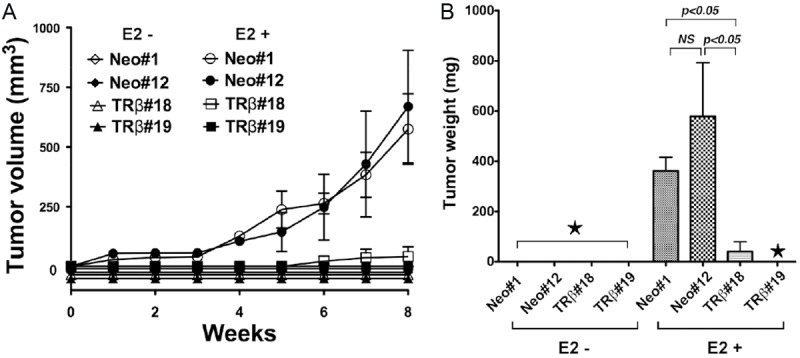
E2-dependent xenograft tumors induced by MCF-7 cells are blocked by the expression of TRβ. A: Two clones each of MCF-7-Neo and MCF-7-TRβ were inoculated into athymic mice without or with E2 as described in Materials and Methods. No tumors were induced by MCF-7 cells in the absence of E2 treatment. B: Tumor weight was determined at the endpoint for the MCF-7-Neo (clones #1 and #12) and MCF-7-TRβ (clones #18 and #19). ★represents no tumors. The data are expressed as mean ± SE (n=3). The p values are indicated.
TRβ expression reduces tumor growth by activating apoptosis of tumor cells
To understand how TRβ expression inhibited tumor growth in vivo, we evaluated the pathohistological features of tumors induced by MCF-7 cells without (Figure 3A-a, 3A-b) or with (Figure 3A-c, 3A-d) the expression of TRβ. H & E-stained cells revealed that the tumors induced by MCF-7-Neo cells demonstrated features typical of an aggressive malignant phenotype including marked nuclear pleomorphism, minimal apoptosis, and numerous mitotic figures (M marked by arrows; Figure 3A-a, 3A-b). Brisk mitotic activity is indicative of rapid unregulated cell cycling and tumor expansion, as expected for a highly malignant phenotype. In contrast, tumors with stably expressed TRβ (panels c and d) showed limited mitotic figures and numerous examples of apoptotic cell death, with cellular and nuclear fragments (Figure 3A-c, 3A-d; marked by arrows). These alterations are consistent with an interruption of cell cycling and induction of the apoptotic cascade, with resultant slowed tumor growth.
Figure 3.
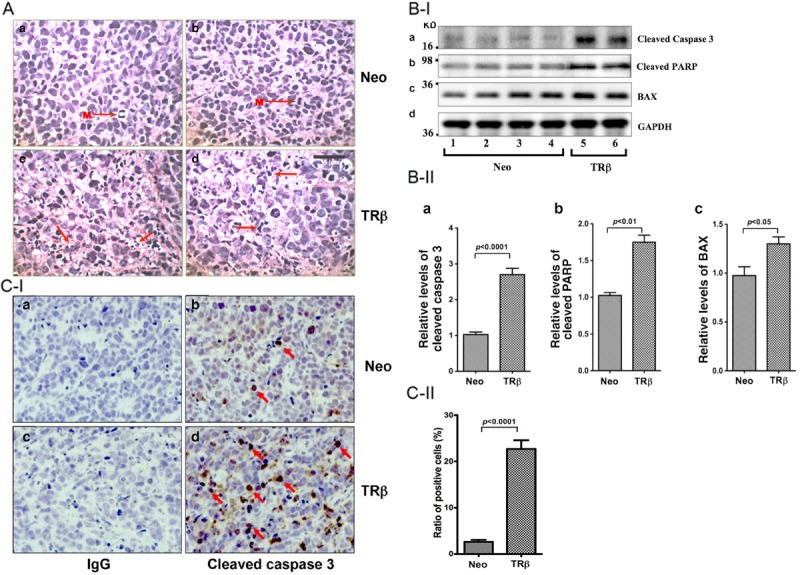
Induction of apoptosis by TRβ in tumors derived from MCF-7-TRβ cells. A: Representative histological features of hematoxylin and eosin (H & E) stained sections of tumors derived from MCF-7-Neo#1 (panel a), MCF-7-Neo#12 cells (panel b), MCF-7-TRβ#18 (panel c and d). Pronounced mitoses (M) were detected in the Neo tumors (a and b) and apoptotic remnants were detected in TRβ tumors (c and d, shown by arrows). Magnification x 300. B-I: Western blot analysis of apoptotic key regulators in tumors. Tumors were excised from the injection sites (hind flanks) of athymic nude mice, and the Western blot analysis was carried as described in Materials and Methods. Lanes 1-4 were apoptotic key regulators detected in extracts from tumors derived from MCF-7-Neo cells, and lanes 5-6 were from tumors derived from MCF-7-TRβ cells. The proteins (cleaved caspase 3, cleaved PARP, BAX, and GAPDH) detected are as marked. B-II: The band intensities of the proteins detected in B-I were quantified and compared. The data are shown as mean ± SE (n=2-4). C-I: Analysis of the protein expression of cleaved caspase 3 by immunohistochemistry. Sections of tumors derived from MCF-7-Neo cells (panels a and b) and MCF-7-TRβ cells (panels c and d) were treated with control anti-IgG (panel a and c) or with anti-cleaved caspase 3 antibodies (panels b and d) as described in Materials and Methods. The cleaved caspase 3-positively stained cells are indicated by arrows. C-II: The cleaved caspase 3-positive cells were counted and expressed as percentage of cleaved caspase 3-positive cells versus total cells. The data are expressed as mean ± SE (n=3). The p values are shown.
These histological findings prompted us to evaluate how the expression of TRβ altered the expression of key regulators in apoptotic pathways in tumors induced by MCF-7-Neo cells. Lanes 5 and 6 in Figure 3B-I show more protein abundance of cleaved caspase 3 (panel a, lanes 5 and 6), cleaved poly-ADP ribose polymerase (PARP) (panel b), and Bcl-associated X protein (BAX) (panel c) in tumors induced by MCF-7-TRβ cells than in control MCF-7-Neo cells (lanes 1-4). The band intensities shown in Figure 3B-I were quantified and summarized in Figure 3B-II, indicating significant increased protein levels of these apoptotic key regulators. Increased cleaved caspase 3 and cleaved PARP are indicative of elevated apoptotic activity [27,28] and elevated BAX acts to promote apoptosis via activating caspases [29]. These data indicated that TRβ could act to increase the expression of BAX to promote apoptosis via activating caspases.
That the cleaved caspase 3 was elevated was further confirmed by immunohistochemical analysis. Only a few cells in the MCF-7-Neo-tumors were stained with anti-cleaved caspase 3 antibodies (Figure 3C-I, panel b); whereas more cells in the MCF-7-TRβ-tumors were intensely stained with anti-cleaved caspase 3 antibodies (Figure 3C-I, panel d). Panels a and c were the negative controls using IgG only. The positively stained cells were counted and the quantitative data are shown in Figure 3C-II, indicating an 80% increase in cells stained with anti-cleaved caspase 3 antibodies from cells derived from MCF-7-TRβ tumors. Taken together, these data indicated that TRβ could act to increase the expression of BAX to promote apoptosis via activating caspases.
TRβ expression inhibits tumor cell proliferation by attenuating JAK-STAT signaling
In addition to promoting increased apoptosis, TRβ expressed in MCF-7 cells could also act to decrease cell proliferation, resulting in the decreased tumor growth shown in Figure 2. We therefore first ascertained whether cell proliferation was affected in tumors induced by MCF-7 expressing TRβ by staining the nuclear proliferation marker, Ki-67, using immunohistochemical analysis. The intense staining of Ki-67 in cells was clearly visible in tumors induced by MCF-7-Neo cells (arrows, Figure 4A-I-b). In contrast, there were markedly fewer cells stained with Ki-67 in tumors induced by MCF-7-TRβ cells (Figure 4A-I-d) than those induced by MCF-7-Neo cells. Panels a and c show the negative controls in which no primary antibodies were used. The Ki-67 positively stained cells were counted and the quantitative data are shown in Figure 4A-II, revealing a 65% reduction of Ki-67 stained cells. These data indicate that the expression of TRβ results in the inhibition of tumor cell proliferation. Thus, the inhibition of tumor growth results from increased apoptosis as well as decreased proliferation of tumor cells.
Figure 4.
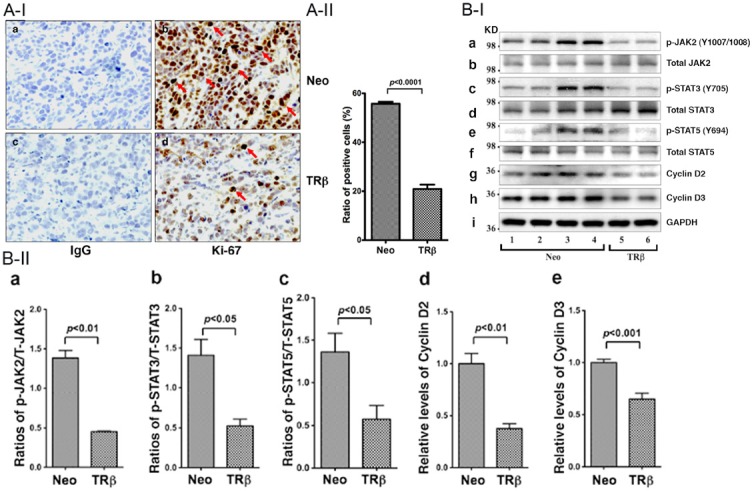
Inhibition of cell proliferation by TRβ in tumors derived from MCF-7-TRβ cells. A-I: Immunohistochemical analysis of protein abundance of the nuclear proliferation marker, Ki-67, in tumors. Sections of tumors derived from MCF-7-Neo cells (panels a and b) and MCF-7-TRβ cells (panels c and d) were treated with control anti-IgG (panel a and c) or with anti-Ki-67 antibodies (panels b and d) as described in Materials and Methods. The Ki-67 positively stained cells are indicated by arrows. A-II: The Ki-67-positive cells were counted and expressed as percentage of Ki-67-positive cells versus total cells. The data are expressed as mean ± SE (n=3). B-I: Western blot analysis of key regulators and effectors in JAK2-STAT signaling pathway in tumors. Tumors were excised from the injection sites (hind flanks) of athymic nude mice, and the Western blot analysis was carried as described in Materials and Methods. Lanes 1-4 were key regulators and effectors detected in extracts from tumors derived from MCF-7-Neo cells, and lanes 5-6 were from tumors derived from MCF-7-TRβ cells. The proteins (p-JAK, total JAK, p-STAT3, total STAT3, p-STAT5, total STAT5, cyclin D2, cyclin D3, and GAPDH) detected are as marked. B-II: The band intensities of the proteins detected in B-I were quantified and compared. The data are shown as mean ± SE (n=2-4). The p values are shown.
To elucidate the pathways by which TRβ acted to inhibit cell proliferation, we hypothesized that this anti-proliferative effect could be mediated through the Janus kinase-signal transducers and activators of transcription (JAK-STAT)-signaling pathway. This hypothesis was based on the findings that E2 up-regulates JAK2 expression in MCF-7 cells and also activates STAT transcriptional activity [30,31]. Moreover, constitutively activated STAT3 is frequently found in breast cancer cell lines and patients with advanced breast disease [32-34]. We therefore investigated whether the JAK-STAT signaling pathway was affected by the expression of TRβ (Figure 4). We found that the activated JAK2-STAT3 signaling in MCF-7-Neo cell-induced xenografted tumors (lanes 1-4, Figure 4B-I) was de-activated as evidenced by the decreased phosphorylated JAK (Y1007/1008) (lanes 5-6, panel a), phosphorylated STAT3 (Y705) (lanes 5-6, panel c), and phosphorylated STAT5 (Y694) (lanes 5-6, panel e). However, no apparent changes in the total JAK2 (panel b), STAT3 (panel d), or STAT5 (panel f) were detected, indicating the signaling was via the phosphorylation cascades. The decreased signaling led to the lowered protein abundance of key cell cycle regular cyclin D1 (compare lanes 1-4 with lanes 5-6, panel g) and D2 (compare lanes 1-4 with lanes 5-6, panel h). The changes are clearly shown in the quantitative evaluation of the band intensities of the Western blots (Figure 4B-II). These results support our hypothesis that the TRβ-mediated decreased proliferation of tumor cells induced by MCF-7-Neo cells is, at least in part, acting through the JAK2-STAT signaling.
Discussion
Previous studies demonstrating a close association of reduced expression and somatic mutations of the THRB gene with several human cancers support the hypothesis that TRβ could act as a tumor suppressor [3-10]. Direct in vivo evidence to support the tumor suppressor role of TRβ in breast cancer came from the studies of a mouse model harboring a dominant negative TRβ mutant (denoted as PV) in both alleles (ThrbPV/PV mice) with haplodeficiency in the phosphatase and tensin homologue deleted from chromosome 10 (Pten; ThrbPV/PVPten+/- mice). The ThrbPV mutation further markedly augments the risk of mammary hyperplasia in Pten+/- mice, which are highly susceptible to mammary tumor development [35]. Importantly, the ThrbPV mutation increases the activity of STAT5 to increase cell proliferation and the expression of the STAT5 target gene encoding β-casein in the mammary gland [36]. Moreover, cell-based studies in T47D cells, a breast cancer cell line, also showed that T3 represses STAT5 signaling in TRβ-expressing cells through decreasing STAT5-mediated transcription activity and target gene expression, whereas sustained STAT5 signaling was observed in TRβPV-expressing cells [36]. These findings clearly support the idea that TRβ could function as a tumor suppressor in breast cancer development in vivo and in vitro.
In the present study, we adopted the gain-of-function approach to further explore the tumor suppressor role of TRβ in an E2-dependent breast cancer cell line. The expression of TRβ in MCF-7 cells led to inhibition of in vivo tumor development by induced apoptosis and decreased proliferation of tumor cells. We further elucidated the pathways by which TRβ blocked the E2-stimulated cell proliferation by attenuating the JAK-STAT signaling. Thus, the present study provides additional in vivo evidence to support the idea that TRβ could act as a tumor suppressor in breast cancer development and progression.
At present, the detailed mechanisms by which TRβ antagonized the aberrantly activated JAK-STAT signaling in MCF-7 cells are not clear. However, studies have shown that cyclin D1 is the downstream target of JAK-STAT activation, leading to increased cell proliferation of MCF-7 cells and other cancer cells [37-41]. In the present studies, we found that protein abundance of cyclin D2 and D3 was lowered in tumors derived from MCF-7-TRβ as a result of the TRβ-mediated attenuation of JAK-STAT signaling (see Figure 4). Previously, we have shown that TRβ represses the STAT5 signaling through decreasing STAT5-mediated transcriptional activity [36]. Thus, one mechanism by which TRβ antagonized the activated JAK-STAT signaling in MCF-7 cells was mediated at the transcriptional level. Other potential mechanisms that could underlie the TRβ-mediated anti-proliferative action await further studies in the future.
Breast cancer is the most common neoplasia and the second-leading cause of cancer deaths in women in western countries [42]. Genetic mutations, either inherited or sporadic, as well as dysregulation of ovarian hormone signaling are known to contribute to the development and progression of breast cancers. However, many studies have investigated the potential association of hypothyroid or hyperthyroid disorders with breast cancer in the last decades, but without clear conclusions [43-47]. The reasons for the controversial findings among these studies are not immediately apparent. TRs have been shown to express in normal mammary gland [48] and breast tumors [49,50]. However, silencing of the THRB gene by promoter hypermethylation, or the expression of truncated TRβ, has been reported in human breast cancers [6,19]. Dysregulation of the TRs was reported to trigger breast cancer development [50]. These findings suggested the critical role of TRs in breast cancer development. The present studies demonstrate that in the presence of T3, the expression of the THRB gene blocked tumor development in MCF-7 cells in vivo. Thus, our findings would support T3’s being considered therapeutically favorable for ER-positive breast cancer.
Acknowledgments
The present research was supported by the Intramural Research Program at the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
References
- 1.Yen PM. Physiological and molecular basis of thyroid hormone action. Physiol Rev. 2001;81:1097–1142. doi: 10.1152/physrev.2001.81.3.1097. [DOI] [PubMed] [Google Scholar]
- 2.Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010;31:139–170. doi: 10.1210/er.2009-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin KH, Shieh HY, Chen SL, Hsu HC. Expression of mutant thyroid hormone nuclear receptors in human hepatocellular carcinoma cells. Mol Carcinog. 1999;26:53–61. doi: 10.1002/(sici)1098-2744(199909)26:1<53::aid-mc7>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 4.Puzianowska-Kuznicka M, Nauman A, Madej A, Tanski Z, Cheng S, Nauman J. Expression of thyroid hormone receptors is disturbed in human renal clear cell carcinoma. Cancer Lett. 2000;155:145–152. doi: 10.1016/s0304-3835(00)00416-x. [DOI] [PubMed] [Google Scholar]
- 5.Kamiya Y, Puzianowska-Kuznicka M, McPhie P, Nauman J, Cheng SY, Nauman A. Expression of mutant thyroid hormone nuclear receptors is associated with human renal clear cell carcinoma. Carcinogenesis. 2002;23:25–33. doi: 10.1093/carcin/23.1.25. [DOI] [PubMed] [Google Scholar]
- 6.Silva JM, Dominguez G, Gonzalez-Sancho JM, Garcia JM, Silva J, Garcia-Andrade C, Navarro A, Munoz A, Bonilla F. Expression of thyroid hormone receptor/erbA genes is altered in human breast cancer. Oncogene. 2002;21:4307–4316. doi: 10.1038/sj.onc.1205534. [DOI] [PubMed] [Google Scholar]
- 7.Safer JD, Colan SD, Fraser LM, Wondisford FE. A pituitary tumor in a patient with thyroid hormone resistance: a diagnostic dilemma. Thyroid. 2001;11:281–291. doi: 10.1089/105072501750159750. [DOI] [PubMed] [Google Scholar]
- 8.Ando S, Sarlis NJ, Oldfield EH, Yen PM. Somatic mutation of TRbeta can cause a defect in negative regulation of TSH in a TSH-secreting pituitary tumor. J Clin Endocrinol Metab. 2001;86:5572–5576. doi: 10.1210/jcem.86.11.7984. [DOI] [PubMed] [Google Scholar]
- 9.Puzianowska-Kuznicka M, Krystyniak A, Madej A, Cheng SY, Nauman J. Functionally impaired TR mutants are present in thyroid papillary cancer. J Clin Endocrinol Metab. 2002;87:1120–1128. doi: 10.1210/jcem.87.3.8296. [DOI] [PubMed] [Google Scholar]
- 10.Kim WG, Cheng SY. Thyroid hormone receptors and cancer. Biochim Biophys Acta. 2013 Jul;1830:3928–36. doi: 10.1016/j.bbagen.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leduc F, Brauch H, Hajj C, Dobrovic A, Kaye F, Gazdar A, Harbour JW, Pettengill OS, Sorenson GD, van den Berg A, et al. Loss of heterozygosity in a gene coding for a thyroid hormone receptor in lung cancers. Am J Hum Genet. 1989;44:282–287. [PMC free article] [PubMed] [Google Scholar]
- 12.Sisley K, Curtis D, Rennie IG, Rees RC. Loss of heterozygosity of the thyroid hormone receptor B in posterior uveal melanoma. Melanoma Res. 1993;3:457–461. doi: 10.1097/00008390-199311000-00009. [DOI] [PubMed] [Google Scholar]
- 13.Chen LC, Matsumura K, Deng G, Kurisu W, Ljung BM, Lerman MI, Waldman FM, Smith HS. Deletion of two separate regions on chromosome 3p in breast cancers. Cancer Res. 1994;54:3021–3024. [PubMed] [Google Scholar]
- 14.Ling Y, Xu X, Hao J, Ling X, Du X, Liu X, Zhao X. Aberrant methylation of the THRB gene in tissue and plasma of breast cancer patients. Cancer Genet Cytogenet. 2010;196:140–145. doi: 10.1016/j.cancergencyto.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 15.Iwasaki Y, Sunaga N, Tomizawa Y, Imai H, Iijima H, Yanagitani N, Horiguchi K, Yamada M, Mori M. Epigenetic inactivation of the thyroid hormone receptor beta1 gene at 3p24.2 in lung cancer. Ann Surg Oncol. 2010;17:2222–2228. doi: 10.1245/s10434-010-0956-9. [DOI] [PubMed] [Google Scholar]
- 16.Joseph B, Ji M, Liu D, Hou P, Xing M. Lack of mutations in the thyroid hormone receptor (TR) alpha and beta genes but frequent hypermethylation of the TRbeta gene in differentiated thyroid tumors. J Clin Endocrinol Metab. 2007;92:4766–4770. doi: 10.1210/jc.2007-0812. [DOI] [PubMed] [Google Scholar]
- 17.Conzen SD. Minireview: nuclear receptors and breast cancer. Mol Endocrinol. 2008;22:2215–2228. doi: 10.1210/me.2007-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mittra I. Mammotropic effect of prolactin enhanced by thyroidectomy. Nature. 1974;248:525–526. doi: 10.1038/248525a0. [DOI] [PubMed] [Google Scholar]
- 19.Li Z, Meng ZH, Chandrasekaran R, Kuo WL, Collins CC, Gray JW, Dairkee SH. Biallelic inactivation of the thyroid hormone receptor beta1 gene in early stage breast cancer. Cancer Res. 2002;62:1939–1943. [PubMed] [Google Scholar]
- 20.Ali IU, Lidereau R, Callahan R. Presence of two members of c-erbA receptor gene family (c-erbA beta and c-erbA2) in smallest region of somatic homozygosity on chromosome 3p21-p25 in human breast carcinoma. J Natl Cancer Inst. 1989;81:1815–1820. doi: 10.1093/jnci/81.23.1815. [DOI] [PubMed] [Google Scholar]
- 21.Soule HD, Vazguez J, Long A, Albert S, Brennan M. A human cell line from a pleural effusion derived from a breast carcinoma. J Natl Cancer Inst. 1973;51:1409–1416. doi: 10.1093/jnci/51.5.1409. [DOI] [PubMed] [Google Scholar]
- 22.Ying H, Furuya F, Zhao L, Araki O, West BL, Hanover JA, Willingham MC, Cheng SY. Aberrant accumulation of PTTG1 induced by a mutated thyroid hormone beta receptor inhibits mitotic progression. J Clin Invest. 2006;116:2972–2984. doi: 10.1172/JCI28598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin KH, Willingham MC, Liang CM, Cheng SY. Intracellular distribution of the endogenous and transfected beta form of thyroid hormone nuclear receptor visualized by the use of domain-specific monoclonal antibodies. Endocrinology. 1991;128:2601–2609. doi: 10.1210/endo-128-5-2601. [DOI] [PubMed] [Google Scholar]
- 24.Toma S, Emionite L, Scaramuccia A, Ravera G, Scarabelli L. Retinoids and human breast cancer: in vivo effects of an antagonist for RAR-alpha. Cancer Lett. 2005;219:27–31. doi: 10.1016/j.canlet.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 25.Guigon CJ, Zhao L, Lu C, Willingham MC, Cheng SY. Regulation of beta-catenin by a novel nongenomic action of thyroid hormone beta receptor. Mol Cell Biol. 2008;28:4598–4608. doi: 10.1128/MCB.02192-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guigon CJ, Zhao L, Willingham MC, Cheng SY. PTEN deficiency accelerates tumour progression in a mouse model of thyroid cancer. Oncogene. 2009;28:509–517. doi: 10.1038/onc.2008.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 28.Soldani C, Lazze MC, Bottone MG, Tognon G, Biggiogera M, Pellicciari CE, Scovassi AI. Poly(ADP-ribose) polymerase cleavage during apoptosis: when and where? Exp Cell Res. 2001;269:193–201. doi: 10.1006/excr.2001.5293. [DOI] [PubMed] [Google Scholar]
- 29.Bernal NP, Stehr W, Coyle R, Erwin CR, Warner BW. Epidermal growth factor receptor signaling regulates Bax and Bcl-w expression and apoptotic responses during intestinal adaptation in mice. Gastroenterology. 2006;130:412–423. doi: 10.1053/j.gastro.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 30.Gupta N, Grebhardt S, Mayer D. Janus kinase 2--a novel negative regulator of estrogen receptor alpha function. Cell Signal. 2012;24:151–161. doi: 10.1016/j.cellsig.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 31.Bjornstrom L, Sjoberg M. Signal transducers and activators of transcription as downstream targets of nongenomic estrogen receptor actions. Mol Endocrinol. 2002;16:2202–2214. doi: 10.1210/me.2002-0072. [DOI] [PubMed] [Google Scholar]
- 32.Garcia R, Yu CL, Hudnall A, Catlett R, Nelson KL, Smithgall T, Fujita DJ, Ethier SP, Jove R. Constitutive activation of Stat3 in fibroblasts transformed by diverse oncoproteins and in breast carcinoma cells. Cell Growth Differ. 1997;8:1267–1276. [PubMed] [Google Scholar]
- 33.Watson CJ, Miller WR. Elevated levels of members of the STAT family of transcription factors in breast carcinoma nuclear extracts. Br J Cancer. 1995;71:840–844. doi: 10.1038/bjc.1995.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bowman T, Broome MA, Sinibaldi D, Wharton W, Pledger WJ, Sedivy JM, Irby R, Yeatman T, Courtneidge SA, Jove R. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc Natl Acad Sci U S A. 2001;98:7319–7324. doi: 10.1073/pnas.131568898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stambolic V, Tsao MS, Macpherson D, Suzuki A, Chapman WB, Mak TW. High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in pten+/- mice. Cancer Res. 2000;60:3605–3611. [PubMed] [Google Scholar]
- 36.Guigon CJ, Kim DW, Willingham MC, Cheng SY. Mutation of thyroid hormone receptor-beta in mice predisposes to the development of mammary tumors. Oncogene. 2011;30:3381–3390. doi: 10.1038/onc.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Catalano S, Giordano C, Rizza P, Gu G, Barone I, Bonofiglio D, Giordano F, Malivindi R, Gaccione D, Lanzino M, De Amicis F, Ando S. Evidence that leptin through STAT and CREB signaling enhances cyclin D1 expression and promotes human endometrial cancer proliferation. J Cell Physiol. 2009;218:490–500. doi: 10.1002/jcp.21622. [DOI] [PubMed] [Google Scholar]
- 38.Sakamoto K, Creamer BA, Triplett AA, Wagner KU. The Janus kinase 2 is required for expression and nuclear accumulation of cyclin D1 in proliferating mammary epithelial cells. Mol Endocrinol. 2007;21:1877–1892. doi: 10.1210/me.2006-0316. [DOI] [PubMed] [Google Scholar]
- 39.Mishra R, Das BR. Activation of STAT 5-cyclin D1 pathway in chewing tobacco mediated oral squamous cell carcinoma. Mol Biol Rep. 2005;32:159–166. doi: 10.1007/s11033-005-0754-9. [DOI] [PubMed] [Google Scholar]
- 40.Zhang F, Li C, Halfter H, Liu J. Delineating an oncostatin M-activated STAT3 signaling pathway that coordinates the expression of genes involved in cell cycle regulation and extracellular matrix deposition of MCF-7 cells. Oncogene. 2003;22:894–905. doi: 10.1038/sj.onc.1206158. [DOI] [PubMed] [Google Scholar]
- 41.Brockman JL, Schroeder MD, Schuler LA. PRL activates the cyclin D1 promoter via the Jak2/Stat pathway. Mol Endocrinol. 2002;16:774–784. doi: 10.1210/mend.16.4.0817. [DOI] [PubMed] [Google Scholar]
- 42.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 43.Tosovic A, Bondeson AG, Bondeson L, Ericsson UB, Manjer J. Triiodothyronine levels in relation to mortality from breast cancer and all causes: a population-based prospective cohort study. Eur J Endocrinol. 2013;168:483–490. doi: 10.1530/EJE-12-0564. [DOI] [PubMed] [Google Scholar]
- 44.Ditsch N, Liebhardt S, Von Koch F, Lenhard M, Vogeser M, Spitzweg C, Gallwas J, Toth B. Thyroid function in breast cancer patients. Anticancer Res. 2010;30:1713–1717. [PubMed] [Google Scholar]
- 45.Tosovic A, Bondeson AG, Bondeson L, Ericsson UB, Malm J, Manjer J. Prospectively measured triiodothyronine levels are positively associated with breast cancer risk in postmenopausal women. Breast Cancer Res. 2010;12:R33. doi: 10.1186/bcr2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Michalaki V, Kondi-Pafiti A, Gennatas S, Antoniou A, Primetis H, Gennatas C. Breast cancer in association with thyroid disorders. J BUON. 2009;14:425–428. [PubMed] [Google Scholar]
- 47.Kuijpens JL, Nyklictek I, Louwman MW, Weetman TA, Pop VJ, Coebergh JW. Hypothyroidism might be related to breast cancer in post-menopausal women. Thyroid. 2005;15:1253–1259. doi: 10.1089/thy.2005.15.1253. [DOI] [PubMed] [Google Scholar]
- 48.Hayden TJ, Forsyth IA. Thyroid hormone binding in rat mammary gland [proceedings] . J Endocrinol. 1977;75:38P–39P. [PubMed] [Google Scholar]
- 49.Alberg AJ, Helzlsouer KJ. Epidemiology, prevention, and early detection of breast cancer. Curr Opin Oncol. 1997;9:505–511. doi: 10.1097/00001622-199711000-00003. [DOI] [PubMed] [Google Scholar]
- 50.Conde I, Paniagua R, Zamora J, Blanquez MJ, Fraile B, Ruiz A, Arenas MI. Influence of thyroid hormone receptors on breast cancer cell proliferation. Ann Oncol. 2006;17:60–64. doi: 10.1093/annonc/mdj040. [DOI] [PubMed] [Google Scholar]